

Abstract
Filamentous phage as a bacteria-specific virus can be conjugated with an anti-cancer drug and has been proposed to serve as a carrier to deliver drugs to cancer cells for targeted therapy. However, how cell-targeting filamentous phage alone affects cancer cell biology is unclear. Phage libraries provide an inexhaustible reservoir of new ligands against tumor cells and tissues that have potential therapeutic and diagnostic applications in cancer treatment. Some of these identified ligands might stimulate various cell responses. Here we identified new cell internalizing peptides (and the phages with such peptides fused to each of ~3900 copies of their major coat protein) using landscape phage libraries and for the first time investigated the actin dynamics when selected phages are internalized into the SKBR-3 breast cancer cells. Our results show that phages harboring VSSTQDFP and DGSIPWST peptides could selectively internalize into the SKBR-3 breast cancer cells with high affinity, and also show rapid involvement of membrane ruffling and re-arrangements of actin cytoskeleton during the phage entry. The actin dynamics was studied by using live cell and fluorescence imaging. The cell-targeting phages were found to enter breast cancer cells through energy dependent mechanism and phage entry interferes with actin dynamics, resulting in reorganization of actin filaments and increased membrane rufflings in SKBR-3 cells. These results suggest that, when phage enters epithelial cells, it triggers transient changes in the host cell actin cytoskeleton. This study also shows that using multivalent phage libraries considerably increases the repertoire of available cell-internalizing ligands with potential applications in targeted drug delivery, imaging, molecular monitoring and profiling of breast cancer cells.
Keywords: Landscape phage library, filamentous actin, cell-internalizing peptide, breast cancer cells
1. Introduction
Breast cancer is one of the most serious death-causing diseases among women. Identification and understanding the detailed mechanism of new breast cancer cell internalizing peptides is potentially beneficial for clinical improvements of cancer imaging and therapy 1. Recently, the search for new cell-penetrating peptides has been very active as they can assist in delivering molecules to a variety of cells. Earlier studies on tat protein from HIV-1 and antennapedia homeodomain from drosophila showed that short proteins and peptides can cross the plasma membrane through translocation process and enter the cells 2. However, cell-penetrating peptides are limited in applications bytheir lack of specificity to a particular cell type 3. Such limitation has led the scientists to search for new cell-internalizing peptides that can show specific binding affinity towards a target cell. The cell-targeting peptides specific to a particular cell type can be taken into the cells through initial electrostatic interactions between their amino acids and plasma membrane components and internalized through endocytosis or energy independent mechanisms 4. Arginine-glycine-aspartic acid (commonly known as RGD) is one of the most extensively studied cell-targeting peptides 5 and its derivatives, such as galacto-RGD, have already been used in therapeutic assays 5. Phage particles displaying cyclic peptide RGD4C can bind surface integrins and be effectively internalized into cells 6. Recently, the heptapeptide LTVSPWY was identified as a potential cell-targeting peptide against the erbB2 receptor 7. However, there is a need for more diverse repertoire of the cell-recognizing peptides that can help in diagnosis and treatment of breast cancer and other tumors. Development and understanding of new short peptide ligands that can specifically recognize the cancer cells could facilitate the design of cell-targeting nanomedicines for breast cancer diagnosis and treatment.
Dynamic actin reorganization during cell migration and other physiological processes plays an important role in both normal and metastatic cells 8. Actin, a 43 Kda cytoskeletal protein plays a significant role in maintaining cell structure and migration 9. Several proteins are associated with polymerization and depolymerization of actin during cell migration 10. Previous in vitro and in vivo studies have highlighted the importance of actin cytoskeleton and its binding proteins in breast cancer development 1, 11. The polymerization of actin near the cell surface is usually dependent on the signals from the surrounding matrix to further stimulate actin assembly. Foreign bodies such as pathogens may modify actin or actin binding proteins, rendering cell signaling pathways leading to actin rearrangements. Since actin plays a vital role in reshaping the plasma membrane during various cell internalization processes such as phagocytosis, macropinocytosis, clathrin and caveolae mediated endocytosis 12, we are interested in understanding the role of actin dynamics during filamentous phage internalization. Even though earlier studies showed the link between the actin cytoskeleton and endocytosis process 12d, 13, to the best of our knowledge, there have been no reports showing the actin dynamics when a filamentous virus enters mammalian epithelial cells. The goal of this part of the study is to initiate more detailed research in this area to further our understanding of cell-phage interaction before the clinical significance of filamentous phage therapy is worked out.
Phage display has been previously demonstrated to be a powerful approach for the selection of short cell-targeting and/or internalizing ligands 14. The principles and applications of phage display for studying protein-ligand interactions were reviewed elsewhere 15. Here we used landscape phage library – a multibillion collection of phages with random octamers fused to each of the ~3900 copies of the major coat protein (called pVIII) (supplementary information, Figure S1) to select new cell internalizing phages against SKBR-3 cells. The multivalent display of the binding peptides on the side walls of landscape phage dramatically increases the affinity of peptide-cell interaction because of avidity effect and increases a repertoire of selected binding clones 16. Furthermore, the fusion pVIII protein comprises a dominant fraction (~95%) of viral mass and can be directly converted into specifically targeted drug- and gene-delivery vehicles 17. The general strategy of the selection of cell-internalizing peptides by using a landscape phage library is outlined in Figure 1 and the detailed selection procedure is described in supporting information. Once the phage was identified, we investigated the role of actin in internalization of phage in SKBR-3 breast cancer cells. We studied actin organization in SKBR-3 cells using live cell imaging and confocal fluorescence imaging where actin filaments were labeled with rhodamine-conjugated phalloidin. Since the SKBR-3 cells are known to overexpress ErbB2 receptors 18, we believe this might be one of the preferred targeting sites for selected phage. The newly identified sequences were compared with natural protein ligands that bind to ErbB receptors and anti-ErbB2 antibody (Table 2 & 3). We propose a hypothetical model which shows the involvement of actin in phage internalization in breast cancer cells based on our study (Figure S1).
Figure 1.

Illustration of the procedure of selecting cancer cell-targeting/internalizing peptides from a landscape phage library (affinity selection). (a) The SKBR-3 breast cancer cells are cultured in 25 cm2 flask. (b) Some phages bind the cancer cells while some don’t after a landscape phage library is added to the cancer cells. (c) non-binding phages are washed away and the cell-binding phages still bind to the cells in the flask; (d) the cell-specific phage is eluted from the flask by using an elution buffer; (e) the eluted phage is amplified by infecting bacteria E. Coli; (f) Amplified phage clones are propagated by PEG/NaCl precipitation and re-dissolved in TBS buffer (pH 7.0); (g) part of phage genome (insert-coding region) is amplified by PCR and sequenced; (h) scheme of a single phage particle, which shows that 5 copies each of pIII and pVI are at one distal end and 5 copies each of pVII and pIX are at the other end of phage whereas ~3900 copies of pVIII form a protein coat wrapping single-stranded DNA. It should be noted that an inserted foreign peptide is inserted in the solvent-exposed end of each of the ~3900 copies of pVIII which constitute the side walls of phage. To identify a cell-internalizing peptide, the procedure is same as above except that after Step (c) the cancer cells are lysed by using a cell lysis buffer to release the cell-internalized phage, which is followed by step (e).
Table 2.
Alignments of selected peptides with ErbB family binding proteins in human
 |
Table 3.
Alignments of selected peptides with anti ErbB2/Her2 proteins in human
 |
2. Experimental Section
2.1. Selection of SKBR3 breast cancer cell-targeting peptides from a landscape phage library
2.1.1 Landscape phage library and cells
We used an 8-mer landscape phage display library (called f8/8) in our selection experiments. 19 The detailed information about the library construction was described elsewhere. Random peptides in this library are fused to N-terminus of the major coat protein pVIII of fd-tet bacteriophage, so that each landscape phage virion displays a unique 8-mer random peptide on each of the 3900 copies of pVIII. The human breast carcinoma SKBR-3 (HER positive) cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). As a control we used normal breast MCF-10A (HER negative) cells purchased from ATCC.
2.1.2 Cell culture
The target SKBR-3 cells and control MCF-10A cells were cultured in 25 cm2 culture flask containing McCoys 5A modified medium and Dulbecco’s modified Eagles medium, respectively, supplemented with 10% ( v/v) fetal bovine serum, 11.2 μg/L penicillin, 11.2 μg/L stepromycin, and 11.2 μg/L ampicillin. The cells were incubated at 37°C with 5% CO2 and the cell culture medium was changed once every two days until the cells reached 90% confluence. The cells were harvested for 5 min incubation at 37°C with a 0.25% trypsin, 1 mM EDTA solution followed by centrifugation at 2100 rpm for 5 min. The cell pellet was dissolved in 5 ml of medium and cell count was determined.
2.1.3 Preparation of the bacterial host starved cells
A fresh loop of E. Coli K91 BlueKan cells was inoculated into three test tubes containing 2 ml NZY/Kanamycin (100 μl/ml) in each tube. The tubes were incubated in a shaking incubator at 220 rpm, 37°C overnight. 300 μl of overnight cultures were transferred into a 300 ml flask containing 20 ml NZY medium. The flask was incubated at 220 rpm, 37° C until OD600 =0.45, followed by gentle shaking (50 rpm) for additional 8 min. The cells in the sterile tube were then centrifuged at 2000g for 10 min at 4°C. The supernatant was discarded and the cells were gently re-suspended in 20 ml of 80 mM NaCl solution. The re-suspended solution was then gently shaken at 50 rpm for 45 min, followed by centrifugation as above. The cell pellet was re-suspended in 1 ml of cold sodium ammonium phosphate buffer (NAP buffer) (80 mM NaCl, 50 mM NaH4PO4 pH=7.0 adjusted with NH4OH) and stored in a refrigerator.
2.1.4 Affinity selection of SKBR-3 cell-binding phage clones
The affinity selection procedure is shown in Figure 1 with a slight modification of our previous protocol.14 Briefly, the f8/8 landscape phage library (~1×1011 virions containing 1×109 unique clones) was first incubated with empty 25 cm2 culture flask at 37°C for 1 h to remove the phages that specifically bound to the culture flask. This process is known as depletion. The resultant depleted library was then used as an input library to the culture flask containing the control MCF-10A cells (HER negative cells). This process is known as negative selection, which was performed to remove phage that binds to the receptors of healthy cells and increase a portion of the phage binding to target SKBR-3 cancer cells. The depleted phage library was transferred to the flask with target SKBR-3 cancer cells (HER positive cells), and incubated at room temperature for 1 h. The flask was washed 10 times with bovine serum albumin (BSA)/Tween washing buffer to remove the unbound phages. The cell surface bound phages were eluted from the culture flask with 800 μl of low pH elution buffer (0.1N HCl, 1 mg/ml BSA and pH adjusted to 2.2 with glycine) for 10 min. The eluate was transferred into a 1.5 ml Eppendorf tube and neutralized by mixing it with 150 μl of 1 M Tris-HCl (pH= 9.1) immediately. At the same time, the cell internalized phages were recovered using a cell lysis buffer (2% sodiumdeoxycholate, 10 mM Tris, 2 mM EDTA, pH 8.0). The tubes were stored at 4°C for further use.
2.1.5 Amplification of eluates
The entire first-round eluate (950 μl) was concentrated to reach a final volume of 150 μl by using a Centricon 100-KDa Ultrafilter (Amicon). 150 μl of concentrated eluate was added to 150 μl of the starved cells and incubated for 15 min at room temperature. The phage-infected cells were transferred to 40 ml of NZY medium with tetracycline (0.2 μg/ml) in a 125 ml flask and incubated for 45 min at 37°C with shaking. The tetracycline concentration was increased to 20 μg/ml and the shaking of the flask was continued at 37°C for 24 h.
2.1.6 Amplification of cell lysate fraction to identify cell-internalizing peptides
The SKBR-3 cell-internalized phage clones were recovered using the cell lysis buffer (2% sodiumdeoxycholate, 10 mM Tris, 2 mM EDTA, pH 8.0). In order to recover internalized phages, the cell monolayer from the cell culture flask was scraped with a sterile pasture pipette and taken into 5 ml of cell culture medium and centrifuged at 130 g for 10 min. Later, the supernatant was removed and 200 μl of the cell lysis buffer was added to the pellet. The fraction was stored at 4°C until amplification. The phages were used for amplification and further selection as stated above.
2.1.7 Propagation and purification of phages
Phage was purified by double polyethylene glycol (PEG) precipitation as described previously 20. Briefly, 40 ml of NZY medium containing 20 μg/ml tetracycline was inoculated with a fresh single colony of phage-infected cells in a 250 ml flask. The culture was incubated in a shaking-incubator at 200 rpm, 37°C overnight. The overnight culture was poured into a 250 ml Beckman centrifuge bottle and centrifuged at 3000 g for 10 min at 4°C. The supernatant was then poured into a fresh tube and centrifuged at 12000 g for 10 min at 4°C. The resultant cleared supernatant was again poured into a fresh tube, to which 6 ml PEG/NaCl solution (100 g PEG 8000, 116.9 g NaCl and 475 ml water) was added. The tube was kept at 4°C overnight for phage precipitation. The precipitated phages were collected by centrifugation at 31000 g for 15 min and the supernatant was removed. The pellet was then dissolved in 1 ml Tris buffered saline (TBS) in a micro-centrifuge tube and centrifuged again at 12000 g for 2 min to remove any undissolved material. 150 μl of the PEG/NaCl solution was added to the supernatant which was then kept on ice overnight. The tube was then centrifuged at 12000 g at 4°C for 10 min and the supernatant was removed. The pellet was dissolved in 200 μl TBS by vortexing.
2.1.8 Tittering of phage
Serial dilutions (10−2 – 10−8) were prepared by using TBS (pH 7.4). For example, a 10−2 dilution was reached by mixing 10 μl of phage, 490 μl of TBS and 500 μl of sterilized water. In a 1.5 ml Eppendorf tube 10 μl of starved cells were mixed with 10 μl of each phage dilution and incubated for 15 min at room temperature. 180 μl NZY with tetracycline (0.2 μg/ml) was added to each tube and the tube was incubated for 45 min at 37° C. Suspensions from the tubes were spread onto NZY plates with tetracycline (20 μg/ml). The plates were incubated overnight at 37° C in an incubator and the number of colonies on the plates was counted the following day. The phage titer was calculated as follows:
2.1.9 Second-fourth rounds of affinity selection
The purified phage after each round was used as an input library for the next round of selection (Figure 1), which was proceeded similar to the first round as described above. After the fourth round of the selection, the eluted as well as lysed phages were not amplified. Instead, they were titered and 40 random colonies were picked up for sequencing in order to determine the SKBR-3-binding peptides that were fused to the major coat protein pVIII of the selected phages.
2.1.10 Sequencing of selected phage colonies
Each colony of phage infected E. Coli K91 BlueKan cells on the plates was inoculated into a tube containing 2 ml NZY/Tetracycline (20 μg/ml) and the tubes were kept in the shaking incubator at 220 rpm, 37° C overnight. 1 ml culture from each tube of phage infected K91 BlueKan cells and 30 μl of 830 μM related primer (5′ –CAAAGCCTCCGTAGCCGTTG-3′) were sent to MC Lab Inc. for sequencing. The nucleic acid sequences of the insert-coding region of the phage DNA were translated to the corresponding peptide sequences using EB1 tool Transeq.
2.1.11 Binding assessment of affinity selected phage to SKBR-3 using phage capture ELISA
Phages that specifically interact with the SKBR-3 cells due to the presence of specific peptides fused to the major coat protein were revealed using phage capture ELISA. The target cells (SKBR-3) and control cells (MCF-10A) (~1×104 cells/well) were applied to the 24 well ELISA plate and incubated at 37°C overnight. All the wells were blocked with 100 μl of 0.1% bovine serum albumin (BSA) and incubated at room temperature for 1 h. Later each well was washed five times with 200 μl of TBS containing 0.5% Tween 20, and 50 μl (1×1010 virions/ml) of purified phage preparation was applied to SKBR-3 cell-coated wells, MCF-10A cell-coated wells and a control well treated with medium, which were then incubated at room temperature for 1h with gentle mixing. After incubation the plates were washed with 200 μl of TBS buffer and supplemented with 40 μl of anti-fd bacteriophage IgG. The plates were then incubated at room temperature for 1 h, washed with TBS buffer and incubated with 40 μl anti rabbit IgG conjugated with alkaline phosphatase at room temperature for 1 h. After a final washing step, alkaline phosphatase substrate, p-nitrophenyl phosphate, was added to the wells and the absorbance was measured at 405 nm using a plate reader. The p-nitrophenyl phosphate is a substrate, which turns yellow when cleaved by alkaline phosphatase. A relative amount of phage bound to the cells was determined by measuring the rate of color change in each well. As shown in Figure S2, many of the isolated phage clones bound to SKBR-3 cells show higher signals than the control wild type phage, which confirms their binding specificity to SKBR-3 cells. In order to confirm that the ELISA results were not due to contaminants in the cell culture, we also performed phage capture ELISA on empty culture flask containing medium without any cells. As expected, we did not observe any signal (data not shown), which confirms that the ELISA signal in Fig. 2C resulted from the phage specifically bound to the SKBR-3 cells.
Figure 2.
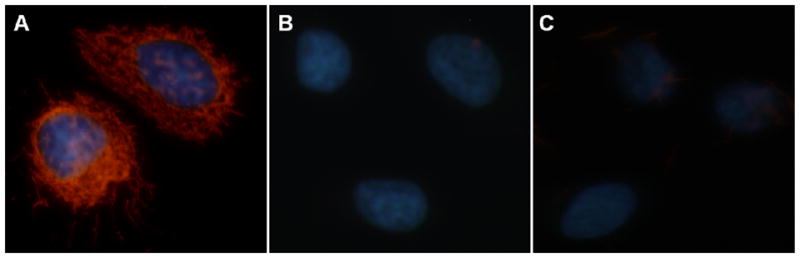
Energy dependency of phage internalization. (A) L1 phage incubated with SKBR-3 cells at 37°C (in the absence of ATP inhibitors). (B) L1 phage incubated with SKBR-3 cells at 4°C. (C) L1 phage incubated with SKBR-3 cells at 37°C (in the presence of ATP inhibitors, sodium azide and sucrose).
2.1.12 Blocking of L1 phage uptake by 12 mer synthetic peptide
Target SKBR-3 cells were cultured in 25 cm2 tissue culture flask and incubated with 2 ml of VSSTQDFPDPAK synthetic peptide (Neopeptide, Cambridge, Massachusetts) in Mc Coy’s 5A medium at a concentration of 0.25mM, 0.75mM and 1.25mM. The synthetic peptide at these concentrations was incubated with the SKBR-3 cells for 1 h at 4°C as previously described. 21 After incubation, 1×107 pfu of L1 phage in 1ml of blocking buffer (0.1% bovine serum albumin, 0.1% Tween 20 in Mc. Coy’s 5A medium) was added to the cells and incubated for 1 h at 37°C. Unbound phage was removed using sterile pasteur pipette and the cells were thoroughly washed five times for 5 min each with cold washing buffer. The phage associated with cells in the presence of synthetic peptide was recovered with lysis buffer as described above. The amount of phage bound was quantified using phage titration method.
2.2 Immunofluorescence studies of phage-cell interactions
The SKBR-3 cells were cultured on cover glasses in a six well plate. Two days after seeding, the medium was removed and the cells were incubated in serum-free medium for 2 h. The medium was removed and 200 μL of phage suspension (approximately 1×1010 virions/ml in TBS) was added to each well and incubated at 37° C for 1 h. The cells were washed three times with PBS and fixed in 4% paraformaldehyde in PBS for 15 min. The fixed cells were washed three times in PBS and permeabilized with 0.5% Triton X-100 and incubated for 1 h in room temperature with 1:10 dilution of Alexa Fluor conjugated anti–pIII antibody. The detailed procedure for conjugation of dye and antibody and purification of dye-conjugated antibody from free dye can be obtained from Invitrogen Alexa Fluor® 488 monoclonal antibody labeling Kit (cat# A-20181). The cells on cover glasses were washed 5 times with PBS and then incubated with DAPI, a nuclear staining dye, for 5 min. The cover glasses were again washed for 5 more times with PBS and mounted on a microscope slides. The cells were examined using a Zeiss Universal epiflourescence microscope equipped with Olympus oil immersion DApo UV objectives. Digital images of cells were collected using Olympus DP-70 camera and software.
2.3 Energy dependent mechanism of phage entry
In order to determine the energy-dependency of phage entry, SKBR-3 cells were incubated with phage clone, labeled with rhodamine B at either 37°C (in presence and absence of inhibitors) or 4°C. Briefly, rhodamine B labeling of engineered phages was performed by mixing 0.2 g of a coupling agent, 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (DEC), and 2.5 mg of rhodamine B in 10 mL of affinity selected phage stock solution (1×1012 pfu) in phosphate buffer (pH 5.6). This procedure results in permanent attachment of the dye through DEC assisted caging. The rhodamine-labeled phages were extensively dialyzed for 6 days under stirring to remove unbound dye and small organic molecules. The modified phages were stored at 4°C in dark until further use. SKBR-3 cells were seeded 48 h prior to phage incubation on cover glasses in the six-well plates (8000 cells/well). The growth medium was replaced with 200 μl of rhodamine conjugated phage in PBS (1×109 cfu/ml), and cells were incubated for 1 h, either at 37°C (in the absence and presence of ATP inhibitors) or at 4°C (all the solutions were prechilled to 4°C). Later the cells were extensively washed three times with PBS at corresponding temperature and fixed in 4% paraformaldehyde in PBS for 15 min at 37°C. After incubation the cells were washed twice with PBS and stained with 250 μl of DAPI (1 μg/ml) for 5 min, the cover slips were again washed for 5 more times with PBS and mounted on a microscope slide. The cells were examined under Zeiss Universal epiflourescence microscope equipped with Olympus oil immersion DApo UV objectives. Digital images of cells were collected using Olympus DP-70 camera and software.
2.4 Live cell imaging of landscape phage treated SKBR-3 cells using video microscopy
SKBR-3 cells cultured on 25-mm cover glasses were transferred to a perfusion filming chamber. The chamber was placed on Zeiss Standard microscope stage maintained at 37°C with an air curtain incubator. The cell images were recorded with Hamamatsu C2400 camera and Scion software. Cells were recorded for 15 min in McCoy’s 5a medium and then 1×1010 pfu/ml selected phage was perfused into the chamber from a 2.5-cc syringe driven by a syringe pump (Sage model 341 A) and recorded for additional 45 min.
2.5 Immunofluorescence of labeled F-actin with phage internalization
The SKBR-3 cells were cultured on the cover glasses in a six well plate. Two days after seeding, the medium was removed and the cells were incubated in serum-free medium for 2 h. The medium was removed and 200 μL of phage suspension (approximately 1×1010 virions/ml in TBS) in cell culture medium was added to each well and incubated at 37° C for 15 min, 30 min, and 45 min respectively. The cells were washed three times with D-PBS and fixed in 4% paraformaldehyde in D-PBS for 15 min. The fixed cells were washed three times in D-PBS and incubated with 2 μM rhodamine-phalloidin in D-PBS for 15 min in dark at room temperature. The cover glasses were washed 5 times with D-PBS and mounted on a microscope slide. The cells were examined using an Olympus fluoview FV 500 confocal laser scanning microscope and the images were acquired at constant PMT, gain, offset, magnification (60× objective) and resolution.
2.6 Statistical analysis
All the statistical analyses were performed using student unpaired two-sided t-test analysis according to MBEC recommendations as stated before 22. P values were considered to be statistically significant with p≤ 0.0002.
3. Results
3.1 Selection of cell internalizing phage against SKBR-3 cells
Phage clones that bound to SKBR-3 cells were selected from the landscape phage library f8/8 19 through successive rounds of affinity selection procedure, in which the phage library was incubated with SKBR-3 cells cultured in a 25 cm2 tissue culture flask. During the affinity selection process, the unbound phages were washed away and the cell surface bound phages were eluted with mild acid treatment. It should be noted that phage library was allowed to interact with normal breast cells first in order to get rid of phages that might bind to normal cells before interacting with breast cancer cells. During each round of affinity selection, after cell-surface-bound phages were eluted, the cells were lysed to release the cell-internalized phages to obtain cell-internalizing peptides. The cell-internalized phages were recovered from cell lysates. Phages that bound to SKBR-3 breast cancer cell surface and internalized in the initial selection procedure were amplified separately by infecting bacteria and used as the input for the next round of affinity selection. This procedure was repeated four times. After the fourth round of affinity selection, 65 clones were randomly picked from both the eluate and lysate fraction and a segment of genomic DNA encoding the octapeptide (i.e., insert-coding region) was sequenced. The cell-internalizing peptides (denoted as L) and cell-surface-targeting ones (denoted as S) and their frequencies were shown in Table 1 and S1, respectively. A comparison between them indicates that two peptides (DGSIPWST and VSSTQDFP) are both surface-targeting and cell-internalizing.
Table 1.
New SKBR-3 cell internalizing peptide clones recovered from landscape phage library by using a lysis buffer. L: denotes cell internalization fraction.
| New Cell-internalizing clones | ||
|---|---|---|
| Clone ID | Motif sequence | Frequency |
| L1 | VSSTQDFP | 13 |
| L2 | DGSIPWST | 8 |
| L3 | AFSEAAQT | 5 |
| L4 | ADAPSNGW | 1 |
| L5 | DNDNYAFP | 1 |
| L6 | DYADYSDL | 1 |
| L7 | AADYDPLA | 1 |
3.2 Characterization of selected peptides and phages
Selected peptides were analyzed using the REceptor Ligand Contacts (RELIC) program, which is a bioinformatics tool for combinatorial peptide analysis and identification of protein-ligand interaction sites 23. A complete review of RELIC and its principle and application in analyzing affinity selected peptides can be found in the literature 23. We used two RELIC programs (RELIC/POPVID and RELIC/INFO) to analyze the binding affinity of our selected peptides towards SKBR-3 cancer cells. RELIC/POPVID program uses a statistical sampling method to estimate the sequence diversity of a combinatorial peptide library based on the sequence obtained from a limited number of randomly sampled members of the library. According to this program, affinity selected peptides towards target SKBR-3 cells should have a less diversity than an equivalent number of randomly selected peptides from the native library. It has been demonstrated that the decrease in the diversity of the sequences obtained from successive rounds of selection is an indicator of affinity selection 24. We calculated the diversities of both affinity selected peptides and random peptides from the native library by using RELIC/POPVID program. Our affinity selected peptides yielded a diversity value of 0.00402 whereas the same number of randomly selected peptides from a native library yielded a diversity value of 0.00913. Compared to randomly selected peptides, our affinity selected peptides have a less diversified population, which is a clear evidence for successful selection of peptides with binding affinity towards SKBR-3 cells. In order to verify that the selected peptides were from affinity selection and not from amplification noise, we used RELIC/INFO program because the noise from amplification has regular and measurable patterns 24b, c. This program was designed to initially calculate the noise pattern and then subtract the noise pattern from the affinity pattern to obtain information content 23. Information content is defined as −ln(PN), where PN is the probability of randomly finding a given peptide. The principle of RELIC/INFO helps in determining the occurrence of information content of the affinity selected peptides and thereby confirms the binding affinity of selected peptides after affinity selection. A larger information content (−lnPN) of a peptide indicates a greater chance of observing this peptide in affinity selection due to specific binding to the target. Indeed, from Figure S2A the normalized distribution of the information content for our SKBR-3 selected peptides is positively shifted towards higher information content in comparison to that of the peptides selected from native phage library. This positive shift of red curve in Figure S2A suggests that our SKBR-3-selected peptides results from affinity selection. Both observed result are in accordance with the principle of RELIC/POPVID and RELIC/INFO 23.
Phage capture ELISA shows that the selected cell-targeting phage has high affinity against SKBR-3 cells (Figure S2B). Immunoflourescence microscopy further shows that wild type phage (labeled by green dye Alexa Fluor through using anti-pIII phage antibody as a linker) cannot enter SKBR-3 cells but cell-internalizing phage (labeled by the same dye) displayed with L1 and L2 peptides against SKBR-3 can enter and stain the cells (Figure S3). Figure S3 indicates that the cell-targeting phage is localized in the cytoplasm and nuclei. These results show that the identified cell-targeting phage can recognize SKBR-3 cells and be internalized by the SKBR-3 cells due to the presence of the cell-internalizing peptides displayed on the major coat of the phage.
To see the motif similarities between peptide inserts from selected phages and natural ligands binding to ErbB family receptors, we used RELIC/MATCH program to align our selected peptide sequences with the sequences of the proteins that were reported to recognize SKBR-3 cells. The alignment results are shown in Table 2 and 3 and suggest that our selected peptides show similarity with some domains on the proteins, which are known to recognize SKBR3 cells, including antibodies.
3.3 Mechanism of phage entry into SKBR-3 cells
In order to understand the mechanism of phage entry into SKBR-3 cells, initial experiments were conducted to determine the energy dependency of L1 phage uptake because endocytosis can be strongly inhibited by lowering the temperature 25. For this purpose, selected phages were labeled with rhodamine as described earlier 26. Phages conjugated to rhodamine were then incubated with SKBR-3 cells at 37°C (in the absence of ATP inhibitors), at 4°C and at 37°C (in the presence of ATP inhibitors, sucrose and sodium azide). As shown in Figure 2A, the phage uptake is only active at 37°C in the absence of inhibitors. In contrast, lowering the temperature completely or in the presence of inhibitors reduced the phage uptake as shown in Figure 2B and C. The phage internalization efficiency was found to be less than 5% at 4°C and in the presence of inhibitors as compared to phage internalization efficiency at 37°C. These findings confirm that selected L1 phage enters the SKBR-3 cells by energy-dependent uptake mechanism. The detailed fluorescent images are shown in supplementary information (Figure S4).
3.4 Competitive binding studies using synthetic peptide
SKBR-3 cell internalizing phage clone, L1 bears VSSTQDFP peptide sequence on its N-terminus. In order to test whether L1 phage internalization takes place through a VSSTQDFP recognition motif, we performed competitive assay using a synthetic VSSTQDFPDPAK peptide. This synthetic peptide is the solvent-exposed domain of pVIII with the selected VSSTQDFP recognition motif terminated. As described in section 2.1.12, synthetic peptide was incubated with the cells prior to the incubation of L1 phage. Figure S5 shows that incubation of synthetic peptide reduced the L1 phage binding to the SKBR-3 cells in a concentration dependent manner. As the concentration of peptide is increased, less number of L1 phage was bound to the cells. This confirms that L1 phage internalization is competitively inhibited in the presence of synthetic peptide.
3.5 Actin dynamics during the phage internalization into SKBR-3 breast cancer cells
In order to understand the mechanism of selected phage internalization into SKBR-3 cells, we first performed video microscopy to observe the behavior of SKBR-3 cells before and after the treatment of cells with internalizing phage (L1). SKBR-3 cells are polygonal cells and display cycles of protrusions and withdrawals of small lamellipodia at the cell edges forming small ruffles at random places around their periphery. The dorsal (upper) cell surface is relatively quiescent, with occasional small ruffles (Figure 3a,b). After injection of internalizing phage into the filming chamber, SKBR-3 cells showed very intense ruffling in multiple places around the cell periphery. Cells were also rapidly forming numerous ruffles on their dorsal surfaces. As shown in Figure 3d–f, cell edges and dorsal surface stay very active over the entire time of recording (45 min). In the control, no significant changes were observed in the behavior of cells before and after the addition of control wild type phage Figure 4a–f. The representative still images of SKBR-3 cells before and after the addition of selected phage and WT phage are shown in Figures 3 and 4 and the original videos are provided as supporting information. These videos clearly demonstrate that there are significant changes in the cell behavior in regard to activity of cell edges and dorsal surface during and after the internalizing phage entry into SKBR-3 cells. It is well established that formation of protrusions in a variety of cells is associated with dynamic changes in cortical actin.
Figure 3.

Live SKBR-3 cells recorded using digital video microscopy in the presence of selected SKBR-3 cell internalizing phage (L1): (a–c) before and (d–f) after phage injection. Red arrows indicate some areas of increased ruffling at the cell edges in the presence of internalizing phage.
Figure 4.

Live SKBR-3 cells recorded using digital video microscopy in the presence of control wild type fd-tet phage (a–c) before and (d–f) phage injection. There was essentially no cell response to the presence of the wild type phage in the environment.
In order to better understand the involvement of actin cytoskeleton in phage internalization, we labeled F-actin with rhodamine-conjugated phalloidin and examined actin distribution using confocal imaging. SKBR-3 cells were treated with cell internalizing phage for 15, 30 and 45 minutes and immediately after incubation, were fixed and labeled with rhodamine-phallodin. All fluorescence images were obtained at constant PMT, gain and offset. Figure 5 shows the fluorescence images of labeled F-actin in SKBR-3 cells incubated with phage and fixed at different time points as stated above. F-actin in control, untreated cells was most diffusely distributed throughout the cell, and also thin bundles of filaments and occasional brighter granular structures were present. After 15 min incubation with internalizing phage, the bright granular structures and diffuse staining greatly diminished and thin actin bundles stain less intensely. At 30 and 45 min of incubation, small disperse granular structures became brighter and clearly visible all over the cells, while thin filament bundles were still less visible than in untreated cells. The F-actin was further quantified using fluorescence microplate reader. For all the quantitative measurements, a constant number of cells (1×104) were placed in all the wells, treated with phage and labeled with rhodamine-phallodin. For fluorescence measurements, the rhodamine was excited at 560 nm and the emission was collected between 590–600 nm. The quantitative measurements showed significant decrease in the amount of polymerized actin in cells incubated with phage for 15 min, followed by gradual increase in F-actin content, but still remaining lower than in untreated cells 45 min after introduction of internalizing phage into cell culture (Figure 6).
Figure 5.
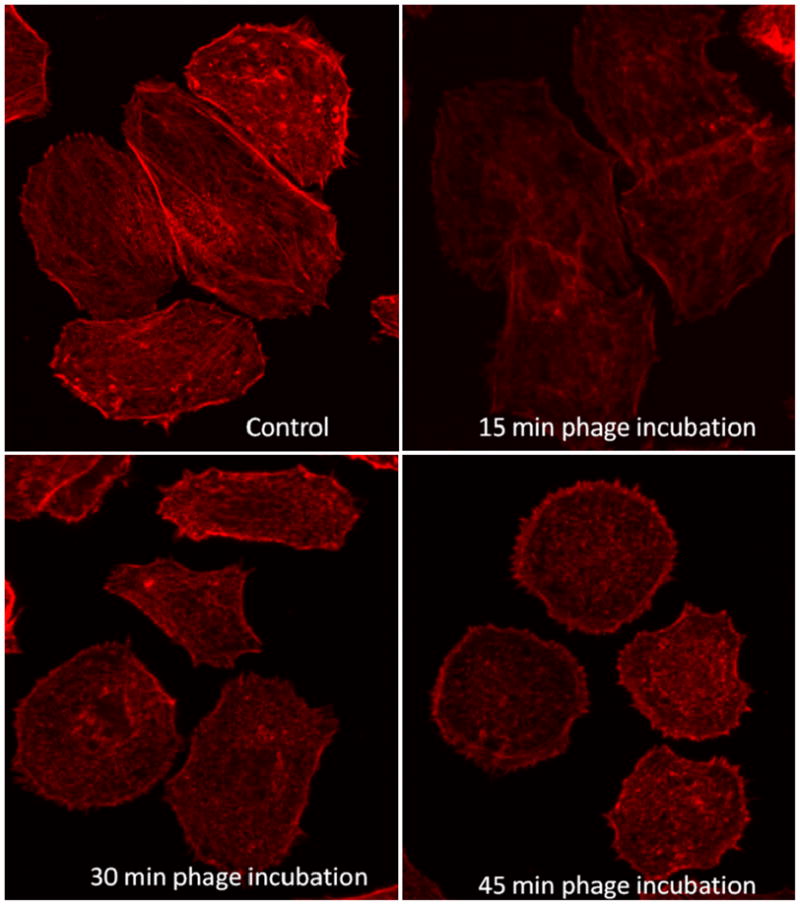
F-actin distribution before and during internalization of phage (L1) into SKBR-3 cells. Control: SKBR-3 without phage treatment; SKBR-3 cells treated with selected phage for 15min, 30min, and 45min (the actin is labeled with rhodamine conjugated phalloidin). F-actin organization in cells changes during incubation with L1 phage.
Figure 6.

Quantitative estimation of F-actin labeled with rhodamine-phalloidin in SKBR-3 cells treated with selected phage clone (L1) at different time points (control, 15min, 30min and 45min) using microplate reader.
4. Discussion
There is a continuing demand for short peptide ligands such as cell internalizing peptides that can serve as cancer-specific diagnostic and therapeutic probes 3. Phage display has been proven to be successful in exploring protein-protein interactions between the ligand and the receptor 27. In this work, we used landscape phage display technique as a tool to identify such cell internalizing peptides specific to SKBR-3 cells and analyzed their similarity to the natural protein ligands of the breast cancer cells. Since earlier studies indicated that ErbB2 is overexpressed on the SKBR-3 cancer cells 28, we compared our selected peptide sequences with protein ligands and anti-ErbB2 antibody that bind to receptors of the ErbB family. As expected, our newly identified sequences show homology with natural ligands such as human EGF, amphiregulin, neuregulin, β-cellulin, heregulin and epigen and anti-ErbB2 antibody (Tables 2 and 3). Our results are also in agreement with earlier reports in which breast-tumor-binding peptides (SSPWSAY and YSSPTQR) were selected by using a pIII phage display library (C7C from New England Biolab) 29. Obviously, our selected sequences (DGSIPWST and VSSTQDFP) with highest frequency (Table 1) have at least four amino acids in common with the previously described tumor-binding peptides (SSPWSAY and YSSPTQR). Based on the occurrence and phage capture ELISA results (Table 1 and Figure S2), we categorize the peptides VSSTQDFP and DGSIPWST as the best binders to SKBR-3 cells under stringent conditions. It is remarkable that the sequences of these peptides differ from the earlier discovered binding peptide sequences, showing that the landscape libraries used in this work can provide a more diverse repertoire of the cancer cell-binding probes 18. It is still unclear why and how the phages displaying VSSTQDFP and DGSIPWST (our selected cell internalizing peptides) can enter the SKBR-3 cells. From previous studies, proline and charged amino acids were found to be involved in cell internalization 30. Thus one possible explanation could be the presence of proline residue in these peptides in conjunction with charged amino acid residues in these peptides.
To further investigate the possible mechanism of phage penetration into SKBR-3 cells, we have studied the effect of energy depletion. The phage internalization was abolished at 4°C (Figure 2B) and in the presence of ATP inhibitors such as sucrose and sodium azide (Figure 2C), indicating that the cellular uptake is an active process requiring energy supply. Our energy depletion studies and preliminary studies using specific inhibitors against individual cell internalization mechanisms such as phagocytosis, pinocytosis and endocytosis show that the phage is entering SKBR-3 cells through endocytosis or specifically through receptor mediated endocytosis. One possible reason for the internalization of phage displaying L1 or L2 sequence could be the intermolecular forces prevailing between the exposed peptides and amino acid residues in EGF receptor. There could be weak hydrogen bond formation between the side chains of threonine in selected peptide and asparagine residue in EGFR leading to the receptor mediated endocytosis. Another possibility could be the formation of hydrophobic interactions between the isoleucine and phenyl alanine residues from selected peptides with that of leucine residues in the EGF receptor. Nonetheless, the detailed mechanism of phage entry displaying these peptides is still under investigation.
While studying the possible mechanism of phage internalization we also focused on the actin dynamics when selected filamentous phage (L1) is internalized into SKBR-3 cells. We hypothesize that phage is internalized using endocytosis mechanism as stated by previous studies 6. Multiple studies indicate that actin reorganization is involved in the process of endocytosis 12–13. We believe that in the process of endocytosis, when the phage comes in contact with the cell membrane receptor, invagination of the membrane is initiated and might result in the formation of vesicles. Although the contact of phage with the cell membrane is the key step in the endocytosis, the exact underlying mechanism is not well understood. Since one possibility could be the involvement of cell cytoskeleton and its associated proteins in the process of phage internalization, we focused our study on the actin cytoskeleton dynamics during phage entry into breast cancer cells. We used video microscopy followed by confocal fluorescence imaging to study cell behavior and visualize actin filaments before and after the phage entry to further our understanding of the mechanism of phage entry. We observed two significant changes in cell motile activity when phage is internalized into SKBR-3 breast cancer cells. One is the change in the behavior of the leading edges of SKBR-3 cells and another is the change in the dorsal ruffling of SKBR-3 cells. Lamellipodia, thin protrusive structures that arise from the cell cortex undergo a dynamic fluctuation at the leading edges of cells. They are protruding and withdrawing in rather cyclical manner in SKBR-3 cells prior to the introduction of the phage into the cell environment. After the introduction of the phage, the characteristic increased ruffling of SKBR-3 cells was observed for the duration of the experiment (45 min). This increased rate of the lamellipodia formation both at the cell periphery and at the dorsal surface is a sign of dynamic reorganization of mostly cortical actin in the cells, since it is well known that actin polymerization and depolymerization is a driving force for the formation of protrusions and the ruffling of the cell edge 31. The changes in F-actin pattern of distribution observed in fluorescence images in Figure 5 are characteristics of changes in the state of actin polymerization. Microspikes, filopodia and lamellipodia are shaped by dense core of actin filaments and changes in actin state of polymerization and distribution reflects the dynamic reorganization of actin during phage entry.
Inside the cell, actin exists in two forms either as polymerized helical form commonly known as filamentous actin or F-actin and as unpolymerized globular form known as G-actin. In order to investigate the changes in the state of polymerization of actin during phage internalization, the cells were treated with phage for different times and immediately fixed with paraformaldehyde and incubated with rhodamine-phalloidin to label F-actin. The relative fluorescence intensities of rhodamine in Figure 6 represent the amount of polymerized actin in phage treated cells. During incubation of cells with phages for up to 45 min, we observed a net depolymerization of actin cytoskeleton in the initial 15 min after the introduction of phage into the cells’ environment. Interestingly, 15 min was the earliest time we observed phages bound to SKBR-3 cells using scanning electron microscopy in our previous study 32. Thus the time of increased actin depolymerization correlates with the time during which the process of the phage entry into the cells takes place. The possible rearrangement of actin in SKBR-3 cells when landscape phage is internalized is shown in Figure S1. There have been reports indicating that actin dynamic reorganization is involved in the entry of the Kaposi’s sarcoma herpes virus into endothelial cells 33 and influenza virus into epithelial cells 34. Also it has been recently reported that the uptake of charged polystyrene nanoparticles into HeLa cells is mediated by the reorganization of actin 35. Our results also show that entry of filamentous phage into the SKBR-3 cells involves the reorganization of actin cytoskeleton and mechanism similar to the entry of viruses into cells may be used by the phage to enter breast cancer cells. More detailed biochemical investigations are necessary to understand the possible signaling pathway/s and associated actin binding proteins involved in phage internalization into epithelial breast cancer cells.
In summary, we identified new cell internalizing peptides that are already fused to ~3900 copies of pVIII protein on each phage body, enabling the pVIII protein to become cell targeting and internalizing vehicle. Our studies confirm that actin plays a major role in the reshaping of plasma membrane when phage is internalized into the cells. One of the potential applications of our findings is that the cell targeting/internalizing fusion protein pVIII, which can be easily purified from one phage particle, can be conjugated with the cell-killing drugs to form a new type of anti-cancer agents. Such anti-cancer drugs can not only target cancer cells, and be internalized into the cancer cells, but also kill the cells. For example, chemotherapeutic drugs such as Doxorubicin usually do not have natural capabilities of recognizing cancer cells. They can be loaded into a liposome. The identified cancer cell targeting/internalizing pVIII can be conjugated with the liposomes to make them specific cell-targeting/internalizing agents 17b. Therefore, our work shows that the use of landscape phage library can identify short cell targeting/internalizing peptides as well as phage displaying such peptides, which can find applications in targeted cancer treatment.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (R21EB009909-01A1) and Department of Defense Breast Cancer Research Program (W81XWH-07-1-0572). We also thankfully acknowledge the financial support from National Science Foundation (DMR-0847758, CBET-0854414, CBET-0854465), National Institutes of Health (R03AR056848-01, R01HL092526-01A2) and Oklahoma Center for the Advancement of Science and Technology (HR06-161S). V.A.P. is supported by NIH Grant 5R01CA125063-03.
References
- 1.Jiang P, Enomoto A, Takahashi M. Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett. 2009;284:122–130. doi: 10.1016/j.canlet.2009.02.034. [DOI] [PubMed] [Google Scholar]
- 2.(a) Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]; (b) Vives E. Present and future of cell-penetrating peptide mediated delivery systems: “is the Trojan horse too wild to go only to Troy?”. J Control Release. 2005;109:77–85. doi: 10.1016/j.jconrel.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 3.Vives E, Schmidt J, Pelegrin A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim Biophys Acta. 2008;1786:126–138. doi: 10.1016/j.bbcan.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik VL, Lebleu B. Cell-penetrating peptides: a reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 5.Beer AJ, Haubner R, Sarbia M, Goebel M, Luderschmidt S, Grosu AL, Schnell O, Niemeyer M, Kessler H, Wester HJ, Weber WA, Schwaiger M. Positron emission tomography using [18F]Galacto-RGD identifies the level of integrin alpha(v)beta3 expression in man. Clin Cancer Res. 2006;12:3942–3949. doi: 10.1158/1078-0432.CCR-06-0266. [DOI] [PubMed] [Google Scholar]
- 6.Hart SL, Knight AM, Harbottle RP, Mistry A, Hunger HD, Cutler DF, Williamson R, Coutelle C. Cell Binding and Internalization by filamentous phage displaying a Cyclic Arg-Gly-Asp-containing peptide. J biol chem. 1994;269:12468–12474. [PubMed] [Google Scholar]
- 7.Shadidi M, Sioud M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003;17:256–258. doi: 10.1096/fj.02-0280fje. [DOI] [PubMed] [Google Scholar]
- 8.Meira M, Masson R, Stagljar I, Lienhard S, Maurer F, Boulay A, Hynes NE. Memo is a cofilin-interacting protein that influences PLCγ1 and cofilin activities, and is essential for maintaining directionality during ErbB2-induced tumor-cell migration. J Cell Sci. 2008;122:787–797. doi: 10.1242/jcs.032094. [DOI] [PubMed] [Google Scholar]
- 9.(a) Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat Mater. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]; (b) Insall RH, Machesky LM. Actin dynamics at the leading edge: From simple machinery to complex networks. Dev Cell. 2009;17:310–322. doi: 10.1016/j.devcel.2009.08.012. [DOI] [PubMed] [Google Scholar]; (c) Pollard TD, Cooper JA. Actin a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Cooper JA. The role of actin polymerization in cell motility. Annu Rev Physiol. 1991;53:585–605. doi: 10.1146/annurev.ph.53.030191.003101. [DOI] [PubMed] [Google Scholar]; (b) Kueh HY, Charras GT, Mitchison TJ, Brieher WM. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J Cell Biol. 2008;182:341–353. doi: 10.1083/jcb.200801027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Mælan AE, Rasmussen TK, Larsson LI. Localization of thymosin β10 in breast cancer cells: relationship to actin cytoskeletal remodeling and cell motility. Histochem Cell Biol. 2007;127:109–113. doi: 10.1007/s00418-006-0208-z. [DOI] [PubMed] [Google Scholar]; (b) Azios NG, Krishnamoorthy L, Harris M, Cubano LA, Cammer M, Dharmawardhane SF. Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia. 2007;9:147–158. doi: 10.1593/neo.06778. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kalra J, Warburton C, Fang K, Edwards L, Daynard T, Waterhouse D, Dragowska W, Sutherland BW, Dedhar S, Gelmon K, Bally M. QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res. 2009;11:1–16. doi: 10.1186/bcr2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]; (b) Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: From yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]; (c) Merrifield CJ. Seeing is believing: imaging actin dynamics at single sites of endocytosis. Trends Cell Biol. 2004;14:352–358. doi: 10.1016/j.tcb.2004.05.008. [DOI] [PubMed] [Google Scholar]; (d) Kaksonen M, Toret CP, Drubin DG. Harnessing actin dynamics for clathrin-mediated endocytosis. Nature Rev Mol Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kaksonen M, Sun Y, Drubin DG. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 2003;115:475–487. doi: 10.1016/s0092-8674(03)00883-3. [DOI] [PubMed] [Google Scholar]; (b) Qualmann B, Kessels MM, Kelly RB. Molecular Links between Endocytosis and the Actin Cytoskeleton. J Cell Biol. 2000;150:111–116. doi: 10.1083/jcb.150.5.f111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brigati JR, Samoylova TI, Jayanna PK, Petrenko VA. Current Protocols in Protein Science. Vol. 51. John Wiley & Sons; 2008. Phage display technique for generating peptide reagents; pp. 18.9.1–18.9.27. [DOI] [PubMed] [Google Scholar]
- 15.(a) Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97(2):391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]; (b) Kehoe JW, Kay BK. Filamentous phage display in the new millennium. Chem Rev. 2005;105(11):4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- 16.Samoylova TI, Petrenko VA, Morrison NE, Globa LP, Baker HJ, Cox NR. Phage probes for malignant glial cells. Mol Cancer Ther. 2003;2:1129–1137. [PubMed] [Google Scholar]
- 17.(a) Mount JD, Samoylova TI, Morrison NE, Cox NR, Baker HJ, Petrenko VA. Cell targeted phagernid rescued by preselected landscape phage. Gene. 2004;341:59–65. doi: 10.1016/j.gene.2004.07.006. [DOI] [PubMed] [Google Scholar]; (b) Jayanna PK, Torchilin VP, Petrenko VA. Liposomes targeted by fusion phage proteins. Nanomedicine. 2009;5:83–89. doi: 10.1016/j.nano.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Petrenko VA. Evolution of Phage Display: From Bioactive Peptides to Bioselective Nanomaterials. Expert Opinion on Drug Delivery. 2008;5:825–836. doi: 10.1517/17425247.5.8.825. [DOI] [PubMed] [Google Scholar]
- 18.Shukla GS, Krag DN. Phage display selection for cell-specific ligands: Development of a screening procedure suitable for small tumor specimens. J Drug Target. 2005;13:7–18. doi: 10.1080/10611860400020464. [DOI] [PubMed] [Google Scholar]
- 19.Petrenko VA, Smith GP, Gong X. A library of organic landscapes on filamentous phage. Protein Engi. 1996;9:797–801. doi: 10.1093/protein/9.9.797. [DOI] [PubMed] [Google Scholar]
- 20.Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993;217:228–257. doi: 10.1016/0076-6879(93)17065-d. [DOI] [PubMed] [Google Scholar]
- 21.Samoylova TI, Cox NR, Morrison NE, Globa LP, Romanov V, Baker HJ, Petrenko VA. Phage matrix for isolation of glioma cell-membrane proteins. Biotechniques. 2004;37:2–7. doi: 10.2144/04372RR02. [DOI] [PubMed] [Google Scholar]
- 22.Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 1999;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. RELIC - A bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 2004;4:1439–1460. doi: 10.1002/pmic.200300680. [DOI] [PubMed] [Google Scholar]
- 24.(a) Carter DM, Gagnon JN, Damlaj M, Mandava S, Makowski L, Rodi DJ, Pawelek PD, Coulton JW. Phage display reveals multiple contact sites between FhuA, an outer membrane receptor of Escherichia coli, and TonB. J Mol Biol. 2006;357:236–251. doi: 10.1016/j.jmb.2005.12.039. [DOI] [PubMed] [Google Scholar]; (b) Makowski L, Soares A. Estimating the diversity of peptide populations from limited sequence data. Bioinformatics. 2003;19:483–489. doi: 10.1093/bioinformatics/btg013. [DOI] [PubMed] [Google Scholar]; (c) Rodi DJ, Soares AS, Makowski L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J Mol Biol. 2002;322:1039–1052. doi: 10.1016/s0022-2836(02)00844-6. [DOI] [PubMed] [Google Scholar]
- 25.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 26.Gitisa V, Adina A, Nasserb A, Guna J, Lev O. Fluorescent dye labeled bacteriophages—a new tracer for the investigation of viral transport in porous media: Introduction and characterization. Water Research. 2002;36:4227–4234. doi: 10.1016/s0043-1354(02)00163-x. [DOI] [PubMed] [Google Scholar]
- 27.Nooren IMA, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roskoski R. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319:1–11. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 29.Shukla GS, Krag DN. Selection of tumor- targeting agents on freshly excised human breast tumors using a phage display library. Oncol Rep. 2005;13:757–764. [PubMed] [Google Scholar]
- 30.(a) Fernandez-Carneado J, Kogan MJ, Castel S, Giralt E. Potential peptide carriers: Amphipathic proline-rich peptides derived from the n-terminal domain of gamma-zein. Angew Chem Int Ed. 2004;43:1811–1814. doi: 10.1002/anie.200352540. [DOI] [PubMed] [Google Scholar]; (b) Foerg C, Ziegler U, Fernandez-Carneado J, Giralt E, Rennert R, Beck-Sickinger A, Merkle H. Decoding the entry of two novel cell-penetrating peptides in HeLa cells: Lipid raft-mediated endocytosis and endosomal escape. Biochemistry. 2005;44:72–81. doi: 10.1021/bi048330+. [DOI] [PubMed] [Google Scholar]; (c) Fernandez-Carneado J, Kogan MJ, Mau NV, Pujals S, Lopez-Iglesias C, Heitz F, Giralt E. Fatty acyl moieties: improving Pro-rich peptide uptake inside HeLa cells. J Pept Res. 2005;65:580–590. doi: 10.1111/j.1399-3011.2005.00253.x. [DOI] [PubMed] [Google Scholar]; (d) Pujals S, Fernandez-Carneado J, Kogan MJ, Martinez J, Cavelier F, Giralt E. Replacement of a proline with silaproline causes a 20-fold increase in the cellular uptake of a pro-rich peptide. J Am Chem Soc. 2006;128:8479–8483. doi: 10.1021/ja060036c. [DOI] [PubMed] [Google Scholar]
- 31.(a) Pollard TD. Actin Curr Opin Cell Biol. 1990;2:33–40. doi: 10.1016/s0955-0674(05)80028-6. [DOI] [PubMed] [Google Scholar]; (b) Copper JA. The role of actin polymerization in cell motility. Annu Rev Physiol. 1991;53:585–605. doi: 10.1146/annurev.ph.53.030191.003101. [DOI] [PubMed] [Google Scholar]; (c) Heath J, Holifield B. Actin alone in lamellipodia. Nature. 1991;352:107–108. doi: 10.1038/352107a0. [DOI] [PubMed] [Google Scholar]; (d) Theriot JA, Mitchison TJ. Actin microfilament dynamics in locomoting cells. Nature. 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]; (e) Stossel T. On the crawling of animal cells. Science. 1993;260:1086–1093. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]; (f) Zigmond SH. Recent quantitative studies of actin filament turnover during cell locomotion. Cell Motil Cytoskeleton. 1993;25:309–316. doi: 10.1002/cm.970250402. [DOI] [PubMed] [Google Scholar]; (g) Welch MD, Mallavarapu A, Rosenblatt J, Mitchison TJ. Actin dynamics in vivo. Curr Opin Cell Biol. 1997;9:54–61. doi: 10.1016/s0955-0674(97)80152-4. [DOI] [PubMed] [Google Scholar]; (h) Safiejko-Mroczka B, Bell PB. Distribution of cytoskeletal proteins in neomycin-induced protrusions of human fibroblasts. Exp Cell Res. 1998;242:495–514. doi: 10.1006/excr.1997.3871. [DOI] [PubMed] [Google Scholar]; (i) Safiejko-Mroczka B, Bell PB. Reorganization of the actin cytoskeleton in the protruding lamellae of human fibroblasts. Cell Motil Cytoskeleton. 2001;50:13–32. doi: 10.1002/cm.1038. [DOI] [PubMed] [Google Scholar]
- 32.Abbineni G, Safiejko-Mroczka B, Mao C. Development of an optimized protocol for studying the interaction of filamentous bacteriophage with mammalian cells by fluorescence microscopy. Microsc Res Tech. 2010;73:548–554. doi: 10.1002/jemt.20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene W, Gao SJ. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009;5:1–18. doi: 10.1371/journal.ppat.1000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun X, Whittaker GR. Role of the actin cytoskeleton during influenza virus internalization into polarized epithelial cells. Cell Microbiol. 2007;9:1672–1682. doi: 10.1111/j.1462-5822.2007.00900.x. [DOI] [PubMed] [Google Scholar]
- 35.Dausend J, Musyanovych A, Dass M, Walther P, Schrezenmeier H, Landfester K, Mailander V. Uptake mechanism of oppositely charged fluorescent nanoparticles in HeLa cells. Macromol Biosci. 2008;8:1135–1143. doi: 10.1002/mabi.200800123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.