

Abstract
This review describes the use of cryoreduction/annealing EPR/ENDOR techniques for determining the active oxidizing species in reactions catalyzed by heme monooxygenases. The three candidate heme states are: ferric peroxo, ferric hydroperoxo, and Compound I intermediates. The enzymes discussed include cytochromes P450, nitric oxide synthase and heme oxygenase.
Keywords: cytochrome P450, nitric oxide synthase, heme oxygenase, ENDOR, EPR, cryoreduction, dioxygen activation
The heme monooxygenases are a superfamily of heme iron enzymes, mostly thiolate-ligated such as cytochromes P450 and NOS, but also the histidyl-ligated heme oxygenase (HO), that catalyze reductive activation of molecular oxygen for the insertion of single oxygen atom into a wide variety of substrates [1-5]. In this process the other oxygen atom is reduced to water. Heme monoxygenases are critical to many biological processes, including steroid hormone biosyntesis, synthesis of NO, drug metabolism and the detoxification of xenobiotics and found in most classes organisms including bacteria, fungi, plants, insects and mammals [1-10]. The reaction cycle of P450 has been the object of considerable research for over three decades. The classic cytochrome P450-type catalytic cycle is initiated by catalytic reduction of the resting-state heme iron (III) (1) to the ferrous state (2) by NAD(P)H-dependent reductases (Scheme 1), followed by binding of molecular oxygen to give the ferrous dioxy complex (3), which has been observed and characterized. It has long been thought that addition of the second electron to the ferrous oxy complex leads initially to a ferric peroxo intermediate (4)(FeIII-O22-). In the classical scheme, protonation of the distal oxygen then forms a ferric hydroperoxo intermediate (5) (FeIII-OOH) [1, 3, 4]. A second protonation of the distal oxygen then leads to heterolytic O-O bond cleavage with loss of water and generation of iron(IV)oxo porphyrin π-cation radical (Fe(IV)=O P+), known as Compound I (6), which was proposed to be the catalytically active species in substrate hydroxylation. However, it also has been proposed that either the 4 or 5 intermediates, as well as 6, can act as the active species in some enzymes and/or with specific classes of substrates [1-5, 9-15]. In support of this idea, studies with some model heme systems showed that the reactivity of intermediates 4-6 can depend significantly on substrate identity and heme model [16-18]. Obviously, contribution of the intermediates in each monooxygenase process should depend on their life time and reactivity which in turn are determined by substrate and enzyme identities. Another way to view the issue is to consider the reduction of 3 as initiating a process in which successively more reactive species (4-6) can form, with the particular one that actually carries out catalysis being determined by the environment of the particular enzyme and the substrate itself.
Scheme 1.

(no caption)
The short life time of the intermediates 4-6 makes difficult their characterization using conventional kinetic and spectroscopic techniques under physiological conditions and until recently, the ferrous dioxygen complex was the last observable intermediate in the heme-monooxygenases cycles. This limitation has been overcome by application of radiolytic cryoreduction at 77K, which both generates and traps one-electron reduced ferrous oxy heme monooxygenase complexes at 77K that catalytically competent, as shown by annealing to higher temperatures [19-26]. The progress in cryogenic stabilization of some of these intermediates has made possible the direct spectroscopic and structural characterization of intermediate 4 and 5, and in some cases 6 [20-33], as well as the measurement of the kinetics of their interconversion and the formation of product. We have developed cryoreduction/annealing and EPR/ENDOR approaches that identifiy the oxidizing species involved in conversion of bound substrate to product, to date having revealed instances where the classical scheme applies and 6 is the hydroxylating species, but also enzymes and substrates where 5 and either 4/5 are the active intermediate (see for example [20, 22-26]).
Methods of Identification of active oxygen intermediates in cryoreduction experiments
The fact that all the intermediates 4-6 that arise during reductive activation of molecular oxygen are paramagnetic makes EPR and ENDOR techniques uniquely applicable in the studies of these monooxygenase catalysis. Although 3 is not paramagnetic, when the observed primary product of 77K cryoreduction is 4, this state is trapped in the geometry of the parent 3, and thus gives information about its structure, as well. The absence of any interference from the diamagnetic precursor 3, and the high sensitivity of EPR permit all experiments to be conducted at relatively low concentration of the cryogenerated species, and thus allows the use of sufficiently low dose to avoid formation and interference by two-electron reduced species [28, 34].
Oxyhemoproteins in frozen solutions at temperatures below 77K are reduced by the mobile electrons generated by ionizing irradiation (gamma and X-ray, or high energy electrons from accelerator or 32P). Although the initial product is the ferric peroxo intermediate 4, in the majority of cases follow-up protonation to form 5 occurs without accumulation of 4. Intermediate 4 exhibits a characteristic rhombic EPR signal with low g-value anisotropy (∼2.26, 2.17, and 1.96) associated with a low spin ferriheme with end-on bound peroxide. The g-anisotropy in an individual protein was shown to be determined by the strength of H-boding interaction between the ligand and the nearest H-bond donor [35]. Protonation of the peroxo ligand to form 5 leads to a noticeable increase in the g-anisotropy (≥2.29, 2.18, 1.93). Of considerable interest, the properties of 4 and 5 as revealed by EPR are almost independent of the identity of the proximal axial ligand, being virtually the same for histidine and cysteinate as axial ligands. Proton delivery to 5 leads to heterolytic cleavage of the O-O bond, with release of H2O and formation of 6. The EPR spectrum of 6 has been characterized for enzymes with a histidine (peroxidases) [36] tyrosine (catalase) [37] and cysteinate (CPO) axial ligand [38]. In all cases the high-valence heme center of 6 can be described in terms of weak exchange coupling (J) between a ferryl ion (S = 1, Fe(IV)=O) and the porphyrin π– cation radical (S = ½).
The temperature at which 4 converts to 5 depends strongly on the structure of the H-bonding/proton-delivery network linked to the peroxo ligand in the distal heme pocket. This process can occur by tunneling at liquid helium temperatures [39], or can require activation even to temperatures approaching ambient. In the case of the oxy forms of P450, NOS, HO [20, 22-27, 35, 39, 40] the protonation of the basic peroxo ligand occurs at 77K and below because of the presence in the distal pocket of a H-bonded proton delivery network that includes a proton donor remote from the heme and an ordered water molecule that is hydrogen-bonded to the terminal oxygen, with this water likely serving as the mediator of proton transfer to the peroxide ligand [25].
Species 5 is trapped upon 77K cryoreduction of all substrate-free ferrous oxy heme monooxygenases [24, 41]: P450cam [41], P4502B4 [24], NOS [25], P450119 [42] and H369 F P450 2BM3. This indicates that in all these binary oxy complexes the distal pocket contains an efficient proton-delivery network, with an ordered molecule water that is H-bonded to the distal oxygen of the O2 ligand, and that mediates efficient proton transfer to the cryogenerated peroxo ligand at 77K [25]. Perturbation of the proton-delivery network in P450cam by site-specific mutation D251N blocks conversion of 4 to 5 at 77K [20, 22, 39]. Conversely, 77K cryoreduction of oxyglobins, which do not have such a proton delivery network, exclusively generates 4, with the distal histidine NH being H-bonded to the peroxo moiety [43]. In these cases 4 becomes protonated only at T > 170K, a temperature at which a molecule of water might diffuse into the active site [44]. It should be noted that the failure of species 5 to accumulate during annealing of cryogenerated 4 does not necessarily indicate that it is not formed: there are cases where 5 has been shown to form but does not accumulate during reaction of cryogenerated 4 because the subsequent proton-dependent conversion of 5 to 6 is rapid [20, 23, 45]. In a binary complex, intermediate 5 commonly decays only at temperatures above 170K, and this process is accompanied by appearance of the low-spin aqua ferric P450 state. This state is expected to form through conversion of 5 to 6 followed by its reduction by protein residues [28], as shown to occur during reactions of substrate-free cytochromes P450 with alkylperoxides [46-48] or by radicals formed during radiation [28, 30, 49], as Dissociation of H2O2 would instead have formed the nonequilibrium high-spin pentacoordinated ferric P450 state.
As discussed below, when a catalytically active 6 is formed in the presence of substrate, its presence can be unambiguously demonstrated by ENDOR experiments on the primary product state. However, even when 6 is the reactant, as in certain P450 reactions and the hydroxylation of L-Arg by NOS, to date no spectroscopically detectable amounts of 6 are observed during these reactions because of the rapid reaction with bound substrate [20, 21, 23, 25]. Indeed, accumulation of 6 is not detected during annealing of cryoreduced oxy HrP and oxyCPO, both of which are well known to form relatively a stable 6 during reaction with H2O2 [50]. This is interpreted to mean that the highly reactive 6 formed during the annealing of cryoreduced oxy Hrp and CPO is reduced by radical products from radiolysis of the matrix or nearby amino-acid residues. Compound I has been observed in cryoreduction experiments, but only during annealing of cryogenerated ferri peroxo DHP [49]. In this case the Compound I forms at relatively high temperatures (above 200K), and the concentration of radiolytically generated matrix radicals is low [49]. In this context, the assignment to 6 an intermediate formed during X-ray structure determination of cryoreduced oxy P450cam [29] should be viewed with the skepticism, as indeed was noted later by the original authors [5].
If 6 rarely accumulates during cryoreduction/annealing, how can its occurrence be inferred, and how can oxygenations by intermediates 4 and 5 be distinguished from that by 6? These questions are answered by an approach based on EPR/ENDOR analysis of the primary product state forming during annealing of cryoreduced oxy complexes (Scheme 2) that has been developed and successfully applied in studies of P450 and NOS [20, 25, 41]. If the reactive species is 6, the primary product state after insertion of oxygen into a C-H bond of substrate (7) contains the product alcohol bound to the heme iron (III) (Scheme 2) [15, 20, 25, 41]. In this state an isotopic label on the substrate (deuteron in Scheme 2; 15N for NOS, see below) can be detected directly by ENDOR; also, the hydroxyl proton originates from substrate and its ENDOR signal is detectable. If reaction occurs in D2O with protonated substrate, the product should be ROH, not ROD, [20] and conversely when deuterated substrate is used in H2O. In the relaxed product state arising during annealing at higher temperatures the hydroxyl proton of substrate then can exchange with solvent. The source of the exchangeable H or D in the bound product can be determined by ENDOR spectroscopy [20, 25, 41]. If instead, 4 or 5 is the reactive species, the primary product would contain a water coordinated to the heme and scrambled hydroxyl proton (Scheme 2). In this case the protons of coordinated water should show signals with significant hyperfine coupling, while the remote substrate in this state will show 1,2H ENDOR signals with small hyperfine coupling.
Scheme 2.
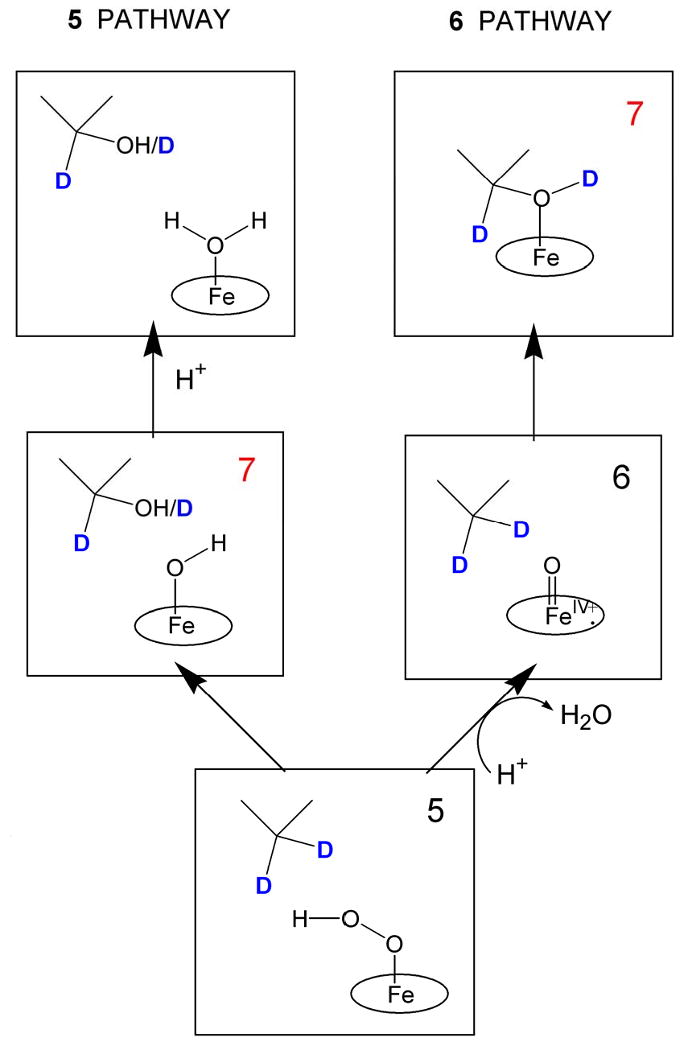
Alternative reaction pathways during hydroxylation of 5-d2-camphor by 5 and 6.
Identification of the active species can be considerably aided by measurements of the solvent kinetic isotope effect (sKIE) on the reaction of trapped intermediates and on product formation, and of the pH dependence of the reaction kinetics [25, 51]. When the rate-limiting step in a reaction is proton transfer, a significant pH dependence and substantial sKIE are expected [25, 51]. Thus, a significant sKIE is expected when the rate-limiting step of monooxygenation is protonation that converts 4 to 5 or 5 to 6. Measurement of the sKIE is of particular importance when the trapping of 4 upon 77K cryoreduction of 3 opens the possibility of its involvement as an active species in monooxygenase process. Annealing may show only that the reaction of intermediate 4 is accompanied by the appearance of product without accumulation of 5 or 6, yet this does not necessarily mean that 4 is the reactive species. It is possible that protonation of 4 to form 5, and possibly of 5 to form 6, occurs very rapidly, as does the reaction of 5 or 6 with substrate [23, 49]. To test for direct reaction of 4, one must measure the sKIE of the loss of 4; the sKIE should be negligible for direct reaction of 4 with substrate. Correspondingly, when 5 is the primary cryogenerated species, this narrows the possible candidates for active species to 5 or 6. However in this case, both the direct reaction of 5 as oxygenating agent and its conversion to 6 involve O-O bond cleavage that is likely to be proton assisted, and thus to display an sKIE.
Application of optical absorption spectroscopy in such studies is of limited use in distinguishing active species because of the strong overlap of the spectra of the 4-6 and the parent 3. Because of the overlap, this technique requires a high radiation dose to achieve a substantial reduction of 3, and such studies can be complicated by formation of two-electron reduced species at high dose. In addition, the high sensitivity of optical spectra of a heme moiety to all structural changes can complicate the identification of intermediates. Thus an optical study of the decay of 5 during annealing of cryoreduced oxy HRP suggested that 5 decays to Fe(III)HRP and H2O2, whereas the EPR spectrum of a cryoreduced/annealed sample that had undergone a second cryoreduction showed that the annealing had generated compound II, thereby revealing that 6 had been formed and reduced by radicals in the matrix [50, 52]. However, optical studies are helpful for more complete characterization of the primary cryoreduced species and when it is difficult to prepare the high concentration of 3 needed for EPR/ENDOR. In addition, knowledge of the optical spectra of the cryogenerated intermediates can be quite useful for identifying the state of a crystal during X-ray diffraction measurements [29-33], and of course is central in spectroscopic techniques with high time resolution at ambient temperature (stopped flow, flash photolysis, pulse radiolysis) [53]. RR was shown to be very helpful for studying structural changes during protonation of the Fe(III)[O2]2- moiety [54].
Finally we have found that application of Mossbauer spectroscopy allows to identify some EPR silent species forming during annealing the cryogenerated intermediates [28].
Compound I (6) as active intermediates in hydroxylation of substatrate by P450 and NOS
The commonly accepted mechanism for P450 catalyzed monooxygenations involves hydrogen abstraction by the ferryl ion of 6 and subsequent oxygen rebound to the alkyl radical of the substrate (see rev [2, 4, 10]). When applied to such reactions as the hydroxylation of camphor by P450cam, the rebound mechanism accounts for key experimental data; the partial loss of stereochemistry and geometrical rearrangement data, as well as the observed primary kinetic isotope effect (KIE) when the activated C–H bond is replaced by C–D [10]. Results of theoretical studies are consistent with this mechanism [15].
Despite numerous attempts to accumulate the intermediate 6 for a P450 under physiological conditions has proven to be experimentally unattainable presumably because of its high reactivity with substrate. Short-lived Compound I-like intermediates with characteristic UV spectra were detected during reactions of substrate free ferric P450s with alkyl peroxides [47, 48]; these subsequently oxidize a nearby protein residue and convert into compound ES with a ferryl neutral porphyrin and protein radical [46, 47]. Newcomb and coworkers presented a new method for preparing P450 compound I-like (probably the perferryl (Fe(V)=O porphyrin)) species based on laser flash photolysis of the compound II generated by reaction ferric P450 with peroxonitrite, and have studied their reactivity [55]. However this species appears to be puzzlingly stable, decaying at ambient temperature with lifetime much longer than expected from other experiments. Thus it remains unclear how the oxidizing species generated by reaction with alkyl peroxide or photochemically are related to the oxidizing species in enzymatic reaction.
Cryoreduction/annealing studies have established that Compound I is the hydroxylating intermediate in a number of key enzymatic monooxygenase reactions. The most detailed studies were carried out with cytochrome P450cam [20, 41, 56] and gsNOS [25] which form moderately stable oxycomplexes. This reaction cycle for P450cam is likely to be shared by most member of the P450 family, and this protein commonly is considered as prototypical [2]. NO formation by NOS occurs by two successive monooxygenase reactions; the first stage is the hydroxylation of L-Arg to form N-hydroxy arginine (NOHA). The second stage, the conversion of NOHA to NO and atrulline, is discussed below.
P450cam
Cryoreduction of the ternary complex of ferrous oxy P450cam with camphor at 77K produces 5 [20]. When cryoreduction is performed at helium temperatures, the product trapped is 4, and protonation of the peroxo ligand to generate 5 occurs at temperatures below 55K [20]. During extended annealing at ∼ 180K the EPR signal of 5 disappears and is replaced by a signal from the primary product state of camphor hydroxylation 7, a non-equilibrium conformer of the complex of ferric P450cam with 5-exo hydroxycamphor. The decay of 5 slows down by factor of sKIE ∼ 2 in deuterated solvent [56], which indicates that the rate-limiting step for reaction of this species involves activation of the hydroperoxy moiety by transfer of the “second “ proton of catalysis. The primary product 7 converts to a relaxed conformer during annealing at ∼ 220 K; at low protein concentration the bound 5-exo hydroxycamphor then dissociates at temperatures above 240K.
1H ENDOR spectra of 7 in experiments with 1H-camphor in 2H2O demonstrate that the hydroxyl proton of the bound hydroxycamphor originates from the substrate C5-H rather than from solvent, precisely as expected for the rebound mechanism with 6 as active species (Scheme 2)) [20]. Conversely, when experiments are carried out with 5-2H2-camphor in 1H2O, 7 carries a deuteron on the hydroxyl as well as on the bound hydroxy-camphor [57]. In the relaxed product complex this proton is exchanged with solvent DFT/MM calculations support this conclusion, suggesting that the initially observed nonequilibrium state 7 is the hexacoordinated product complex with a short Fe-O distance (∼ 2 A), and that the heme pocket then relaxes to accommodate the longer Fe-O bond of 2.67 A found in the crystal structure [15]. Cryoreduction/annealing EPR/ENDOR studies thus unambiguously confirm that in the P450cam catalytic cycle 6 is the active oxidizing species.
The replacement of Asp 251 with Asn in P450cam leads to the trapping of 4 upon 77K cryoreduction, indicating that the mutation inhibits the protonation of the peroxo ligand [20]. However, the primary peroxo species converts to 5 at 170K which in turn reacts at 190K. ENDOR established that this reaction also is accompanied by formation of a non-equilibrium form of the hydroxy-camphor product-enzyme complex as the primary product state, thereby establishing that this occurs through formation and reaction of 6 [20].
Cryoreduction/annealing of the ternary oxy T252A P450cam complex with camphor produces a quite different result [20]. The 77 K cryoreduction generates 5 that is spectroscopically similar to that for wt P450cam, but this intermediate does not react with bound camphor, although it does epoxidize methylene camphor (see below). This confirms that 5 is not active in enzymatic camphor hydroxylation.
NOS:Stage I
gsNO
Cryoreduction/annealing- EPR/ENDOR experiments with the arginine-bound oxy NOS from Geobacillus stearothermophilus confirm that 6 is the reactive heme species in the first stage of NO production, the hydroxylation of L-arginine to N-hydroxy–L-arginine (NOHA) [25]. Cryoreduction of ferrous oxy gsNOS complex with Arg (or N-Me-Arg) at 77K generates intermediate 5. Subsequent annealing of 5 at 145K generates the primary product state of reaction, 7. This was determined by EPR/ENDOR spectroscopy to have NOHA coordinated to the ferric heme, thereby establishing that 6 indeed is the reactive species (Scheme 3). The decisive experiment was a 15N ENDOR measurement of 7 prepared with Arg isotopically labeled with 15N at the terminal guanidino nitrogens. It revealed the 15N ENDOR signal from NOHA coordinated to Fe that identified 6 [25]. Figure 1 presents a view of this process based on crystal structures of gsNOS. Upon annealing at temperatures above 220K, 7 relaxes to the equilibrium pentacoordinate state in which NOHA remains bound to the protein, but not as a ligand to the ferric heme.
Scheme 3.
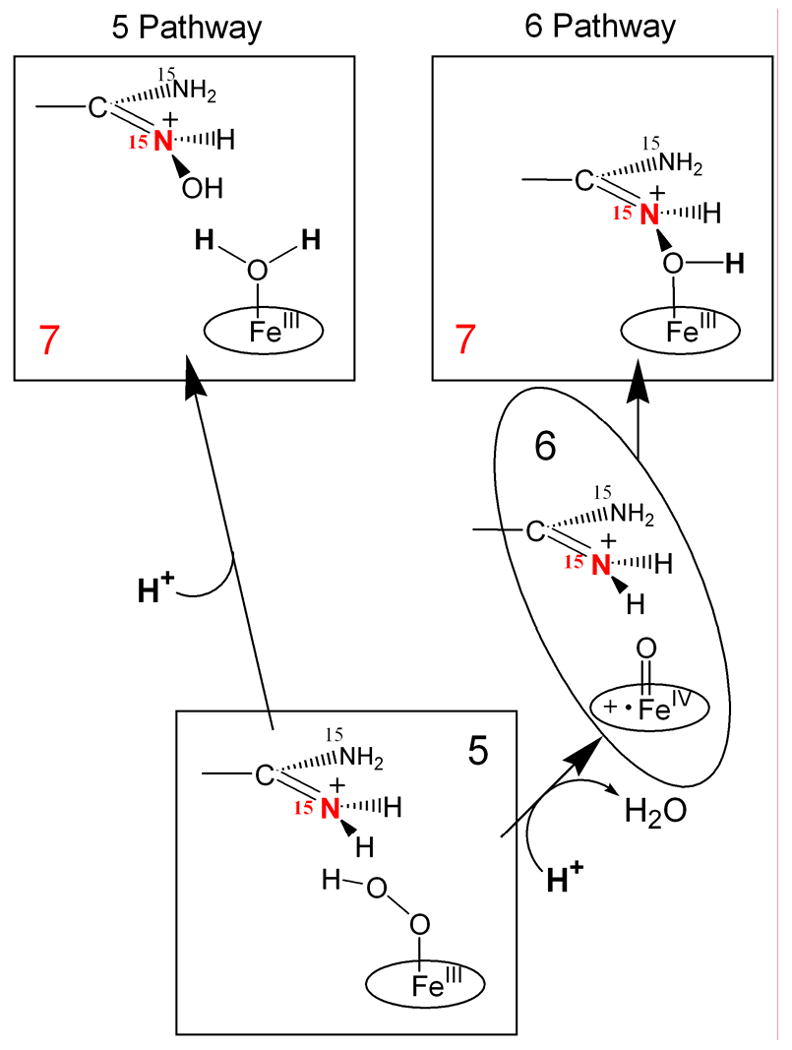
Alternative reaction pathways during reaction of bound Arg with 5 and 6.
Fig. 1.
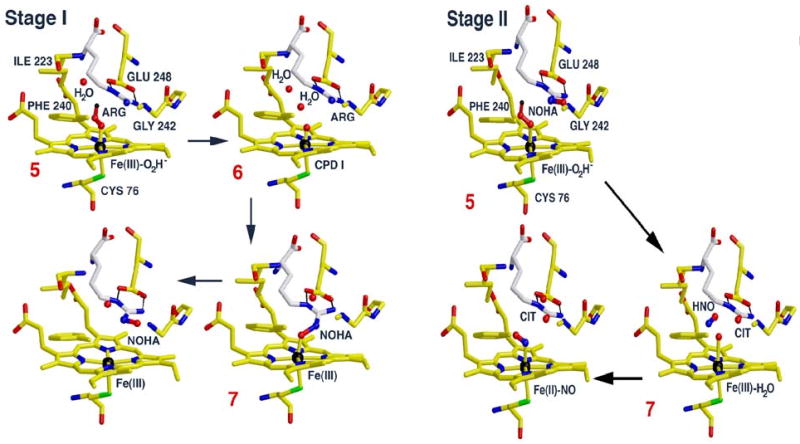
Catalytically active states and primary products of reaction for Stages I and II of the gsNOS catalytic cycle as revealed by cryo-annealing EPR/ENDOR studies. These representations are based on crystallographic structures of bacterial NOS active centers with substrates and heme ligands bound, (PDB codes: 1M7V, 1M7Z, 2FC1, 2FBZ), but are purely figurative, and not meant to capture the yet uncharacterized conformational changes that likely accompany these states.
The very low temperature at which hydroxylation of L-Arg by gsNOS occurs implied that the second proton for catalysis is provided by the side chain COOH group of highly conserved Glu residue that H-bonds to the substrate guanidinium group, which is in close vicinity to the heme coordination site. The conversion of 5 to 6 shows a large solvent kinetic isotope effect, sKIE ≅8, and accelerates at lower pH, which indicates that rate-limiting step in this process is a proton assisted O-O bond cleavage.
eNOS
Although the active site of gsNOS is similar to that of eNOS, cryoreduction of the termary complex of oxy-eNOS with Arg produces only 4, not 5, and 5 does not accumulate during conversion to product NOHA. EPR/ENDOR spectra of the primary product state 7 formed after annealing at 165K are very like those for gsNOS, suggesting that 6 is the active species for both. This difference in the observed product of 77K cryoreduction likely reflects differences between the H-bond networks that provide proton delivery to the oxyheme moiety in e- and gsNOSs. It was suggested that in the oxy eNOS –Arg complex there is no ordered water adjacent to the peroxy ligand and the structural relaxation during annealing at 165K allows a water to approach the peroxy heme center and convey a proton to the ligand, forming 5. This does not accumulate at 165 K because of rapid conversion to 6
P450 2B4
For completeness, we note that the cryoreduction/annealing procedure of examining the 7 to test the roles of 5 and 6 does not work if 7 is unstable to dissociation of hydroxylated product at the annealing temperatures required, which happens with P450 2B4. As with P450cam, cryoreduction of the ternary oxy 2B4 complex with butylated hydroxytoluene as substrate generates 5, and subsequent annealing quantitatively forms product, in this case hydroxymethyl BHT and 3-hydroxy-tert-butyl BHT. However, the annealing reaction occurs at unusually high temperatures, above 175K, and as a result, the primary product state 7 immediately relaxes with the release of product, so that one observes direct formation of the high-spin pentacoordinate ferric P4502B4 [24]. However, the fact that cryoreduction of substrate-free oxy complex generates 5, and that only the low-spin aqua ferric P450 is formed during annealing of 5, suggests that 6 is most likely the active species, because as noted above, this state only forms upon reduction of 6 by a nearby protein residue or radiolytically generated radicals.
Ferric peroxo/hydroperoxo species (4/5) as active oxygen intermediates in heme monooxygenase cycle
As noted above, when 4 is trapped during 77K cryoreduction of oxy heme monooxygenases, then 4, 5, or 6 are candidates for the oxygenating intermediate; when 5 is trapped then the list is limited to 5 or 6. We now discuss the cases in which cryoreduction/annealing has shown that 6 is not the reactive species, then cases that are yet to be resolved. The potential reactivity of 4 and 5 as alternatives to 6 as the oxygenating intermediate has been considered in numerous reviews [1, 3-5, 8, 10, 11, 17, 58-62]. It has been proposed that the nucleophilic ferric peroxy anion of 4 is involved in P450 catalyzed 14α-demethylation of lanosterol, in the deformylation of 19-aldo-AD to estrone by aromatase, and in CYP17-catalyzed 17α, 20 bond scission that converts 17α-hydroxyprogesterones into androgens (see reviews [3, 5, 10]). Coon and coworkers suggested that the catalytic elimination of the aldehyde group from a series of aliphatic aldehyde by CYP2B4, with formation of the corresponding olefins, can occur through reaction of 4 [63]. More recently, it was proposed the intermediate 4 of NOS is involved in the conversion of NOHA to citrulline and NO in the second stage of NO formation [6, 7, 9].
Intermediate 5 was considered as a possible alternative to 6 in hydroxylation of hydrocarbons [10, 61]. However DFT computations and experimental studies with mutant enzymes in which conversion of 5 to 6 is blocked suggest that 5 is much more sluggish than 6 [15, 17]. For example, the T252A mutant of P450cam, in which Thr252 plays a key role in the conversion of 5 to 6, does not hydroxylate camphor, but is capable of epoxidation of the double bond of 5-methylenyl camphor, albeit more sluggishly than 6 of the WT enzyme [18]. Curiously, however, the double mutant of P450cam, T252A/D251N, in which the protonation machinery has been disrupted by mutations of both Thr252 andAsp251, is able to hydroxylate camphor [5] despite the T252A mutation that should have presumably blocked the formation of 6. This implies either that 5 has a variable reactivity in different mutants that include the T → A mutation, or that variable quantities of 6 may be present even in the case of the different T → A mutant. Thus, while the participation of 5 in oxidation is still a clouded issue, the data behave as though more than one oxidant is available to P450.
Heme oxygenase degrades heme, and the first step in the process is a monooxygenase reaction of an unusual kind, the self-hydroxylation of its heme to form α-meso-hydroxyheme, using the histidyl-ligated heme group as both a prosthetic group and substrate [33, 64]. Among the evidence for 5 as the mesohydroxylating species in HO catalysis, H2O2 was found to replace NADPH/O2 in supporting the first step in heme oxidation, while acyl hydroperoxides that generate 6 were incompetent [64].
HO
EPR/ENDOR, cryoreduction/annealing studies provided direct evidence for reaction by 5 during normal catalytic turnover of HO [22, 51]. 77K cryoreduction of rat heme oxygenase-1 (HO) was shown to produce 5, which synchronously converts to the α-meso-hydroxyheme product during progressive annealing at 214K. This conversion exhibits an sKIE = 2.3, which indicates that the synchronous bond formation between the distal oxygen and α-meso-carbon is activated by proton transfer to the Fe3+-OOH moiety through the distal pocket H-bond network, likely from a carboxyl group of Asp 140 [51]. Disruption of the H-bonding network within the distal pocket of HO by alanine mutation of residue D140 prevents product formation. The state 5 of D140A HO instead undergoes heterolytic cleavage of the O-O bond, ultimately yielding the EPR silent compound II species that does not form product [22].
NOS, Stage II
In the second stage of NOS catalysis, NOHA is converted to citrulline and NO. In the enzymatic cycle of mammalian NOSs under physiological conditions, a tetrahydrobiopterin cofactor reduces the ferrous oxy NOS/NOHA complex to 4/5, forming the BH4 radical. The resulting enzyme intermediate goes on to form Fe(III)NOS, citrulline, and NO-/HNO; the latter in turn is oxidized by the BH4 radical to the final product, NO [6]. When the electron supplied by BH4 instead is delivered externally, through use of H2O2 as oxidant or through cryoreduction, the final product of reaction is expected to be the Fe(II)NO complex, formed by reaction of the Fe(III) heme with NO-/HNO. In support of the assignment of 4/5 as the stage II reactive species, not 6, Marletta and coworker presented evidence that during the shunt reaction of mammalian NOS with hydrogen peroxide, cyanoornithine and inorganic nitrogen oxides are products when 6 is expected to be the reactant with NOHA, whereas citrulline and Fe(II)NO are the products when 5 is expected to be the reactant [65, 66].
Cryoreduction at 77K of ternary complexes of both oxy gsNOS and oxy eNOS with bound NOHA yields 4, even though cryoreduction of oxy gsNOS form 5 both in the absence and presence of Arg. This difference indicates that binding of NOHA perturbs the proton delivery network. Annealing of the 4/NOHA complex for gsNOS gave convincing evidences for 5 as the active oxidant in the second half of nitric oxide synthase reaction (Scheme 4). The gsNOS, 4/NOHA intermediate protonates at 160 K, with a large sKIE to form 5/NOHA, which reacts further, yielding a product state with H2O bound to the ferriheme. Above 170K the ferriheme signal in turn disappears and that of NO-Fe(II)gsNOS appears, indicating that HNO is one product of the reaction of 5; chemical analysis shows that the other product is citrulline, with no detectable amounts of cyanoornithine. Taken together with the observations of Marletta and coworkers that when 6 is formed it reacts with NOHA to generate cyanoornithine, these results clearly implicate 5 as the oxidant in the second step of the NOS reaction. In the H2O2 shunt reactions and in cryoreduction/annealing, as well as in the physiological reaction, HNO and citrulline are formed by two-electron oxidation of NOHA. Physiologically, formation of NO occurs through the additional oxidation of NO-/HNO by the BH4 radical formed in the initial reduction of 3. In the other two processes, the NO-/HNO reacts with the ferriheme to generate Fe(II)NO. Figure 1 also presents a representation of stage II catalysis.
Scheme 4.
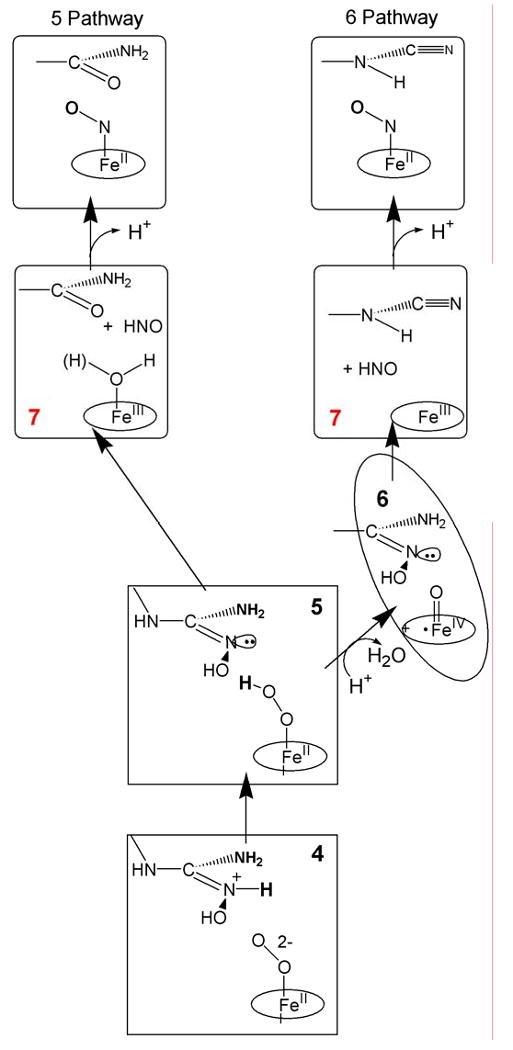
(no caption)
Given that 77K cryoreduction of eNOS produces 4, not 5, it is possible that eNOS has 4 as the active species, not 5. The key experiment for establishing whether 4 in fact is the reactant for eNOS, or whether 4 protonates to form 5, will be to measure the sKIE in the decay of 4/NOHA for eNOS.
It should be noted that theoretical estimations made by Thieles and coworkers [67] suggest the redox potential for the Fe(II)O2/4 couple is E ≈ -1 V, which is much lower than that for physiological electron donors in the enzymatic cycle. This suggests that under physiological conditions the oxy ferrous heme can only be reduced by proton-coupled ET to form 5. On the other hand, although 5 is formed in the reaction sequence, the interaction of the hydroperoxo moiety of 5 with bound NOHA might involve its deprotonation, as proposed for the last step in the aromatase cycle - deformylation of 19-aldoandrostenedione (19-aldo-AD) [58, 68].
Cyp 19
The annealing of cryoreduced ternary oxy CYP 19-androstenedione (AD) reported by Sligar and coworkers [26] was performed to test whether that conversion of AD into 19-OH-AD catalyzed by aromatase utilizes 4 as the active species. Aromatase metabolism of AD proceeds in three steps: in the first, the C19 methyl of AD is hydroxylated forming 19-hydroxy-AD. A second hydroxylation at C19 produces the diol, which is dehydrated to yield 19-aldo AD. 19-aldo AD is deformylated in the third step, forming estrone. It was proposed that in two first steps 6 is the active species, while in deformylation of 19-aldo AD nucleophilic 4 directly reacts with the substrate's aldehyde functionality. The 77K cryoreduction of the ternary oxy P450-substrate complex indeed yielded intermediate 4. The authors interpreted their data as indication that 4 converts directly into the ferric P19-19-OH-AD complex after annealing at 210K without formation of 5 whose EPR signal was not observed [26]. As discussed above, however, the failure to see accumulation of 5 during annealing is not sufficient to rule out either 5 or 6 as the active species. Careful examination of the annealing pattern for cryoreduced oxy cyP19-substrate presented in this publication discloses that the decay of 4 is complete at 195K without the appearance of any new EPR-active species. This suggests that 4 does not directly convert to 7 during this process, which forms later and at higher temperature (210K). Reexamination of this process likely will reveal a new signal. If 4 or 5 is the reactive species, one would expect to observe aquo-ferriheme primary product, while measurement of the sKIE for decay of 4 would show whether 4 is protonated to form 5, or is the active agent.
P450cam mutants/alternate substrates
Cryoreduction/annealing investigations confirmed an earlier proposal that 5 is the active species in epoxidation of 5-methylenyl camphor [18] catalyzed by P450cam and α-hydroxylation of heme in the heme oxygenase reaction. It was suggested that the Asp251-Thr252 dyad working in concert with active-site water molecules, and control of the regiospecificity of protonation of the 4 and 5 intermediates. The T252A mutant does not hydroxylate camphor but is able to epoxidize a double bond, albeit more sluggishly then wt enzyme [14, 18]. This mutant is unable to form 6 due to nonregiospecific protonation of 5 and therefore epoxidations of alkenes was thought to be produced by 5 [14]. Cryoreduction/annealing experiments confirmed this supposition. As formed cryoreduction of the ternary complexes of ferrous oxy wt and T252A P450 with 5-methylenyl camphor produced 5 with similar EPR and ENDOR properties, both formed the epoxide upon annealing product [41, 56]. However, the primary product state 7 trapped during annealing the wt 5 has the epoxide product bound to the ferriheme, as shown by the presence of strongly–coupled, non-exchangeable proton ENDOR signals, and thus implies that 6 is the intermediate that epoxidizes 5-methylenyl camphor (Scheme 2). In contrast, 7 formed upon epoxidation of the substrate by 5 of T252A P450 shows an exchangeable proton ENDOR signal from water coordinated to ferric heme which indicates that the reactive species in the mutant is the 5 (Scheme 2) [18].
Substrate control of the active species in heme monooxygenase catalysis
Many of the cytochrome P450s have a broad spectrum of possible substrates for individual enzyme. In some sense the action of these enzymes may be better described by a version of Koshland's model of “induced fit” according to which different substrates generate conformational changes that may modulate the reactivity of the active site. This is indicated by Scheme 5, where Y represents the heme-pocket component whose properties are modulated by substrate. Substrate effects have been reported for cytochrome P450s and NOS, where it was found out that substrate can modify the structure and reactivity of the enzyme active site in the monooxygenase cycle [24, 25, 27, 41, 69-71]. The results of cryoreduction/annealing studies show directly that the active oxidizing species in monooxygenase cycles depends not only on enzyme identity which is expected but also on the substrate itself for the same enzyme. As one example, given above, in the formation of NO by NOS, the Stage I oxidation of Arg to NOHA is carried out by 6, while the Stage II conversion of NOHA to citrulline and NO is catalyzed by 5. Analogously, the hydroxylation of camphor by P450cam used 6 as the oxidant, whereas in the T252A mutant, 5 epoxidizes methylenylcamphor. To be tested are the proposals that multiple oxidants are involved in multiple reactions steps of steroid hormone biosythesis by P450aromatase (CYP19), lanosterol 14R-demethylase (CYP51) and progesterone 17R-hydroxylase/17,20-lyase (CYP17) (see [3, 4, 8, 10] and ref herein).
Scheme 5.
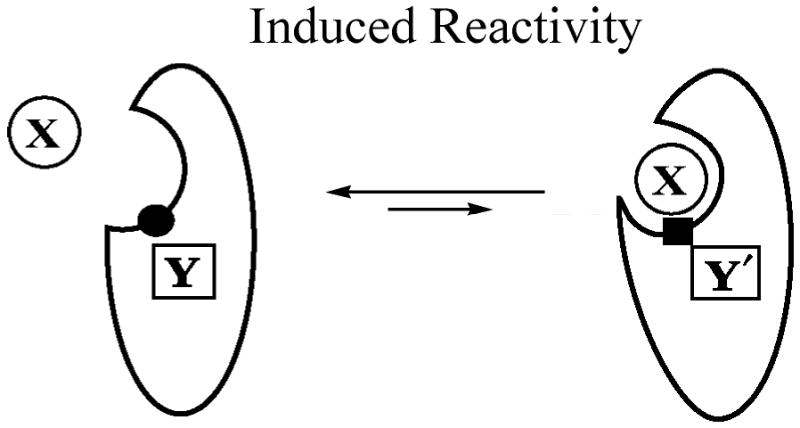
(no caption)
The contribution of the active species in the oxidation of substrate is expected to be determined (i) by the degree of substrate reactivity versus the increasing activities of the three heme oxidants, 4, 5, and 6 (Scheme 1); and (ii) by perturbing effects of bound substrate on proton delivery. The lifetime of the intermediates depends not only on inherent properties of the oxy heme moiety but also on ability of the proton delivery network to provide a proton to the peroxo ligand, and then a second proton to the hydroperoxo ligand. In cases where 77K cryoreduction leads to the formation of 4, it is because protonation of the cryogenerated peroxo ligand has been impeded, by substrate modulation of the proton delivery network.
Model studies with iron(III) meso-tetramesithyl porphyrin complex demonstrate that in general the relative reactivities of the 5 and 6 depend very much on the type of reaction and the substrate nature [16]. Particularily, the studies with the M69A mutant of cytochrome C552 have demonstrated that 5 is not reduced by ascorbate whereas 6 is [17]. Intermediate 5 of P450 101 was shown to produce the epoxidation of 5-methylene camphor with lower rate as compared to that for compound I [14, 18].
Theoretical computation have showed that reactivity and stability of 5 and 6 can be modulated significantly by bound substrate [15, 72]. It was found that substrates of different sizes might dramatically affect the lifetime of 6. Thus, in the absence of the substrate (camphor) the lifetime of 6 was extremely short while in the presence of camphor derivatives of increasing size, the lifetime increased by as much as 80-fold [41].
Summary
Difficulties in characterizing the intermediates that occur during monooxygenations catalyzed by heme enzymes have been largely overcome by application of cyroreduction/annealing techniques. These not only allow us to generate and trap these intermediates for characterization by EPR/ENDOR, but also enable measurements of the kinetics of their interconversion and the formation of product. This review has described the approach and some of the key findings regarding the reactive intermediate for each of a number of key heme monooxygenases.
Research highlights.
EPR/ENDOR spectroscopy of radiolytically cryoreduced oxy heme monooxygenases has
-
identified the active oxidizing species in:
cytochrome P450 reactions
the two stages by which NOS generates NO
meso-heme hydrohylation by heme oxygenases
demonstrated the influence of substrate in modulating monooxygenase activity
Acknowledgments
This work has been supported by the NIH (HL 13531, BMH). It would not have been possible without the efforts of our many collaborators whose work is referenced. It further relied on the support of our colleagues, Mr. Clark Davoust and Peter Doan.
Abbreviations
- P450
cytochrome P450
- NOS
niric oxide synthase
- eNOS
endothelial NOS
- gsNOS
NOS from Geobacillus stearothermophilus
- NOHA
Nω-hydroxy-L-arginine
- BH4
tetrahydropterin
- CPO
chloroperoxidase
- HrP
horseradisch peroxidase
- HO
heme oxygenase
- ENDOR
electron nuclear double resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- 1.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz de Montellano PR. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortiz de Montellano PR, De Voss JJ. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 4.Hlavica P. Eur J Biochem. 2004;271:4335–4360. doi: 10.1111/j.1432-1033.2004.04380.x. [DOI] [PubMed] [Google Scholar]
- 5.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 6.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 7.Marletta MA, Hurshman AR, Rusche KM. Curr Opin Chem Biol. 1998;2:656–663. doi: 10.1016/s1367-5931(98)80098-7. [DOI] [PubMed] [Google Scholar]
- 8.Bernhardt R, Waterman MR. Met Ions Life Sci. 2007;3:361–396. [Google Scholar]
- 9.Zhu Y, Silverman RB. Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 10.Perera R, Jin S, Sono M, Dawson JH. Met Ions Life Sci. 2007;3:319–359. [Google Scholar]
- 11.Guengerich FP. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 12.Guengerich FP, Vaz ADN, Raner GN, Pernecky SJ, Coon MJ. Mol Pharmacol. 1997;51:147–151. doi: 10.1124/mol.51.1.147. [DOI] [PubMed] [Google Scholar]
- 13.Isin EM, Guengerich FP. Biochimica et Biophysica Acta. 2007;1770:314–329. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 14.Jin S, Bryson TA, Dawson JH. JBIC, J Biol Inorg Chem. 2004;9:644–653. doi: 10.1007/s00775-004-0575-7. [DOI] [PubMed] [Google Scholar]
- 15.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 16.Fertinger C, Hessenauer-Ilicheva N, Franke A, van Eldik R. Chem--Eur J. 2009;15:13435–13440. S13435/13431–S13435/13434. doi: 10.1002/chem.200901804. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe Y, Nakajima H, Ueno T. Acc Chem Res. 2007;40:554–562. doi: 10.1021/ar600046a. [DOI] [PubMed] [Google Scholar]
- 18.Jin S, Makris TM, Bryson TA, Sligar SG, Dawson JH. J Am Chem Soc. 2003;125:3406–3407. doi: 10.1021/ja029272n. [DOI] [PubMed] [Google Scholar]
- 19.Davydov R, Kappl R, Hutterman R, Peterson J. FEBS Lett. 1991;295:113–115. doi: 10.1016/0014-5793(91)81398-r. [DOI] [PubMed] [Google Scholar]
- 20.Davydov R, Makris TM, Kofman V, Werst DW, Sligar SG, Hoffman BM. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 21.Denisov IG, Makris TM, Sligar SG. J Biol Chem. 2001;276:11648–11652. doi: 10.1074/jbc.M010219200. [DOI] [PubMed] [Google Scholar]
- 22.Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman B. J Am Chem Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- 23.Davydov R, Ledbetter-Rogers A, Martasek P, Larukhin M, Sono M, Dawson JH, Masters BSS, Hoffman BM. Biochemistry. 2002;41:10375–10381. doi: 10.1021/bi0260637. [DOI] [PubMed] [Google Scholar]
- 24.Davydov R, Razeghifard R, Im SC, Waskell L, Hoffman BM. Biochemistry. 2008;47:9661–9666. doi: 10.1021/bi800926x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davydov R, Sudhamsu J, Lees NS, Crane BR, Hoffman BM. J Am Chem Soc. 2009;131:14493–14507. doi: 10.1021/ja906133h. [DOI] [PubMed] [Google Scholar]
- 26.Gantt SL, Denisov IG, Grinkova YV, Sligar SG. Biochem Biophys Res Commun. 2009;387:169–173. doi: 10.1016/j.bbrc.2009.06.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davydov RM, Chauhan N, Thackray SJ, Anderson JLR, Papadopoulou ND, Mowat CG, Chapman SK, Raven EL, Hoffman BM. J Am Chem Soc. 2010;132:5494–5500. doi: 10.1021/ja100518z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Serres R, Davydov RM, Matsui T, Ikeda-Saito M, Hoffman BM, Huynh BH. J Am Chem Soc. 2007;129:1402–1412. doi: 10.1021/ja067209i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet BM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 30.Berglund GI, Carlsson GH, Smith AT, Szoeke H, Henriksen A, Hajdu J. Nature (London) 2002;417:463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 31.Unno M, Chen H, Kusama S, Shaik S, Ikeda-Saito M. J Am Chem Soc. 2007;129:13394–13395. doi: 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]
- 32.Hersleth HP, Hsiao YW, Ryde U, Goerbitz CH, Andersson KK. Biochem J. 2008;412:257–264. doi: 10.1042/BJ20070921. [DOI] [PubMed] [Google Scholar]
- 33.Matsui T, Iwasaki M, Sugiyama R, Unno M, Ikeda-Saito M. Inorg Chem. 2010;49:3602–3609. doi: 10.1021/ic901869t. [DOI] [PubMed] [Google Scholar]
- 34.Davydov R.,Hoffman BM In preparation
- 35.Davydov R, Satterlee JD, Fujii H, Sauer-Masarwa A, Busch DH, Hoffman BM. J Am Chem Soc. 2003;125:16340–16346. doi: 10.1021/ja037037e. [DOI] [PubMed] [Google Scholar]
- 36.Schulz CE, Devaney PW, Winkler H, Debrunner PG, Doan N, Chiang R, Rutter R, Hager LP. FEBS Lett. 1979;103:102–105. doi: 10.1016/0014-5793(79)81259-4. [DOI] [PubMed] [Google Scholar]
- 37.Benecky MJ, Frew JE, Scowen N, Jones P, Hoffman BM. Biochemistry. 1993;32:11929–11934. doi: 10.1021/bi00095a024. [DOI] [PubMed] [Google Scholar]
- 38.Rutter R, Hager LP, Dhonau H, Hendrich M, Valentine M, Debrunner P. Biochemistry. 1984;23:6809–6816. doi: 10.1021/bi00321a082. [DOI] [PubMed] [Google Scholar]
- 39.Davydov R, Chemerisov S, Werst DE, Rajh T, Matsui T, Ikeda-Saito M, Hoffman BM. J Am Chem Soc. 2004;126:15960–15961. doi: 10.1021/ja044646t. [DOI] [PubMed] [Google Scholar]
- 40.Davydov R, Kofman V, Nocek J, Noble RW, Hui H, Hoffman BM. Biochemistry. 2004;43:6330–6338. doi: 10.1021/bi036273z. [DOI] [PubMed] [Google Scholar]
- 41.Davydov R, Perera R, Jin S, Yang TC, Bryson TA, Sono M, Dawson JH, Hoffman BM. J Am Chem Soc. 2005;127:1403–1413. doi: 10.1021/ja045351i. [DOI] [PubMed] [Google Scholar]
- 42.Denisov IG, Hung SC, Weiss KE, McLean MA, Shiro Y, Park SY, Champion PM, Sligar SG. J Inorg Biochem. 2001;87:215–226. doi: 10.1016/s0162-0134(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 43.Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM. J Am Chem Soc. 1999;121:10656–10657. [Google Scholar]
- 44.Kappl R, Höhn-Berlage M, Hüttermann J, Bartlett N, Symons MCR. Biochim Biophys Acta. 1985;827:327–343. [Google Scholar]
- 45.Shanmugam M, Zhang B, McNaughton RL, Kinney RA, Hille R, Hoffman BM. J Am Chem Soc. 2010 doi: 10.1021/ja106432h. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jung C, Schuenemann V, Lendzian F. Biochem Biophys Res Commun. 2005;338:355–364. doi: 10.1016/j.bbrc.2005.08.166. [DOI] [PubMed] [Google Scholar]
- 47.Spolitak T, Dawson JH, Ballou DP. J Biol Chem. 2005;280:20300–20309. doi: 10.1074/jbc.M501761200. [DOI] [PubMed] [Google Scholar]
- 48.Egawa T, Shimada H, Ishimura Y. Biochem Biophys Res Commun. 1994;201:1464–1468. doi: 10.1006/bbrc.1994.1868. [DOI] [PubMed] [Google Scholar]
- 49.Davydov R, Osborne R, Shanmugam M, Du J, Dawson J, Hoffman B. J Am Chem Soc. 2010 doi: 10.1021/ja1059747. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denisov IG, Makris TM, Sligar SG. J Biol Chem. 2002;277:42706–42710. doi: 10.1074/jbc.M207949200. [DOI] [PubMed] [Google Scholar]
- 51.Davydov R, Matsui T, Fujii H, Ikeda-Saito M, Hoffman BM. J Am Chem Soc. 2003;125:16208–16209. doi: 10.1021/ja038923s. [DOI] [PubMed] [Google Scholar]
- 52.Davydov R., Perera R, Sono, Dawson J.Hoffman BM, In preparation
- 53.Blyumenfel'd LA, Davydov RM. Biochim Biophys Acta. 1979;549:255–280. doi: 10.1016/0304-4173(79)90002-8. [DOI] [PubMed] [Google Scholar]
- 54.Denisov IG, Mak PJ, Makris TM, Sligar SG, Kincaid JR. J Phys Chem A. 2008;112:13172–13179. doi: 10.1021/jp8017875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheng X, Zhang H, Hollenberg PF, Newcomb M. Biochemistry. 2009;48:1620–1627. doi: 10.1021/bi802279d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim SH, Yang TC, Perera R, Jin S, Bryson TA, Sono M, Davydov R, Dawson JH, Hoffman BM. Dalton Trans. 2005;21:3464–3469. doi: 10.1039/b506764m. [DOI] [PubMed] [Google Scholar]
- 57.Davydov R., Perera R., Dawson J, Hoffman BM. In preparation
- 58.Akhtar M, Corina D, Miller S, Shyadehi AZ, Wright JN. Biochemistry. 1994;33:4410–4418. doi: 10.1021/bi00180a039. [DOI] [PubMed] [Google Scholar]
- 59.Coon MJ, Vaz ADN, McGinnity DF, Peng HM. Drug Metab Dispos. 1998;26:1190–1193. [PubMed] [Google Scholar]
- 60.Newcomb M, Chandrasena REP. Biochem Biophys Res Commun. 2005;338:394–403. doi: 10.1016/j.bbrc.2005.08.208. [DOI] [PubMed] [Google Scholar]
- 61.Chandrasena REP, Vatsis KP, Coon MJ, Hollenberg PF, Newcomb M. J Am Chem Soc. 2004;126:115–126. doi: 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- 62.Veeger C. J Inorg Biochem. 2002;91:35–45. doi: 10.1016/s0162-0134(02)00393-8. [DOI] [PubMed] [Google Scholar]
- 63.Roberts ES, Vaz ADN, Coon MJ. Proc Natl Acad Sci U S A. 1991;88:8963–8966. doi: 10.1073/pnas.88.20.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ortiz de Montellano PR. Acc Chem Res. 1998;31:543–549. [Google Scholar]
- 65.Woodward JJ, Chang MM, Martin NI, Marletta MA. J Am Chem Soc. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- 66.Clague MA, Wishnok JS, Marletta MA. Biochemistry. 1997;36:14465–14473. doi: 10.1021/bi971024u. [DOI] [PubMed] [Google Scholar]
- 67.Wang D, Thiel W. J Mol Struct: THEOCHEM. 2009;898:90–96. [Google Scholar]
- 68.Oh SS, Robinson CH. J Steroid Biochem Mol Biol. 1993;44:389–397. doi: 10.1016/0960-0760(93)90242-o. [DOI] [PubMed] [Google Scholar]
- 69.Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman MR, Kitagawa T. Biochemistry. 2006;45:5631–5640. doi: 10.1021/bi060094a. [DOI] [PubMed] [Google Scholar]
- 70.Tosha T, Kagawa N, Arase M, Waterman MR, Kitagawa T. J Biol Chem. 2008;283:3708–3717. doi: 10.1074/jbc.M707338200. [DOI] [PubMed] [Google Scholar]
- 71.Sono M, Perera R, Jin S, Makris TM, Sligar SG, Bryson TA, Dawson JH. Arch Biochem Biophys. 2005;436:40–49. doi: 10.1016/j.abb.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 72.Lai W, Chen H, Cho KB, Shaik S. J Phys Chem A. 2009;113:11763–11771. doi: 10.1021/jp902898s. [DOI] [PubMed] [Google Scholar]