

Abstract
Molecular dynamics (MD) simulations, circular dichroism (CD), and dynamic light scattering (DLS) measurements were used to investigate the aggregation propensity of the eye-lens protein γS-crystallin. The wild-type protein was investigated along with the cataract-related G18V variant and the symmetry-related G106V variant. The MD simulations suggest that local sequence differences result in dramatic differences in dynamics and hydration between these two apparently similar point mutations. This finding is supported by the experimental measurements, which show that although both variants appear to be mostly folded at room temperature, both display increased aggregation propensity. Although the disease-related G18V variant is not the most strongly destabilized, it aggregates more readily than either the wild-type or the G106V variant. These results indicate that γS-crystallin provides an excellent model system for investigating the role of dynamics and hydration in aggregation by locally unfolded proteins.
Introduction
Understanding protein aggregation is important not only in the context of protein conformational diseases and functional amyloids, but also as a fundamental biophysical problem. A wide variety of proteins have been found to misfold and form fibrils both in vitro and in vivo (1). The fact that even extremely soluble proteins, such as lysozyme, can fibrillize under certain solution conditions (2) or as a consequence of disease-related point mutations (3) has led to the hypothesis that amyloid fibrils represent a generally stable state of proteins. The formation of aggregates can be a result of misfolding; however, aggregation need not directly correlate with protein thermodynamic instability. For example, several variants of the human prion protein associated with disease phenotypes are not destabilized relative to the wild-type (WT) protein (4,5), and do not undergo significant changes in overall structure (6) or dynamics (7). Many proteins form aggregates from localized unfolding of native monomers (8–12), as reviewed by Chiti and Dobson (13). Local unfolding can promote aggregation either by direct interaction between the unfolded portions or by exposing residues that are normally protected from exposure to the solvent. Interactions with the solvent influence both the kinetics of aggregation (14) and the structures of the aggregates formed (15,16). For proteins that exist at high concentrations in their cellular milieu, protein-protein interactions in both the native and partially unfolded states may be equally important for maintaining solubility. Here, we investigate the roles of thermodynamic instability and intermolecular interactions via the unfolding and aggregation properties of the human eye lens protein γS-crystallin. This protein and variants thereof provide an excellent model system for investigating the relationships between protein misfolding, hydration, and dynamics because its primary biological function is to remain soluble at high concentration.
Crystallins, which are the major protein components of the eye lens, are not crystalline, but instead form large complexes that exhibit short-range order. This enables packing at very high concentration (>400 mg/mL). Unlike in most tissues, protein turnover is negligible in the eye lens, and thus the crystallins must remain stable and soluble for a lifetime. There are two types of crystallins: βγ-crystallins, which are primarily structural, and α-crystallins, which have an additional function of binding to misfolded proteins and preventing aggregation (17,18). Like the other structural βγ-crystallins, γS is a compact β-sheet protein consisting of two double Greek key domains. Although they are not identical in sequence, the two domains are highly symmetric, making this protein essentially a structural homodimer. The two domains of human γS have different stabilities (19) and stabilize each other via the interdomain interface (20). Biophysical studies have shown that short-range protein-protein interactions are responsible for the continuity of the refractive index (21) and that the βγ-crystallins exhibit attractive interactions despite their very low aggregation propensity (22), implying a delicate balancing act between attractive and repulsive interactions among the crystallins. Intermolecular interactions clearly play an important role in establishing the high concentration of the proteins in the eye lens and thus its high refractive index.
A cataract is formed when high-molecular-weight crystallin aggregates accumulate in the lens, rendering it opaque. Aggregation can be caused by misfolding, as in the case of γD-crystallin and γC-crystallin, which have been shown to form amyloid fibers in vitro (23,24), or by disruption of intermolecular interactions, as in aged α-crystallins, which lose their ability to bind and solubilize damaged proteins (25). Recent advances in x-ray crystallography (26,27), solid-state NMR (28–31), and optical spectroscopy (32,33) have enabled investigators to obtain detailed structural models of many fibrillar systems. However, this level of structural information is not yet available for γS-crystallin aggregates. The high solubility of these proteins also complicates structural analyses of γS-crystallin in its native state, although a crystal structure has been solved for the C-terminal domain (34). A solution-state NMR structure of the highly homologous mouse protein (35) provides a clear view of the monomer but does not address its supramolecular structure or aggregation behavior.
In the absence of a detailed structural model, one way to separate the effects of folding and intermolecular interactions on protein aggregation is to investigate variants with reduced solubility or stability. Many different crystallin mutations can cause autosomal-dominant cataracts of diverse morphologies that affect the α- (36,37), β- (38), and γ- (39–43) crystallins. Investigating clinically discovered mutations is a clear starting point; however, it is essential to make use of site-directed mutagenesis to test hypotheses about the molecular basis of protein solubility. In this work, we used molecular dynamics (MD) simulations and optical measurements to compare the WT protein with its G18V variant, which is associated with early-onset cataract (39), and the symmetry-related G106V variant. Our simulated and experimental results demonstrate that these two superficially similar mutations have quite different effects on folding stability and solubility.
A structural model of human γS-crystallin (Fig. 1) provides a starting point for analyzing structural and dynamical changes that may be induced by the mutations. The overall fold and the positions of the mutated residues are shown in Fig. 1 a. In Fig. 1 b, the two domains are overlaid, demonstrating the similarity of their folds and validating the idea that G18 and G106 occupy symmetry-related positions on the two domains. The backbone alignment between the two domains has a root mean-square deviation (RMSD) of only 1.08 Å. Residues G18 and G106 are located in solvent-exposed loops on either side of the protein, with backbone torsion angles that are only allowable for glycine (the starting torsion angles are Φ = 88.4°, Ψ = –167.6° for G18, and Φ = 92.5° and Ψ = –164.5° for G106). Fig. 1, c and d, show configuration snapshots of these loops taken from the end of a 50 ns MD simulation. Fig. 1, e and f, show how the same loops undergo disruptions in secondary structure in the G18V and G106V variants.
Figure 1.
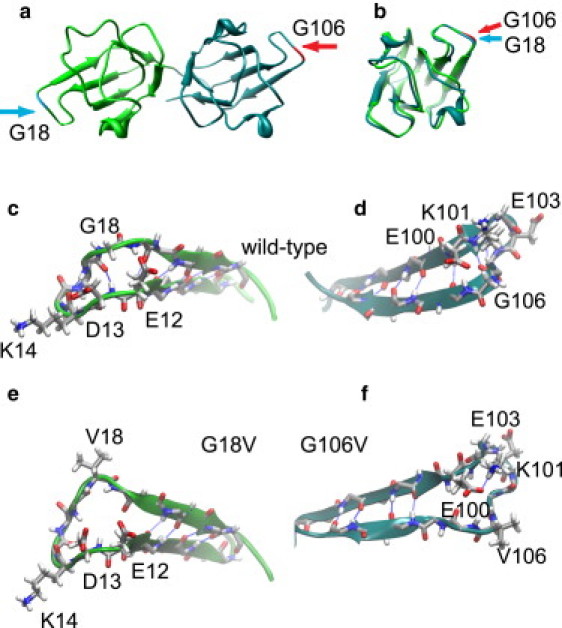
(a) Human γS-crystallin homology model based on the murine γS NMR structure (35). The N- and C-terminal domains are shown overlaid with the mutated residues highlighted. This view shows the highly accessible positions of G18 and G106 and the symmetry of γS-crystallin domains. (b) Structural overlay of a backbone alignment of the N- and C-terminal domains of γS-crystallin. The backbone alignment between the two domains has an RMSD of only 1.08 Å. (c and d) Detail of the loops containing G18 and G106, respectively, from a configuration of the WT MD simulation. In both panels the Gly residues, the charged side chains in their neighborhood, and backbone atoms for the rest of the residue are shown in licorice representation. The backbone hydrogen-bond interactions are also highlighted. (e and f) Configuration snapshot for the same regions of the protein from the MD simulations of the G18V and G106V variants, respectively. In both variants, significant loss of secondary structure in the region occurs, along with opening of each loop. In each case the hydrophobic valine side chain is pointing out into solution because of unfavorable contacts with nearby charged residues.
Although localized loss of secondary structure is suggested by the positions occupied by these glycine residues, and is predicted by the MD simulations, it is also necessary to consider the solubility impact of placing a highly hydrophobic residue in a solvent-exposed position. At pH 7, valine is one of the most hydrophobic residues (44). The addition of hydrophobic side chains could provide additional attractive forces between crystallin monomers even without major structural changes. The abnormal aggregation of the sickle cell variant of hemoglobin is caused by just such a substitution: a glutamic acid to valine point mutation on the surface (45). The variant hemoglobin forms metastable clusters in solution (46). Formation of these clusters depends on long-range interactions, as well as hydration and solution conditions, and the resulting regions of high protein density can provide nucleation sites for aggregation based on local, protein-specific mechanisms (47). Given the extremely high concentration of structural crystallins in the lens, which is similar to the concentration of hemoglobin in red blood cells, reduced solubility due to additional hydrophobic interactions is a plausible mechanism for the increased propensity of G18V to form aggregates. If aggregation results purely from local hydrophobic effects, one would expect G18V and G106V to have nearly identical stability and solubility because the position of the added valine side chain with respect to the protein and the solvent is essentially equivalent. However, if the solubility impact of the G18V variant is primarily due to local protein unfolding, then G106V, which is the analogous mutation to the more stable C-terminal domain, should have either the same or a less disruptive effect on the thermodynamic stability of the entire protein. A recent study showed that G18V is less stable with respect to thermal denaturation (48), but it did not address the contributions of other factors, such as protein-protein interactions, dynamics, and hydration. Our results from simulations and experimental data indicate that aggregation in γS-crystallin is not mediated purely by either thermal instability or the addition of hydrophobic residues to the surface of the protein; rather, it is mediated by a more complex mixture of changes in structure, dynamics, and hydration that depend on the particular amino acid sequences of each domain.
Materials and Methods
MD simulations
We constructed a model of human γS-crystallin with Swiss-Pdb Viewer (49), using the solution structure of the mouse protein (35) (PDB ID 2A5M) as template (96% homology). The initial structure was placed in the center of a 64 × 56 × 68 Å3 box containing water molecules and a single counterion to neutralize the protein charge. Water molecules that overlapped with the protein were removed. The final system consisted of a single protein chain, 5490 waters, and one sodium ion for a total of 20,681 atoms.
The system was subjected to three stages of energy minimization in which harmonic restraints were placed on all heavy atoms with force constants of 10, 5, and 1 kcal/mol·Å, respectively. Each stage consisted of 200 steps of conjugate-gradient energy minimization. The minimization was followed by two 100 ps MD runs at constant volume and a constant temperature of 300 K. Harmonic constraints were applied to the protein backbone heavy atoms during the first of these two runs. The constant-volume simulation was followed by a simulation at constant pressure (1 atm) and temperature (300 K). The variants were generated from the equilibrated WT trajectory (after 20 ns). The initial equilibration for the variant systems consisted of 64 steps of conjugate-gradient energy minimization followed by 20 ps of MD at constant volume and temperature. Constant-pressure and -temperature simulations of all three systems (WT, G18V, and G106V) were run for 50 ns, and analyses were performed over the last 30 ns.
The MD trajectories were generated using the NAMD 2.7 software package (50). The CHARMM22 force field (51) was used for the protein, and the TIP3P model was used for water (52). The smooth particle mesh Ewald method (53,54) was used to calculate the electrostatic interactions. The short-range, real-space interactions were cut off at 11 Å by means of a switching function. A reversible multiple time-step algorithm (55) was used to integrate the equations of motion with time steps of 2 fs for electrostatic forces and short-range nonbonded forces, and 1 fs for bonded forces. The SHAKE algorithm was used to hold all bond lengths involving hydrogen atoms fixed. A Langevin dynamics scheme was used to control temperature, and Nose-Hoover-Langevin pistons were used for pressure control (56,57). The VMD 1.8.7 (58) and UCSF chimera (59) software packages were used for visualization and analysis. The covariance analysis was performed using the R (60) package bio3d (61).
Gene construction, expression, and purification
A plasmid containing the WT human γS-crystallin cDNA sequence was purchased from OpenBiosystems (Huntsville, AL). Oligonucleotides were purchased from Sigma-Aldrich (St. Louis, MO). The γS-crystallin gene was amplified with primers containing flanking restriction sites for NcoI and XhoI, an N-terminal 6x His tag, and a TEV cleavage sequence (ENLYFQG) with the N-terminal methionine of γS-crystallin replaced by the final glycine in the cleavage sequence. The polymerase chain reaction product was cloned into the pET28a(+) vector (Novagen, Darmstadt, Germany), and point mutant constructs were assembled using splice-overlap polymerase chain reaction to mutagenize the WT gene. γS was overexpressed in Rosetta (DE3) Escherichia coli using standard IPTG-induced overexpression protocols at 37°C (WT and G106V) or 16°C (G18V). Cells were allowed to grow for 5-6 h (WT and G106V) or 12–14 h (G18V) postinduction. The cells were lysed by sonication and the cell debris was removed by centrifugation. His-TEV-γS was purified on an Ni-NTA column (Applied Biosystems, Foster City, CA) and cut with the use of a His-tagged TEV protease (produced in-house). The TEV protease and His tag were removed by a second application to an Ni-NTA column, and the pure γS was collected in the column flow-through. γS was dialyzed into 10 mM phosphate buffer, pH 6.9, for all experiments unless otherwise stated.
Circular dichroism
Purified γS was diluted to 0.125 mg/mL in a solution of 10 mM phosphate buffer at pH 6.9 for acquisition of full circular dichroism (CD) spectra, and to 0.25 mg/mL in a solution of 10 mM phosphate buffer, pH 6.9, 150 mM NaCl, and 1 mM DTT for unfolding experiments. Measurements were taken on a J-810 spectropolarimeter (JASCO, Easton, MD) equipped with a thermal controller. For unfolding measurements, the sample was heated at a rate of 2°C/min. For thermal denaturation curves, the CD at 218 nm was monitored and the curves were fit to a two-state equilibrium unfolding model. The temperature lag due to slow equilibration time between the CD thermal controller and the sample was measured with a thermocouple and adjusted for in all thermal unfolding calculations.
Fluorescence spectroscopy
Fluorescence measurements were made on γS-crystallin and variants at a concentration of 0.25 mg/mL in 10 mM phosphate buffer, pH 6.9, 22°C, unless otherwise stated. Absorption-emission fluorescence spectra were obtained with an F4500 fluorescence spectrophotometer (Hitachi, Tokyo, Japan).
Dynamic light scattering
Dynamic light scattering (DLS) measurements were obtained with a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) on WT and both variants of γS at a concentration of 1.0 mg/mL in 10 mM phosphate buffer (pH 6.9). At each temperature, the sample was allowed to equilibrate for 2 min before measurements were obtained, after which scattering measurements were performed in triplicate, resulting in a heating rate of ∼0.5°C every 5 min.
Results and Discussion
MD simulations predict differences in dynamics and hydration
Atomistic MD simulations of each protein in solution were carried out to gain insights into the structural and dynamical effects of the mutations. Upon thermal equilibration in the 10-ns timescale, the Cα RMSD from the initial configuration levels off to ∼2–3 Å in all systems (Fig. S3 in the Supporting Material), validating the homology modeling. Both mutations cause losses in secondary structure (Fig. S4), but because of the sequence differences between the two domains, the changes disrupt the fold in different ways. In the WT protein, residues 18 and 106 each represent the beginning of a sharp turn connecting two strands comprising part of the N- and C-terminal β-sheets, respectively (Fig. 1, a and b). In both variants, addition of the bulky valine side chain truncates the β-strand and enlarges the turn radius. In the simulation of the G18V variant, the hydrophobic valine side chain points away from the protein, due to its unfavorable interaction with the nearby charged residues E12 and D13. In fact, the aspartic acid shifts toward the turn due to a favorable electrostatic interaction with K14 (Fig. 1 e). In the G106 variant, residues E100 and K101 turn toward each other to form a salt bridge (Fig. 1 f). Although both variants display local secondary structure disruption resulting from opening of the loop in the region of the mutated residue, consistent with reduced stability, the disruption of secondary structure is comparable in both domains (Fig. S4).
An examination of the configurational dynamics of each protein reveals significant differences. The introduction of the mutations causes opposing effects in the protein backbone thermal fluctuations, as revealed by the Cα root mean-square fluctuations (RMSFs) (Fig. 2) and covariance analysis (see the Supporting Material). Overall, thermal fluctuations are higher in the G106V protein than in the WT, and the disease-related G18V variant appears to be more rigid than the WT protein. Some of the observed thermal fluctuations are common to all three proteins, including intradomain correlations within turns and helical domains, and between neighboring secondary structure elements. In addition, correlations resulting from internal movements of the β-sheets are present in the N-terminal but not the C-terminal domain. As expected, the β-sheets are the most rigid regions in all cases (Fig. 2). The larger thermal fluctuations within secondary structure elements in the N-terminal domain are consistent with the reported difference in thermodynamic stability between the two domains (19).
Figure 2.
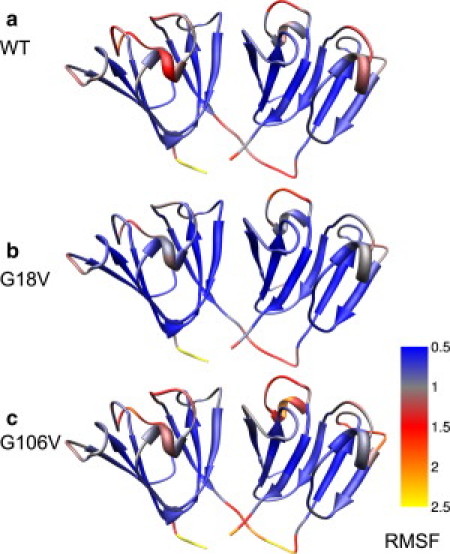
Cα RMSFs mapped onto the γS-crystallin homology model. (a) WT. (b) G18V. (c) G106V. In both variants, changes in conformational dynamics with respect to the WT are not limited to the region around the mutations and are also found in the helices, connecting turns, and loops. Compared with the WT protein, G18V shows diminished thermal fluctuations in these regions. In contrast, G106V appears to be more flexible overall, with increased fluctuations in both surface regions and the β-sheet core.
Experiments show that both γS-crystallin variants are folded and have β-sheet structure at room temperature
Optical spectra were collected to assess potential differences in structure among the WT and variant proteins. A comparison of the CD spectra of the WT, G18V, and G106V crystallins at room temperature shows that all three are folded with a minimum ellipticity at 218 nm, consistent with a primarily β-sheet secondary structure (Fig. 3 a). The spectra of the WT and G106V variants are nearly identical, whereas the 218 nm signal is slightly less intense in the G18V protein, possibly indicating minor disruption of the β-sheet structure. Fluorescence spectra can provide a more sensitive probe of local structural perturbations than CD, which just addresses the overall fold. This should be particularly informative in γ-crystallins, where fluorescence is mediated by four highly conserved tryptophan residues in the core of the protein (24). The specific positioning of the tryptophan side chains has been found to be important in rapid quenching of the UV fluorescence and may act as a UV filter protecting the retina (62,63); thus, large changes in the positions and solvent exposure of these residues should be readily observable in the UV fluorescence spectra. The results for the WT and variant proteins are shown in Fig. 3 b. The fluorescence emission maxima of the WT and G106V spectra are at identical positions at 326 nm, whereas the maximum is shifted slightly to 332 nm for G18V. This red shift is consistent with slight unfolding to partly expose tryptophan to the solvent, but overall the structural differences between the WT and variant proteins at room temperature appear to be minimal, consistent with the MD simulations.
Figure 3.
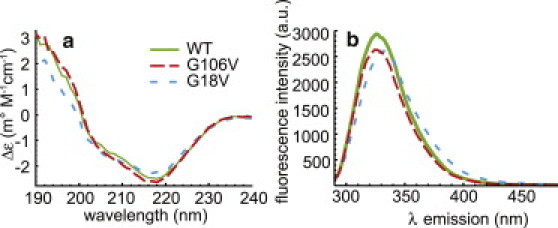
(a) CD spectra of the WT, G18V, and G106V crystallins at a concentration of 0.125 mg/mL in a solution of 10 mM phosphate buffer (pH 6.9) at 20°C. All three display the negative ellipticity at 218 nm that is indicative of β-sheet secondary structure and common to γ-crystallins. (b) Fluorescence emission spectra of WT, G106V, and G18V, 0.25 mg/mL in 10 mM phosphate buffer pH 6.9 at 22°C, collected on a Hitachi F4500 fluorescence spectrophotometer. Both the WT and the G106V variant have a maximum at 326 nm, whereas in the G18V variant it is slightly shifted to 332 nm. Both of these methods indicate little structural difference between the WT and variant proteins at room temperature.
The disease-related G18V variant is not the least stable
The proteins appear structurally similar at room temperature; however, when we heat the protein while monitoring the signal at 218 nm, interesting differences become apparent. Although it is relatively insensitive to local changes in protein conformation, the CD melting curve provides information about the overall stability of the protein fold. As shown in Fig. 4 a, the two mutations do not have the same effect on protein stability, and the disease-related variant is not the least stable. Unexpectedly, the G106V variant unfolds far more readily than G18V. This result indicates that there may be subtle structural changes involving more of the protein than the small loop regions containing G18 and G106. In addition, the fact that both of these point mutations have such significant effects on the thermodynamic stability of γS-crystallin indicates that these glycines and the loops that rely on their dihedral angles are critical for the stability of the crystallin fold.
Figure 4.
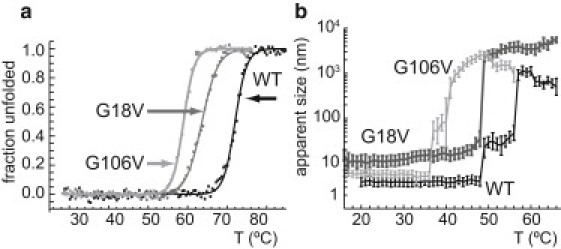
(a) Thermal unfolding curves of WT, G18V, and G106V crystallins measured by monitoring the CD signal at 218 nm, with best-fit unfolding curves. Unfolding measurements were obtained with a JASCO J-810 spectropolarimeter equipped with a thermal controller. The protein concentration was 0.25 mg/mL in the same buffer conditions as in Fig. 3a, but with 150 mM NaCl, and 1 mM DTT (reducing agent). Tm-values for WT, G18V, and G106V are 72.0°C, 65.6°C, and 59.0°C, respectively. All three variants exhibit behavior consistent with two-state equilibrium unfolding. (b) DLS measurement of thermally induced aggregation of WT, G18V, G106V γS. Although the CD unfolding curves show G106V to be the least thermodynamically stable, G18V is found to be the most aggregation-prone variant.
Using nonlinear regression in Mathematica, we fit the CD melting curves to a two-state equilibirum unfolding model described by the following expression:
The results of these measurements and their analysis are shown in Fig. 4 a.
The WT protein had a Tm = 72.0 ± 0.1°C, with ΔH = 161.2 ± 6.1 kcal/mol, and ΔS = 0.467 ± 0.018 kcal mol−1K−1. The melting temperature for G18V was depressed ∼7°, with Tm = 65.6 ± 0.1°C, ΔH = 111.9 ± 3.0 kcal/mol, and kcal mol−1 K−1. The G106V mutation depressed the melting temperature even further, to ∼13° below WT, yielding Tm = 59.0 ± 0.1°C, kcal/mol, and kcal mol−1K−1. The thermodynamic melting temperatures for WT and G18V crystallins determined from the CD unfolding experiment are in agreement with published values (19,48).
The G18V variant aggregates more readily than the WT or G106V
We measured aggregation in the WT and both variants directly by DLS. The results are shown in Fig. 4 b. These measurements were made at low salt concentration in the absence of reducing agents to avoid interfering with any relevant attractive intermolecular forces. We averaged the % abundance by number of the three scans at each temperature and fit the results to a Gaussian function using nonlinear regression. In Fig. 4 b, the average particle size is plotted as a function of temperature. The WT protein is monomeric up to ∼49.0°C, where aggregates 30–50 nm in diameter are formed. At this point, the solution still remains optically clear with no visible light scattering. The estimated diameter of these intermediate-size aggregates remains relatively constant with increasing temperature until 58.5°C. Above 58.5°C, the protein rapidly forms aggregates of up to 1 μg in diameter. At 59–60°C, these become the most numerous aggregates, with virtually all of the observable protein in this state at 60°C. The G106V variant behaves similarly to the WT, although its solubility is reduced, and the transitions occur at lower (by 11–15°C) temperatures. Monomers are not observed in the G18V sample even at the lowest temperatures investigated, and it seems to undergo only one transition, with the large particles appearing at 49°C and most of the protein is in this state at 50°C.
Because the Fourier analysis used to estimate the particle size assumes that particles are roughly spherical and may weight the abundance incorrectly if the forming particles are long fibrils, we observed the morphology of aggregated γS-crystallin using scanning electron microscopy (SEM) to confirm both the size and the morphology of the aggregates. SEM images of the largest aggregates (formed at 70°C) appeared to be composed of interconnected strands of protein roughly the diameter of the intermediate aggregates determined by DLS (Fig. S2). This supports the idea that the crystallin protein has an intermediate stage of aggregation during which it will stably form small particles of a consistent size. A detailed comparison of intermediate aggregates with fibers formed over a longer period of time is needed to ascertain whether the aggregates undergo further conformational change to form amyloid fibers or remain as aggregates of native-like proteins.
The higher critical aggregation temperatures (i.e., formation of large aggregates) in the DLS measurements are in agreement with the Tm-values determined by CD, indicating that this larger hydrodynamic radius state may be related to protein unfolding, as the overall fold of G18V is more stable than G106V. However, the lower critical aggregation temperatures (i.e., formation of smaller aggregates) suggest that G18V has a greater thermal aggregation propensity than G106V in the absence of salt, because it forms particles of intermediate size even below 20°C. This result is surprising given the similarity of the overall folds of the two domains (Fig. 1 b). Previous thermodynamic studies on γS have demonstrated that unfolding experiments require some form of salt (either NaCl or GuCl) to keep the protein soluble up to its thermodynamic unfolding temperature (19,48). Results obtained by repeating the DLS measurements on γS-WT in the presence of varying amounts of salt indicate that the first aggregation may be shielded by the presence of ions that can neutralize charged groups on γS that are either exposed or separated during thermal perturbation (Fig. S3). It is also possible that protein dynamics play an important role in preventing undesirable intermolecular interactions leading to the first stage of aggregation. It is essential to understand the formation of small aggregates, because these intermediate aggregates are thought to be the toxic species in many protein deposition diseases (1,64). Dynamics, along with the avoidance of too-similar surface residues that might form crystal contacts, may be a target of selection pressure on this protein, which has evolved to resist aggregation.
MD simulations show a solvent-exposed cavity in the N-terminal domain of G18V
The CD and UV fluorescence data obtained at room temperature suggest that the addition of V18 or V106 to the folded protein does not unfold the whole Greek key domain; rather, it acts by a more complicated mechanism. The simulations show that G18V becomes more rigid, whereas G106V is more mobile, consistent with its lowest thermodynamic stability. However, the anomalously high aggregation propensity of G18V, in light of the lower thermodynamic stability and higher mobility of G106V, appears to require additional factors related to specific sequence differences. This could involve exposing interior residues to the solvent, or forcing a separation of charge in oppositely charged residues on adjacent loops. The distribution of charged residues on γS is particularly interesting, given their abundance on the surface of the protein despite its expected pI of 6.44, and its high solubility while neutrally charged. It is possible that the distribution and organization of charged surface residues have been tuned to optimize the solubility of γS-crystallin by controlling the hydration.
To quantify protein hydration in the MD simulation trajectories, we computed the number density of waters in the first coordination shell of the protein in a 1 Å/point grid, and identified grid points with a number density of at least twice the bulk value as water sites (65). There is an apparent inverse correlation between the number of water sites and the total solvent-accessible surface area among the three proteins, suggesting the presence of buried waters in the clefts and crevices of the fold (Fig. S12). Consistent with this idea, the largest population of water sites occurs at the interfacial region between domains (Fig. 5 a) for all three proteins. G18V exhibits the largest number of water sites (Fig. S12), which are located in crevices on the surface of the N-terminal domain that are not found in either the WT or the G106V mutant (Fig. 5 a). The formation of these crevices in the G18V N-terminal domain can be traced back to the salt-bridge interactions among the charged side chains in the connecting segments between β-sheets, in particular those involving D78. In the WT protein, D78 alternates salt-bridge interactions with R79 or R36 (Fig. 5 b and Fig. S13 a). Similarly, in G106V there is a persistent salt-bridge interaction between D78 and R36 (Fig. S13 a). In the G18V variant, this pattern is broken: the D78 and R79 side chains are not correctly positioned to form a salt bridge; rather, they form persistent salt bridges with residues in the outer loops (Fig. 5 c and Fig. S13 a), and this new distribution of salt-bridge interactions opens the crevices. There is no analogous charged pair in the same connecting loop of the C-terminal domain. There are a few unique salt-bridge interactions in the C-terminal domain of the G106V mutant (Fig. S13 b), but these are all between neighboring residues, and their effect on the C-terminal hydration, with respect to the WT, is negligible.
Figure 5.
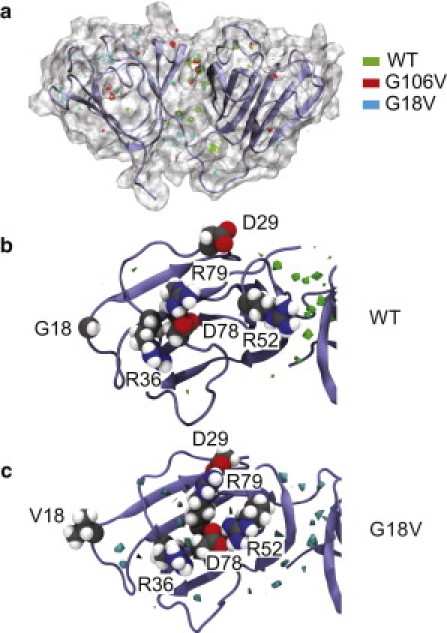
(a) Regions in the first coordination shell of the protein with water density of twice the bulk value are highlighted. The region between the two domains exhibits the largest number of water sites in all three proteins. A significant number of water sites are also observed in the N-terminal loop regions of the G18V variant. A configuration snapshot of the WT protein is shown in secondary structure representation and as a molecular surface. (b and c) Top view of the N-terminal domain in the WT and G18V variant, respectively. The water isodensity surfaces (twice bulk value) are shown in green (WT) and blue (G18V). Crevices are formed in the protein surface of the G18V variant in the region of the mutation and between the inner loops as a key charged pair (D78 and R79) form persistent salt-bridges with residue located in the outer loops. In the WT, in contrast, D78 alternates salt-bridge interactions with R79 and the neighboring R36. In both panels the protein is shown in secondary structure representation and the specific side chains are shown in CPK representation colored by atom (carbon, gray; nitrogen, blue; oxygen, red; hydrogen, white).
Hydration is an important parameter in the aggregation behavior of proteins, and specifically bound or trapped waters play structural and functional roles in many systems (66). Although the densely packed β-sheet structures of amyloid fibrils are relatively dehydrated, previously published two-dimensional infrared measurements suggest that individual bound waters are present between β-strands (15). In the fibril-forming peptide islet amyloid, organic solvents have been shown to dramatically increase the aggregation rate, presumably by altering hydrogen bonding or stabilizing intermediate aggregates (67).
Conclusions
In summary, the initial aggregation propensity in γS-crystallin is not directly correlated with thermodynamic instability. This suggests that locally unfolded states are responsible. If full unfolding were required for aggregation, making the equivalent mutation and concomitant structural perturbation to the more stable C-terminal domain should result in a variant with both intermediate stability and solubility. Instead, the results of this study indicate that although the G106V variant is less thermodynamically stable, it is not as aggregation-prone as the disease-related G18V variant. These results establish that γS-crystallin and its aggregation-prone variants provide an excellent model system for elucidating the structural and dynamic determinants of protein solubility. Although further experiments and simulations will be necessary to more fully characterize both the intermediate and the final aggregates, and measure the kinetics of aggregation, our study shows that in this system, protein dynamics and hydration may be just as important as stability. Equally important is the finding that apparently similar point mutations have dramatically different effects on the dynamics, hydration, and aggregation behavior of this protein.
Acknowledgments
We thank Wytze van der Veer for expert assistance with optical data collection, and Nathan Crawford and Joseph Farran for outstanding computing support. The SEM data were collected in the Zeiss Center for Excellence (University of California, Irvine, CA).
This work was supported by an award from the Camille and Henry Dreyfus Foundation and a National Science Foundation (NSF) CAREER grant (CHE-0847375) to R.W.M., and NSF grant CHE-0750175 and National Institutes of Health grant GM86685 to D.J.T. K.J.G. was supported by a Department of Education GAAN fellowship, and V.M. received support from Chem-SURF, a Research Experience for Undergraduates program supported in part by the NSF.
Supporting Material
References
- 1.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Booth D.R., Sunde M., Pepys M.B. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 3.Pepys M.B., Hawkins P.N., Hsuan J.J. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- 4.Liemann S., Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- 5.Caughey B., Lansbury P.T. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 6.Hosszu L.L.P., Jackson G.S., Collinge J. The residue 129 polymorphism in human prion protein does not confer susceptibility to Creutzfeldt-Jakob disease by altering the structure or global stability of PrPC. J. Biol. Chem. 2004;279:28515–28521. doi: 10.1074/jbc.M313762200. [DOI] [PubMed] [Google Scholar]
- 7.Bae S.-H., Legname G., Dyson H.J. Prion proteins with pathogenic and protective mutations show similar structure and dynamics. Biochemistry. 2009;48:8120–8128. doi: 10.1021/bi900923b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinha N., Tsai C., Nussinov R. A proposed structural model for amyloid fibril elongation: domain swapping forms an interdigitating β-structure polymer. Protein Eng. 2001;14:93–103. doi: 10.1093/protein/14.2.93. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y., Gotte G., Eisenberg D. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 2001;8:211–214. doi: 10.1038/84941. [DOI] [PubMed] [Google Scholar]
- 10.Sanders A., Jeremy Craven C., Staniforth R.A. Cystatin forms a tetramer through structural rearrangement of domain-swapped dimers prior to amyloidogenesis. J. Mol. Biol. 2004;336:165–178. doi: 10.1016/j.jmb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Chow M.K., Ellisdon A.M., Bottomley S.P. Polyglutamine expansion in ataxin-3 does not affect protein stability: implications for misfolding and disease. J. Biol. Chem. 2004;279:47643–47651. doi: 10.1074/jbc.M405799200. [DOI] [PubMed] [Google Scholar]
- 12.Eakin C.M., Attenello F.J., Miranker A.D. Oligomeric assembly of native-like precursors precedes amyloid formation by β-2 microglobulin. Biochemistry. 2004;43:7808–7815. doi: 10.1021/bi049792q. [DOI] [PubMed] [Google Scholar]
- 13.Chiti F., Dobson C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 14.Mukherjee S., Chowdhury P., Gai F. Effect of dehydration on the aggregation kinetics of two amyloid peptides. J. Phys. Chem. B. 2009;113:531–535. doi: 10.1021/jp809817s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim Y.S., Liu L., Hochstrasser R.M. 2D IR provides evidence for mobile water molecules in β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2009;106:17751–17756. doi: 10.1073/pnas.0909888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamley I.W., Nutt D.R., Rodríguez-Llansola F. Influence of the solvent on the self-assembly of a modified amyloid β peptide fragment. II. NMR and computer simulation investigation. J. Phys. Chem. B. 2010;114:940–951. doi: 10.1021/jp906107p. [DOI] [PubMed] [Google Scholar]
- 17.Horwitz J. α-Crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka N., Tanaka R., Hamada D. Amyloid fibril formation and chaperone-like activity of peptides from αA-crystallin. Biochemistry. 2008;47:2961–2967. doi: 10.1021/bi701823g. [DOI] [PubMed] [Google Scholar]
- 19.Wenk M., Herbst R., Jaenicke R. γS-crystallin of bovine and human eye lens: solution structure, stability and folding of the intact two-domain protein and its separate domains. Biophys. Chem. 2000;86:95–108. doi: 10.1016/s0301-4622(00)00161-7. [DOI] [PubMed] [Google Scholar]
- 20.Mills I.A., Flaugh S.L., King J.A. Folding and stability of the isolated Greek key domains of the long-lived human lens proteins γD-crystallin and γS-crystallin. Protein Sci. 2007;16:2427–2444. doi: 10.1110/ps.072970207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaye M., Tardieu A. Short-range order of crystallin proteins accounts for eye lens transparency. Nature. 1983;302:415–417. doi: 10.1038/302415a0. [DOI] [PubMed] [Google Scholar]
- 22.Tardieu A., Vérétout F., Slingsby C. Protein interactions in the calf eye lens: interactions between β-crystallins are repulsive whereas in γ-crystallins they are attractive. Eur. Biophys. J. 1992;21:1–12. doi: 10.1007/BF00195438. [DOI] [PubMed] [Google Scholar]
- 23.Papanikolopoulou K., Mills-Henry I., King J. Formation of amyloid fibrils in vitro by human γD-crystallin and its isolated domains. Mol. Vis. 2008;14:81–89. [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y., Petty S., King J. Formation of amyloid fibrils in vitro from partially unfolded intermediates of human γC-crystallin. Invest. Ophthalmol. Vis. Sci. 2010;51:672–678. doi: 10.1167/iovs.09-3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takemoto L., Ponce A., Sorensen C.M. Age-dependent association of γ-crystallins with aged α-crystallins from old bovine lens. Mol. Vis. 2008;14:970–974. [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz-Avalos R., Long C., Caspar D.L. Cross-β order and diversity in nanocrystals of an amyloid-forming peptide. J. Mol. Biol. 2003;330:1165–1175. doi: 10.1016/s0022-2836(03)00659-4. [DOI] [PubMed] [Google Scholar]
- 27.Sawaya M.R., Sambashivan S., Eisenberg D. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 28.Ferguson N., Becker J., Fersht A.R. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA. 2006;103:16248–16253. doi: 10.1073/pnas.0607815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petkova A.T., Yau W.M., Tycko R. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heise H. Solid-state NMR spectroscopy of amyloid proteins. ChemBioChem. 2008;9:179–189. doi: 10.1002/cbic.200700630. [DOI] [PubMed] [Google Scholar]
- 31.Paravastu A.K., Qahwash I., Tycko R. Seeded growth of β-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. USA. 2009;106:7443–7448. doi: 10.1073/pnas.0812033106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strasfeld D.B., Ling Y.L., Zanni M.T. Strategies for extracting structural information from 2D IR spectroscopy of amyloid: application to islet amyloid polypeptide. J. Phys. Chem. B. 2009;113:15679–15691. doi: 10.1021/jp9072203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Popova L.A., Kodali R., Lednev I.K. Structural variations in the cross-β core of amyloid β fibrils revealed by deep UV resonance Raman spectroscopy. J. Am. Chem. Soc. 2010;132:6324–6328. doi: 10.1021/ja909074j. [DOI] [PubMed] [Google Scholar]
- 34.Purkiss A.G., Bateman O.A., Slingsby C. The X-ray crystal structure of human γS-crystallin C-terminal domain. J. Biol. Chem. 2002;277:4199–4205. doi: 10.1074/jbc.M110083200. [DOI] [PubMed] [Google Scholar]
- 35.Wu Z., Delaglio F., Bax A. Solution structure of (γ)S-crystallin by molecular fragment replacement NMR. Protein Sci. 2005;14:3101–3114. doi: 10.1110/ps.051635205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Litt M., Kramer P., Weleber R.G. Autosomal dominant congenital cataract associated with a missense mutation in the human α crystallin gene CRYAA. Hum. Mol. Genet. 1998;7:471–474. doi: 10.1093/hmg/7.3.471. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y.Z., Zhang X.Y., Shang F. A novel αB-crystallin mutation associated with autosomal dominant congenital lamellar cataract. Invest. Ophthalmol. Vis. Sci. 2006;47:1069–1075. doi: 10.1167/iovs.05-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Litt M., Carrero-Valenzuela R., Maumenee I.H. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human β-crystallin gene CRYBB2. Hum. Mol. Genet. 1997;6:665–668. doi: 10.1093/hmg/6.5.665. [DOI] [PubMed] [Google Scholar]
- 39.Sun H., Ma Z., Shen Y. γ-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J. Med. Genet. 2005;42:706–710. doi: 10.1136/jmg.2004.028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li F.F., Wang S.Z., Ma X. Mutation G61C in the CRYGD gene causing autosomal dominant congenital coralliform cataracts. Mol. Vis. 2008;14:378–386. [PMC free article] [PubMed] [Google Scholar]
- 41.Basak A., Bateman O., Pande J. High-resolution X-ray crystal structures of human γD crystallin (1.25 A) and the R58H mutant (1.15 A) associated with aculeiform cataract. J. Mol. Biol. 2003;328:1137–1147. doi: 10.1016/s0022-2836(03)00375-9. [DOI] [PubMed] [Google Scholar]
- 42.Pande A., Pande J., Benedek G.B. Crystal cataracts: human genetic cataract caused by protein crystallization. Proc. Natl. Acad. Sci. USA. 2001;98:6116–6120. doi: 10.1073/pnas.101124798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pande A., Annunziata O., Pande J. Decrease in protein solubility and cataract formation caused by the Pro23 to Thr mutation in human γ D-crystallin. Biochemistry. 2005;44:2491–2500. doi: 10.1021/bi0479611. [DOI] [PubMed] [Google Scholar]
- 44.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 45.Pauling L., Itano H.A., Wells I.C. Sickle cell anemia a molecular disease. Science. 1949;110:543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 46.Pan W., Galkin O., Vekilov P.G. Metastable mesoscopic clusters in solutions of sickle-cell hemoglobin. Biophys. J. 2007;92:267–277. doi: 10.1529/biophysj.106.094854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan W., Vekilov P.G., Lubchenko V. Origin of anomalous mesoscopic phases in protein solutions. J. Phys. Chem. B. 2010;114:7620–7630. doi: 10.1021/jp100617w. [DOI] [PubMed] [Google Scholar]
- 48.Ma Z., Piszczek G., Hejtmancik J.F. The G18V CRYGS mutation associated with human cataracts increases γS-crystallin sensitivity to thermal and chemical stress. Biochemistry. 2009;48:7334–7341. doi: 10.1021/bi900467a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 50.Kale L., Skeel R., Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 51.MacKerell A.D., Jr., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 52.Jorgensen W., Chandrasekhar J., Klein M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 53.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 54.Batcho P.F., Case D.A., Schlick T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001;115:4003–4018. [Google Scholar]
- 55.Grubmuller H., Heller H., Schulten K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991;6:121–142. [Google Scholar]
- 56.Feller S.E., Zhang Y., Brooks B.R. Constant-pressure molecular-dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 57.Martyna G.J., Tobias D.J., Klein M.L. Constant-pressure molecular-dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 58.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 59.Pettersen E.F., Goddard T.D., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 60.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2008. R: A Language and Environment for Statistical Computing.http://www.R-project.org [Google Scholar]
- 61.Grant B.J., Rodrigues A.P.C., Caves L.S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 62.Chen J., Callis P.R., King J. Mechanism of the very efficient quenching of tryptophan fluorescence in human γD- and γS-crystallins: the γ-crystallin fold may have evolved to protect tryptophan residues from ultraviolet photodamage. Biochemistry. 2009;48:3708–3716. doi: 10.1021/bi802177g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu J., Chen J., Knutson J.R. Femtosecond fluorescence spectra of tryptophan in human γ-crystallin mutants: site-dependent ultrafast quenching. J. Am. Chem. Soc. 2009;131:16751–16757. doi: 10.1021/ja904857t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 65.Paliwal A., Asthagiri D., Paulaitis M.E. Light-scattering studies of protein solutions: role of hydration in weak protein-protein interactions. Biophys. J. 2005;89:1564–1573. doi: 10.1529/biophysj.105.065284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rasaiah J.C., Garde S., Hummer G. Water in nonpolar confinement: from nanotubes to proteins and beyond. Annu. Rev. Phys. Chem. 2008;59:713–740. doi: 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- 67.Padrick S.B., Miranker A.D. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.