

Abstract
Tetrameric ligand binding domains of the family of ionotropic glutamate receptors assemble as dimers-of-dimers. Crystallographic studies of several glutamate receptor subtype isolated core-dimers suggest a single stable dimeric conformation. A binding domain dimer has not been captured in other conformations without the aid of biochemical methods to disrupt a critical dimer interface. Molecular dynamics simulations and continuum electrostatics calculations reveal that the active glutamate bound form of the ligand-binding domain found in typical crystal structures is the preferred energetic state of the isolated core-dimer in the presence of agonist glutamate. A desensitized conformational state is a higher energy ligand-bound state of the core-dimer. The resting apo conformational state is comparatively the least energetically favored conformation and does not contain a single state but a set of energetically equivalent conformational core-dimer states. We hypothesize the energetic balance of an open versus closed transmembrane region must be included to characterize the absolute energetic states of the full receptor, which in the presence of the ligand is believed to be a desensitized state.
AMPA subtype ionotropic glutamate receptors (GluR1–4) mediate fast synaptic transmissions in the forebrain. These ligand-gated cationic channels form the basis for neural plasticity and development of cognitive processes such as learning and memory (1). A crystal structure of GluR2 bound to antagonist solved at 3.6 Å resolves a tetrameric protein topology consisting of an N-terminal domain, ligand-binding domain (LBD), and a transmembrane region (2). Binding of principal excitatory neurotransmitters to the receptor's cytosolic LBD initiates neuronal excitation via cation entry to the cell in a well-controlled manner.
Electrophysiology experiments reveal three definable states of the full receptor:
-
1.
For low neurotransmitter concentrations, the receptor is in a resting (channel closed) state.
-
2.
As the concentration increases, the channel is in an activated (channel open, ligand bound) conducting state.
-
3.
At high concentrations of neurotransmitter, receptor desensitization inhibits ion conduction (3,4).
Mutational analyses and x-ray crystallography suggest channel desensitization may be controlled at the LBD level (5,6).
Although full mechanistic descriptions of any biological system require knowledge of protein structure, at ∼100 kDa in size, GluR is a crystallographically challenging system. Fortunately, GluR is a modular protein; the ligand-binding portion can be separated from the full receptor protein and remain functionally intact (6–9). Although monomeric in solution at low concentrations (6), functional studies of the full-length receptor show ligand binding to two out of four monomers is the minimum requirement to initiate ion conduction (10). Thus, the LBD core-dimer is a functionally relevant system for characterizing energetics.
X-ray crystallography of these isolated LBDs shows a two-lobe (D1, D2) domain (Fig. 1) that forms a cleft for ligand binding (7–9,11). The three functionally representative states have been produced: an active, ligand-bound state defined by a dimer interface formed by D1-D1 favorable monomer interactions (Fig. 1 A); an apo state in the absence of ligand having a similar D1-D1 interface (Fig. 1 C); and a functionally desensitized, structurally desensitized-like state defined by a disrupted D1-D1 interface (Fig. 1 B) (11). Most recent rapid perfusion experiments combined with cysteine cross-linking of the intact receptor indicate the resting state of the ligand-binding domain does not consist of a single state but samples multiple conformations (12). Luminescence resonance energy transfer distance measurements have also indicated a different resting conformational state and suggest this LBD state of the intact receptor is in a desensitized-like conformation (13). Together these experiments suggest the LBD as the major regulatory domain for inducing the functional states of the full receptor.
Figure 1.
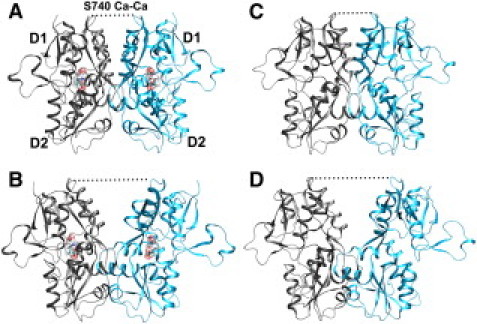
Conformations of the LBD core-dimer. Monomers are shown using a ribbon representation colored gray or blue. Bound glutamate is shown using spacefilling (CPK) representation in (A) the active, glutamate bound and (B) desensitized, glutamate bound. (C) Resting, apo, and (D) A “desensitized-like” state. (Dotted lines) S740 Cα-Cα distances.
Ion channels are complex protein systems made up of many domains whose individual contributions allow functioning of the total machine (i.e., LBD ligand binding and transmembrane ion pore open/close). The modular nature of GluRs allows characterization of these individual regions to be computationally possible. Taking this approach experimentally can be a difficult task (i.e., no measurable functional response of the isolated LBD, and no source for operating the receptor transmembrane region). This study lays the foundation for describing the subtle balance of how LBD regulatory conformational states and ion channel conformational states together define the total functional state of the intact receptor. The following uses molecular dynamics (MD) simulations and continuum electrostatics calculations to provide a conceptual understanding of GluR2 in the realm of how the conformational energetic states of its LBD are organized.
The degree to which two proximal proteins favorably or unfavorably interact can be described with a free energy of interaction ΔGinter. Decomposed into individual energy terms,
| (1) |
where ΔGVDW is the change in intermolecular van der Waals free energy, ΔGCoulomb is the change in intermolecular electrostatic free energy, ΔGRxField is the change in reaction field free energy of the solvent that arises from forming a complex, ΔGSASA is the change in nonpolar contribution to the solvation free energy, T is temperature, and ΔS is change in entropy. All of these quantities can be calculated from a single equilibrium MD simulation using the above decomposition technique, similar to MD/PBSA (14) and have been applied successfully by our group for monomeric AMPA LBDs (15,16). Significant differences in electrostatic free energy between conformations arise from the ΔGVDW, which describes the attraction/repulsion within the complex. The entropic term TΔS was estimated at 0.2 kcal/mol, negligible for this system.
All-atom MD simulations were performed with the AMBER MD package (14) using the force field of Cornell et al. (17). Systems were solvated in a TIP3P water box, minimized, and equilibrated over a period of one nanosecond in the NVT ensemble. Equilibrium molecular dynamics in the NPT ensemble were conducted for six nanoseconds, of which the last four nanoseconds were examined. Four separate LBD core-dimers starting from high-resolution crystal structures were prepared for:
- 1.
- 2.
-
3.
Desensitized-like glutamate-bound GluR2 (PDB ID: 2I3V) (11), and
-
4.
An additional conformation of the core-dimer was produced based on observations from experiment (12,13), which represents a desensitized-like resting apo conformation, by mapping of the apo 1FTO structure onto the desensitized 2I3V structure geometry.
These structures are shown in Fig. 1 and interfacial distances are summarized in Table 1.
Table 1.
Interaction free energies
| Dimer conformation | S740 Cα-Cα distance (Å)∗ | ΔGinter (kcal/mol)∗ |
|---|---|---|
| Active, Glu bound | 18.0 | −16.0 |
| Desensitized Glu bound | 26.0 | −9.5 |
| Resting, apo | 18.0 | −9.0 |
| Desensitized-like Apo | 25.0 | −9.3 |
Distances (S740 Cα-Cα) and energies are averages from 4-ns free simulation time.
Free energy of interaction was calculated as an average from simulation using the SEITRAJ program (18). The free energies listed for each conformation of the LBD core-dimer in Table 1 reflect solvated interaction energy between each monomer in the corresponding conformation. It is the difference between these conformational free energies that is of importance. The active, glutamate-bound conformation is ∼7 kcal/mol more favorable over the desensitized conformation. In the absence of glutamate, the two core-dimer conformations representing possible resting states are only separated by a few tenths of a kilocalorie per mole, and constitute a higher energy state compared to the active, ligand-bound conformation.
The lipid bilayer is an important part of the full GluR receptor as it allows the transmembrane regions to form the ion channel pore. Core-dimer interaction with the membrane most certainly plays a role in receptor functioning. Electrostatic free energy of interaction was calculated using the Poisson solver of HARLEM (19) using a grid size of 201 × 201 × 351 Å3 between a single conformation of the core-dimer and a dielectric membrane as a function of distance along the membrane normal (15),
| (2) |
where the protein, membrane, and solvent are assigned dielectric constants of 2, 4, and 80, respectively. The results shown in Fig. 2 reveal a similar picture as the free energy of interaction between monomers (Table 1) where the active glutamate-bound conformation remains outside the cluster formed by other conformational states of the core-dimer. Albeit these distances are small, the trend remains consistent.
Figure 2.
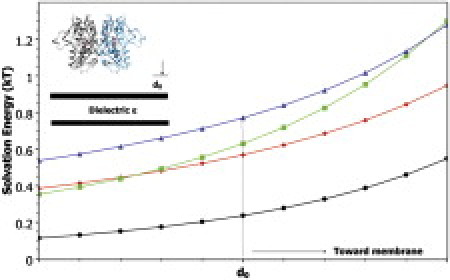
Dimer-membrane electrostatic interaction free energy. The d0 position is the starting distance of 17 Å between P632 Cα and the surface of the dielectric. The dimer was varied from the dielectric calculating the interaction free energy in 1 Å steps. Active (black), desensitized (green), apo (red), and desensitized-like apo (blue).
Note that these results are not an artifact, due to the addition of charge with the presence of the glutamate ligand as the desensitized conformation of the core dimer also has glutamate bound in all calculations.
These results address a number of questions concerning the LBD of the AMPA GluR.
First, the resting or apo conformational state of the core-dimer cannot be defined by a single conformation. The monomer was shown to be very flexible in both theoretical (20) and experimental (21) studies. The results in Table 1 show similar energetics among possible resting states and thus provide rationale for recent experiments suggesting the resting state of the LBD can explore a large set of conformational states yet still result in no functional consequence (12,13). The other conformational states describe two functional states of the full receptor: the active glutamate bound and the desensitized, glutamate bound. The active conformation is favored over the desensitized conformation of the core-dimer and it is this free energy difference that keeps the core-dimer from immediately entering the desensitized state once ligand is bound. These results are consistent with ultracentrifugation experiments that reveal a free energy difference of 6.5 kcal/mol between the wild-type and a nondesensitizing mutant GluR2 LBD (6). However, it is most likely the energetic balance of an open versus closed transmembrane region that defines the absolute energetically favored state of the full receptor, which in the presence of the ligand is believed to be a desensitized state.
Second, the presence of the lipid bilayer may be important for the functional response of the full receptor. We have shown that the active, glutamate-bound conformation of the core-dimer has a unique interaction with the membrane compared to all other conformations. This may be significant when transmitting the mechanical energy from ligand binding to opening the transmembrane channel. Binding of ligand and/or subtle change in charge distribution within the core dimer alters the interaction with the lipid bilayer. This change in interaction would consequently vary the stresses placed on the connecting peptides between the LBD and the transmembrane region leading to different functional states of the full ion channel.
Acknowledgments
This work was supported in part by the National Institutes of Health grant No. R01GM067962 to M.K. and the American Heart Association grant No. 0715286U to M.Y.
References and Footnotes
- 1.Dingledine R., Borges K., Traynelis S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 2.Sobolevsky A.I., Rosconi M.P., Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiskin N.I., Krishtal O.A., Tsyndrenko A.Y. Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci. Lett. 1986;63:225–230. doi: 10.1016/0304-3940(86)90360-5. [DOI] [PubMed] [Google Scholar]
- 4.Mayer M.L., Vyklicky L., Jr. Concanavalin A selectively reduces desensitization of mammalian neuronal quisqualate receptors. Proc. Natl. Acad. Sci. USA. 1989;86:1411–1415. doi: 10.1073/pnas.86.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stern-Bach Y., Russo S., Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y., Olson R., Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong N., Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong N., Sun Y., Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 9.Gouaux E. Structure and function of AMPA receptors. J. Physiol. 2004;554:249–253. doi: 10.1113/jphysiol.2003.054320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li G., Niu L. How fast does the GluR1Qflip channel open? J. Biol. Chem. 2004;279:3990–3997. doi: 10.1074/jbc.M310410200. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong N., Jasti J., Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 12.Plested A.J., Mayer M.L. AMPA receptor ligand binding domain mobility revealed by functional cross linking. J. Neurosci. 2009;29:11912–11923. doi: 10.1523/JNEUROSCI.2971-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez J., Du M., Jayaraman V. Role of dimer interface in activation and desensitization in AMPA receptors. Proc. Natl. Acad. Sci. USA. 2010;107:9891–9896. doi: 10.1073/pnas.0911854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Case D.A., Walker R.C., Kollman P.A. University of California; San Francisco, CA: 2008. AMBER 10. [Google Scholar]
- 15.Mamonova T., Yonkunas M.J., Kurnikova M.G. Energetics of the cleft closing transition and the role of electrostatic interactions in conformational rearrangements of the glutamate receptor ligand binding domain. Biochemistry. 2008;47:11077–11085. doi: 10.1021/bi801367d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Speranskiy K., Kurnikova M. On the binding determinants of the glutamate agonist with the glutamate receptor ligand binding domain. Biochemistry. 2005;44:11508–11517. doi: 10.1021/bi050547w. [DOI] [PubMed] [Google Scholar]
- 17.Cornell W.D., Cieplak P., Kollman P.A. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 18.Cui Q., Sulea T., Purisima E.O. Molecular dynamics-solvated interaction energy studies of protein-protein interactions: the MP1-p14 scaffolding complex. J. Mol. Biol. 2008;379:787–802. doi: 10.1016/j.jmb.2008.04.035. [DOI] [PubMed] [Google Scholar]
- 19.Kurnikov, I. V. 1996. HARLEM—molecular modeling program. Http://harlemprog.org. Accessed December 14, 2010.
- 20.Mamonova T., Speranskiy K., Kurnikova M. Interplay between structural rigidity and electrostatic interactions in the ligand binding domain of GluR2. Proteins. 2008;73:656–671. doi: 10.1002/prot.22090. [DOI] [PubMed] [Google Scholar]
- 21.McFeeters R.L., Oswald R.E. Structural mobility of the extracellular ligand-binding core of an ionotropic glutamate receptor. Analysis of NMR relaxation dynamics. Biochemistry. 2002;41:10472–10481. doi: 10.1021/bi026010p. [DOI] [PubMed] [Google Scholar]