

Abstract
We describe a simulation study of Ca2+ dynamics in mice with cardiomyocyte-specific conditional excision of the sarco(endo)plasmic reticulum calcium ATPase (SERCA) gene, using an experimental data-driven biophysically-based modeling framework. Previously, we reported a moderately impaired heart function measured in mice at 4 weeks after SERCA2 gene deletion (knockout (KO)), along with a >95% reduction in the level of SERCA2 protein. We also reported enhanced Ca2+ flux through the L-type Ca2+ channels and the Na+/Ca2+ exchanger in ventricular myocytes isolated from these mice, compared to the control Serca2flox/flox mice (flox-flox (FF)). In the current study, a mathematical model-based analysis was applied to enable further quantitative investigation into changes in the Ca2+ handling mechanisms in these KO cardiomyocytes. Model parameterization based on a wide range of experimental measurements showed a 67% reduction in SERCA activity and an over threefold increase in the activity of the Na+/Ca2+ exchanger. The FF and KO models were then validated against experimentally measured [Ca2+]i transients and experimentally estimated sarco(endo)plasmic reticulum (SR) function. Simulation results were in quantitative agreement with experimental measurements, confirming that sustained [Ca2+]i transients could be maintained in the KO cardiomyocytes despite severely impaired SERCA function. In silico analysis shows that diastolic [Ca2+]i rises sharply with progressive reductions in SERCA activity at physiologically relevant pacing frequencies. Furthermore, an analysis of the roles of the compensatory mechanisms revealed that the major combined effect of the compensatory mechanisms is to lower diastolic [Ca2+]i. Finally, by using a comprehensive sensitivity analysis of the role of all cellular calcium handling mechanisms, we show that the combination of upregulation of the Na+/Ca2+ exchanger and increased L-type Ca2+ current is the most effective means to maintain diastolic and systolic calcium levels after loss of SERCA function.
Introduction
Mechanical contraction of the heart is initiated by a transient rise in cytosolic Ca2+ concentration ([Ca2+]i), resulting from a small amount of Ca2+ entering the cell through the voltage-gated L-type Ca2+ channels during an action potential (AP), which in turn triggers a large amount of Ca2+ release from the intracellular Ca2+ store within the sarcoplasmic reticulum (SR). Relaxation of contraction occurs as Ca2+ is removed from the cytosol through both Ca2+ uptake through the SR Ca2+ ATPase (SERCA) and Ca2+ extrusion via two mechanisms: the Na+/Ca2+ exchanger (NCX) and the sarcolemmal Ca2+ ATPase (PMCA). Disruption to [Ca2+]i homeostasis resulting from abnormalities in Ca2+ handling is believed to be an important mechanism underlying heart failure (HF) (1–5). In particular, a decrease in the expression level of SERCA2, the cardiac isoform of SERCA, and thus SR Ca2+ transport have been reported in both animals models with pressure overload-induced hypertrophy (1,2) and in failing human myocardium (3–5).
Recently, a genetically modified mouse model with cardiomyocyte-specific, conditional excision of the Serca2 gene (6) has been developed that now allows investigation into the effects of reduced SERCA2 expression with increased specificity. Data from this experimental model show that at four weeks after gene excision (knockout (KO)), cardiac function was only moderately impaired compared to the Serca2flox/flox (flox-flox (FF)) control despite a reduction in SERCA2 protein expression level to <5% of normal values. Given that SR Ca2+ uptake in healthy controls accounts for >90% of Ca2+ removal in the mouse heart under physiological conditions (7), these findings have sparked significant debate over how these animals can survive with such a dramatic reduction in SERCA function. It has been hypothesized that enhanced Ca2+ fluxes through L-type Ca2+ channels, NCX and PMCA act as compensatory mechanisms in the KO myocytes to shift the reliance of the [Ca2+]i transient generation from SR Ca2+ release to sarcolemmal Ca2+ fluxes (6). However, whether all of these compensatory changes are crucial and their relative contributions in maintaining cardiac function remain to be fully investigated. Furthermore, although the rate of decay of the [Ca2+]i transient in the KO is significantly slower compared to FF at a low pacing frequency (1 Hz), it is only minimally affected at physiologically relevant pacing rates (6). These observations motivate further investigation to elucidate the mechanisms underpinning Ca2+ handling in both the FF and KO animal models.
Mathematical modeling is a potentially powerful tool for such an investigation with the capacity to bring together different aspects of the intracellular events required for providing an integrated understanding of the changes that occur in the SERCA2 KO mice (8). Trafford et al. (9) have applied this methodology previously, conducting a preliminary simulation study using the Bondarenko et al. murine AP model (10). Simulation results showed an increased diastolic [Ca2+]i, a decreased rate of decay of the [Ca2+]i transient that becomes less profound at higher frequencies, and finally a prolonged action potential (9). However, Trafford et al. (9) assumed a simple linear correlation between the expression and activity of the proteins, an assumption that deserves further validation. It is also worth noting that the Bondarenko et al. (10) model was developed based on experimental data obtained at room temperature (25°C), whereas the study on SERCA2 KO mice (6) was carried out at 37°C. The formulation of Ca2+ dynamics in the Bondarenko et al. (10) model was derived from a previous model of guinea pig ventricular myocytes (11), which assumes constant SERCA uptake rate at different frequencies, and hence does not account for frequency-dependent acceleration of relaxation observed experimentally in both FF and KO mice (see below). Thus although mathematical modeling provides a potentially promising and quantitative framework for analyzing the mechanisms underlying the observed Ca2+ handling, for the reasons outlined above, the characteristics of the [Ca2+]i transients simulated using the Bondarenko et al. (10) model are unlikely to be a good representation of those observed experimentally by Andersson et al. (6).
In the current study, we have reparameterized a mathematical model of Ca2+ dynamics specifically to experimental measurements obtained from the FF and KO cardiomyocytes, to enable direct comparison between simulation results and experimental data. Using this model, we examine the underlying changes in Ca2+ handling and the relative contributions of the compensatory mechanisms present in the SERCA2 KO mice.
Materials and Methods
A detailed description of the generation of Serca2flox/flox (FF) and Serca2flox/flox Tg(αMHC-MerCreMer) (KO) mice is in the Supporting Material.
Cardiomyocyte experiments
Cardiomyocytes were enzymatically isolated (6,12), plated on laminin-coated coverslips, and placed in a perfusion chamber on the stage of an inverted microscope. [Ca2+]i transient measurements were recorded in fluo-4 AM (Invitrogen Molecular Probes, Eugene, OR) loaded myocytes during field stimulation (3 ms biphasic pulse, 25% above threshold). Cells were perfused with HEPES Tyrode solution containing (in mM): 140 NaCl, 1 CaCl2, 0.5 MgCl2, 5.0 HEPES, 5.5 glucose, 0.4 NaH2PO4 and 5.4 KCl (pH 7.4, 37°C), and an LSM 510 microscope (Zeiss GmbH, Jena, Germany) was used to record [Ca2+]i transients in line-scan mode (12).
Patch-clamp experiments were conducted with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA), pCLAMP software (Axon Instruments), and low resistance pipettes (1–2 MΩ) L-type Ca2+ current was recorded during 200 ms voltage steps from -50 mV to a range of potentials, with an internal solution containing (in mM) 130 CsCl, 0.33 MgCl2, 4 Mg-ATP, 0.06 EGTA, 10 HEPES, and 20 TEA, and an extracellular solution containing (in mM) 20 CsCl, 1 MgCl2, 135 NaCl, 10 HEPES, 10 D-glucose, 4 4-aminopyridine, and 1 CaCl2 (6,13). Diastolic [Ca2+]i was measured by whole-cell photometry (Photon Technology International, Monmouth Junction, NJ) in cells pipette-loaded with a solution containing (in mM): 0.1 fura-2 salt (Invitrogen Molecular Probes), 120 K-aspartate, 0.5 MgCl2, 10 NaCl, 0.06 EGTA, 10 HEPES, 10 glucose, 25 KCl and 4 K2-ATP (pH 7.2) (6,13). Fura-2 fluorescence ratios (F340/F380) were calibrated to [Ca2+]i, and calculated diastolic values were then used to calibrate [Ca2+]i transients recorded in fluo-4 loaded cells (6,13,14).
SR Ca2+ content was measured by rapidly switching the extracellular solution to one containing 10 mM caffeine, and measuring either the magnitude of the elicited [Ca2+]i transient or the integral of the elicited Na+-Ca2+ exchange current (6,13). The SR-dependent component of the [Ca2+]i transient was estimated by comparing the magnitude of [Ca2+]i transients in the presence and absence of caffeine (6,13).
Model derivation
Following the same approach developed and applied in our previous study of Ca2+ dynamics in wild-type murine ventricular myocytes (15), we reparameterized the mathematical framework using experimental measurements obtained from the FF and KO cardiomyocytes. NCX and PMCA were parametrized based on the decay of the caffeine-induced [Ca2+]i transient, which then allowed parameterization of SERCA from the decay of the field-stimulated [Ca2+]i transients at 1 and 6 Hz. The fitted maximum activity of NCX in the KO model was found to be three times higher than the FF value, consistent with our previous finding of an increased NCX activity in the KO cardiomyocyte. The process of parameterizing SERCA in the FF and KO models is shown in Fig. 1. Representative recordings of [Ca2+]i transients, paced at 1 and 6 Hz, are shown in Fig. 1, A and C, respectively. The corresponding fluxes of Ca2+ through SERCA (JSERCA) at 1 and 6 Hz are shown in Fig. 1, B and D, respectively. At 1 Hz, JSERCA in the KO cardiomyocytes was measurable over only a limited range of [Ca2+]i due to the small magnitude of its [Ca2+]i transient. For the same level of [Ca2+]i, JSERCA in the KO was on average 67% lower than the FF value at 1 Hz (Fig.1 B, inset). During parameterization, the reduction in JSERCA was found to be associated with a >60% decrease in the maximum uptake activity of the pump (Vup) and an increase in the rate of SR Ca2+ leak (Vleak) consistent with the increased Ca2+ spark frequency observed experimentally in the KO at an earlier time point (16). No significant difference was found in the affinity of SERCA to intracellular Ca2+ (Km,up), i.e., allowing Km,up to change between the FF and KO did not improve the quality of the fit. Therefore, Km,up was assumed to be unchanged. At the higher pacing frequency of 6 Hz, JSERCA increased approximately threefold in both the FF and KO, indicating that frequency-dependent acceleration of relaxation (FDAR) is present in both experimental models. The decay of the [Ca2+]i transients predicted from the model using the fitted values of SERCA, NCX and PMCA were superimposed to the experimental recordings in Fig. 1, A and C.
Figure 1.
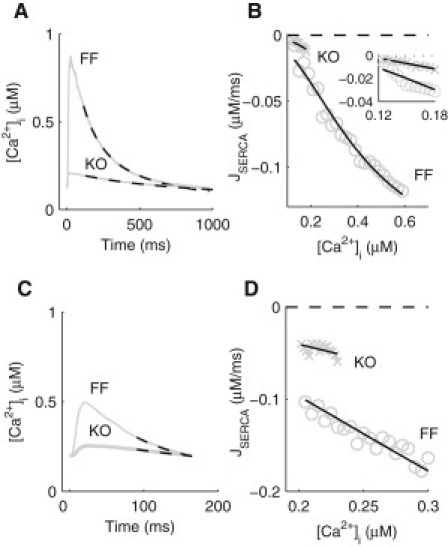
Parameterization of SERCA in the FF and KO models. (A) Average experimentally recorded [Ca2+]i transients, paced at 1 Hz, in isolated ventricular myocytes from the SERCA2 FF and KO mice as indicated. Simulated decay of the [Ca2+]i transients (dashed lines) using the fitted parameter values for SERCA (1 Hz), NCX, and PMCA are superimposed. (B) Ca2+ flux through SERCA (JSERCA) at 1 Hz, plotted as a function of [Ca2+]i, for FF (circles) and KO (crosses) mice. Fitted (JSERCA) are superimposed. Inset: an enlarged view comparing SERCA activity in the KO and FF. (C) Average experimentally recorded [Ca2+]i transients, paced at 6 Hz, in isolated ventricular myocytes from the SERCA2 FF and KO mice as indicated. Simulated decay of the [Ca2+]i transients (dashed lines) using the fitted parameter values for SERCA (6 Hz), NCX, and PMCA are superimposed. (D) Ca2+ flux through SERCA (JSERCA) at 6 Hz, plotted as a function of [Ca2+]i, for FF (circles) and KO (crosses) mice. Fitted (JSERCA) are superimposed.
The formulation for the L-type Ca2+ current (ICaL) was parameterized based on the current-voltage relationship and the time course of the current during a voltage clamp at -10 mV. The time to half inactivation of the simulated ICaL during a voltage clamp at -10 mV was 4.09 ms in the FF model and 8.89 ms in the KO model, compared to the experimental values of 4.79 ± 0.26 and 9.10 ± 0.84 ms in the FF and KO cardiomyocytes respectively. The integrals of the aforementioned simulated currents were 0.092 and 0.23 pC/pF in the FF and KO models respectively, compared to 0.105 ± 0.0006 and 0.205 ± 0.02 pC/pF in the FF and KO cardiomyocytes measured experimentally.
Detailed descriptions of the parameterization process for each of the Ca2+ handling mechanisms above and a full list of parameter values are provided in the Supporting Material. For all the simulation results, the model was run for at least 100 s or until the limit cycle was reached.
Results
Model validation
The Ca2+ dynamics models for the FF and KO cardiomyocytes described above were coupled with the sarcolemmal ionic currents from the previously developed murine ventricular myocyte electrophysiology model (15). To validate the models, we compared simulation results with experimentally measured field-stimulated [Ca2+]i transients, caffeine-induced [Ca2+]i transients and experimentally estimated SR function (Table 1), as shown in Fig. 2. Detailed description of model validation can be found in the Supporting Material.
Table 1.
Comparison between experimental measured and simulated [Ca2+]i transients
| [Ca2+]i Transient characteristics | FF experiment | FF model | KO experiment | KO model |
|---|---|---|---|---|
| Magnitude at 1 Hz (μM) | 0.786 ± 0.145 | 0.720 | 0.098 ± 0.031 | 0.090 |
| Magnitude at 6 Hz (μM) | 0.290 ± 0.029 | 0.323 | 0.066 ± 0.007 | 0.085 |
| Diastolic [Ca2+]i at 1 Hz (μM) | 0.139 ± 0.024 | 0.110 | 0.127 ± 0.014 | 0.128 |
| Diastolic [Ca2+]i at 6 Hz (μM) | 0.204 ± 0.036 | 0.219 | 0.190 ± 0.025 | 0.207 |
| Time to half decay at 1 Hz (μs) | 131 ± 10 | 128 | 285 ± 18 | 269 |
| Time to half decay at 6 Hz (μs) | 54 ± 1 | 48 | 65 ± 2 | 60 |
Figure 2.
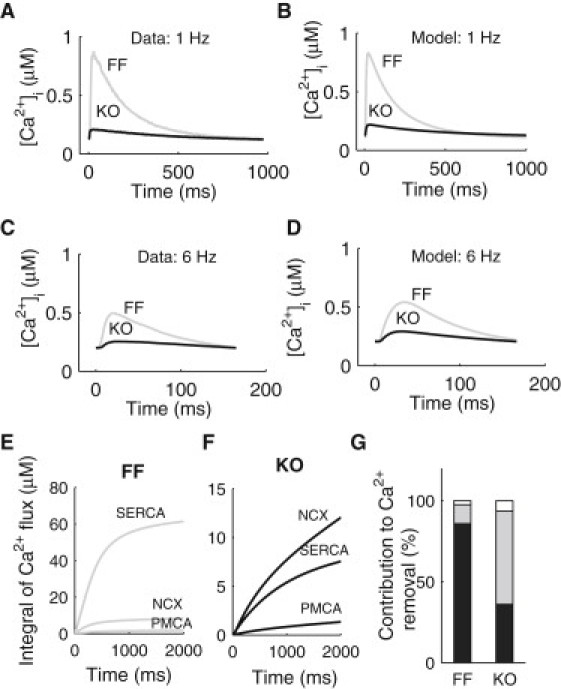
Comparison between experimentally measured and simulated [Ca2+]i transients. (A) Average experimentally measured [Ca2+]i transients at 1 Hz in the FF (gray) and KO (black) myocytes. (B) Simulated [Ca2+]i transients at 1 Hz in the FF (gray) and KO (black) models. (C) Average experimentally measured [Ca2+]i transients at 6 Hz in the FF (gray) and KO (black) myocytes. (D) Simulated [Ca2+]i transients at 6 Hz in the FF (gray) and KO (black) models. (E) Time courses of the integrals of the Ca2+ fluxes through SERCA, NCX, and PMCA in the FF model over one cardiac cycle at 0.5 Hz. (F) Time courses of the integrals of the Ca2+ fluxes through SERCA, NCX, and PMCA in the KO model over one cardiac cycle at 0.5 Hz. (G) Percentage contributions of SERCA, NCX, and PMCA to Ca2+ removal in the FF and KO models.
Quantitative analysis of changes in Ca2+ handling with SERCA2 KO
A quantitative analysis of the differences in Ca2+ handling between the FF and KO models revealed small increases in action potential durations (APD50 and APD90) at 1 Hz in the KO model (Fig. S3), in agreement with a tendency toward AP prolongation reported previously at this 4-week time point after SERCA2 knockout (13). Simulated ICaL in the KO model had a greater magnitude and slower inactivation kinetics compared to the FF model, resulting in a much greater Ca2+ influx during an AP (33% and 49% higher at 1 and 6 Hz, respectively). Simulated Ca2+ uptake through SERCA was significantly decreased during both systole and diastole, and the integral of the flux over each heart beat was severely reduced (84% and 73% lower at 1 and 6 Hz, respectively, than FF values). Simulated Ca2+ flux through NCX in the KO model showed an approximately fourfold increase in Ca2+ entry through NCX in reverse mode, although the magnitude was relatively small compared to Ca2+ influx through ICaL. During systole, Ca2+ extrusion through NCX was only moderately reduced at 1 Hz and slightly higher at 6 Hz compared to the FF values, despite a 73% decrease in the peak of the [Ca2+]i transient. During diastole, simulated Ca2+ extrusion through NCX was increased 2.5-fold in the KO model at both 1 and 6 Hz. A full analysis of the frequency-dependent changes in ICaL, Ca2+ flux through NCX, and AP kinetics are provided in the Supporting Material.
Simulated [Na+]i at 1 Hz in the KO model was 9.9 mM and rose to 13.7 mM at 6 Hz, compared to values of 9.3 and 13.2 mM, respectively, in the FF model. The relatively small increase in [Na+]i between the FF and KO models are consistent with experimental measurements, in which no statistically significant difference in [Na+]i between the FF and KO mice was observed (13). [Na+]i is regulated primarily by the Na+/K+ ATPase (NKA). The formulation of NKA used in our model has previously been parameterized to data from mouse ventricular myocytes at 37°C (15,17). Experimental data on NKA activity plotted as a function of [Na+]i show that NKA is half-activated at [Na+]i equal to 16.6 mM. At the [Na+]i levels predicted by our model, NKA activity lies in the steep part of the curve, indicating a high level of sensitivity that will quickly compensate changes in [Na+]i. Furthermore, as shown in Fig. S3, despite the over threefold increase in the maximum exchange rate of NCX, the actual NCX activity over one cardiac cycle did not increase significantly in the KO mice due to the dramatic reduction in peak [Ca2+]i. Therefore, the increase in Na+ influx through NCX in the KO mice was limited.
Graded reduction of the maximum uptake activity of SERCA
To better understand the changes that occur in the Ca2+ dynamics of the KO cardiomyocytes, we carried out a series of simulation studies based on our model. We first tested the sensitivity of the peak and diastolic [Ca2+]i to a series of graded reductions in SERCA uptake activity in both the FF and KO model. The maximum uptake rate of the pump (Vup) was reduced from 95% of its FF value (calculated from the decay of the experimentally measured [Ca2+]i transient in the FF at 1 and 6 Hz) to 0 (or until alternans appeared) in 5% increments. In all simulations, the L-type Ca2+ channels, NCX and all other parameters were kept unchanged from their original values.
At 1 Hz (not shown), decreases in SERCA activity decreased peak [Ca2+]i and increased diastolic [Ca2+]i in both the FF and KO model. With increasing pacing frequency to 6 Hz (Fig. 3), peak [Ca2+]i appeared to be insensitive to decreases in SERCA activity, whereas diastolic [Ca2+]i, increased dramatically with decreasing SERCA activity in both the FF and KO models. With the FF model, diastolic [Ca2+]i almost tripled when SERCA activity was reduced to zero. In the KO model, a similar trend was observed, although the extent of increase in diastolic [Ca2+]i was smaller. These results suggest that at a physiologically relevant pacing frequency, a reduction in SERCA activity has a much stronger effect on diastolic [Ca2+]i than on peak [Ca2+]i.
Figure 3.
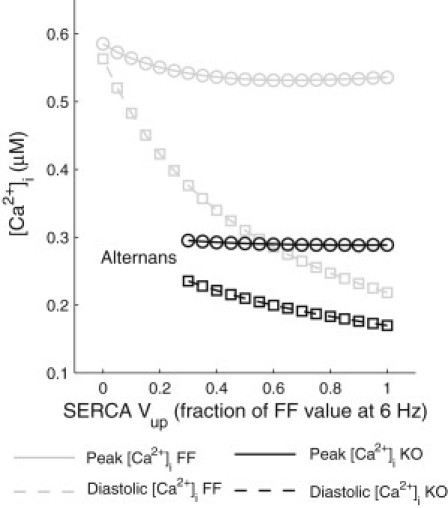
Changes in peak (circles and solid lines) and diastolic [Ca2+]i (squares and dashed lines) in response to graded reductions in the maximum uptake activity of SERCA in the FF (gray) and KO (black) models.
Contributions of the compensatory mechanisms
Experimental observations have suggested that in SERCA2 KO cardiomyocytes, trans-sarcolemmal Ca2+ fluxes are enhanced to compensate for the dramatically impaired SR function (6,13). Specifically, an enhanced ICaL may increase the amount of Ca2+ entry into the cell, and increased Ca2+ extrusion through up-regulated NCX and PMCA may facilitate Ca2+ removal during the decay of the [Ca2+]i transient. These compensatory mechanisms are thought to shift the reliance of the [Ca2+]i transient generation toward sarcolemmal Ca2+ fluxes and away from SR Ca2+ release (6,13).
A quantitative investigation into the contribution of each compensatory mechanism to Ca2+ handling in the KO mice was carried out using in silico perturbations of our models. The first in silico perturbation involved simulating the KO model with ICaL clamped to have the same time course as that seen in the FF model, which will be referred to as the KO-ICaL model. The second perturbation involved reducing the maximum activity of NCX in the KO model to the same level as that in the FF model, which will be referred to as the KO-NCX model. The third perturbation, in which both the ICaL and the NCX were adjusted to the levels in the FF model, was designed to test the combined effects of these two compensatory mechanisms and will be referred to as the KO-ICaL-NCX model.
The resulting Ca2+ dynamics from these three perturbations were then compared to those observed in the control KO model, as shown in Fig. 4. These simulations were carried out at 1 Hz, as opposed to 6 Hz. Simulations at 6 Hz resulted in unstable responses, which were attributed to the nonphysiological combinations of parameter values in these models. It can be seen that in the absence of an increased ICaL, simulated Δ[Ca2+]i and SR Ca2+ content decreased by 28% and 15%, respectively, from the KO values. Furthermore, diastolic [Ca2+]i decreased by 9% and RT50 increased by 6%. In the absence of an upregulated NCX, the simulated [Ca2+]i transient was characterized by a 58% increase in diastolic [Ca2+]i, a 2.6-fold increase in Δ[Ca2+]i and a twofold increase in the SR Ca2+ content. There was a small decrease of 2% in the RT50.
Figure 4.
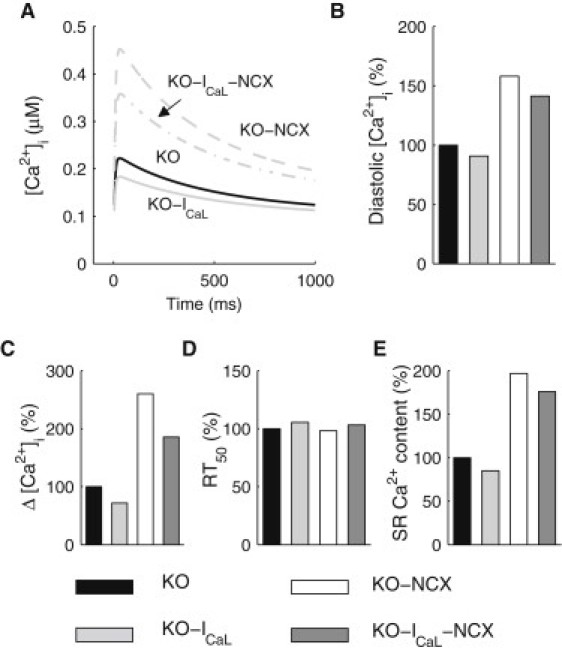
In silico perturbations to the KO model. (A) Simulated [Ca2+]i transients at 1 Hz in the KO, KO-ICaL and KO-NCX and KO-ICaL-NCX models. (B–E) Diastolic [Ca2+]i level (B), magnitude of the [Ca2+]i transient (ΔCai) (C), time to half relaxation of the transient (RT50) (D), and SR Ca2+ content (E) in the KO, KO-ICaL, and KO-NCX models.
In the absence of both compensatory mechanisms, Δ[Ca2+]i, diastolic [Ca2+]i and SR Ca2+ content all remained higher than the KO values, by 86%, 41%, and 76%, respectively. There was a small 3% increase in RT50, indicating a minimal effect of the perturbation on the rate of decay of the [Ca2+]i transient. The above results suggest that whereas increasing ICaL increased systolic [Ca2+]i and NCX upregulation decreased diastolic [Ca2+]i, the overall effects of the coexistence of these two compensatory mechanisms appeared to be decreasing diastolic [Ca2+]i whereas also decreasing systolic [Ca2+]i. Furthermore, as the RT50 of the transient was not found to be significantly increased in either the KO-NCX or the KO-ICaL-NCX model, these compensatory mechanisms may not be as critical for facilitating the decay of the transient as hypothesized previously (6).
Efficiency of other possible compensatory mechanisms
We have shown in the previous section that the increase in the L-type Ca2+ current is an important compensatory mechanism for enhancing systolic [Ca2+]i. Whether changes in systems other than upregulation the NCX could also maintain diastolic [Ca2+]i to the level observed experimentally in the KO remains to be investigated. Possible candidates included: 1), a decrease in the persistent Na+ current (INab), which would reduce [Na+]i and in turn provide a greater thermodynamic drive for Ca2+ extrusion through NCX to lower diastolic [Ca2+]i; 2), a decrease in SR Ca2+ leak that could potentially decrease diastolic [Ca2+]i, enhance SR Ca2+ load, and thus increase systolic [Ca2+]i; and 3), an increase in the activity of the Na+/K+ ATPase (NKA) that would lower [Na+]i hence enhancing NCX activity and lowering diastolic [Ca2+]i.
To test these alternative mechanisms at 6 Hz stimulation frequency, we set up the control model that was the KO model but without upregulation of NCX. Then for each of the above candidate mechanisms, we varied the conductance of the persistent Na+ channel (GNab), the rate of SR Ca2+ leak (Vleak), the maximum NKA pump current (), respectively, as well as the maximum exchanger rate (VNCX) for comparison, whereas all other components were kept unchanged. We then examined the peak and diastolic [Ca2+]i as a function of the relative change in the conductance, as shown in Fig. 5.
Figure 5.
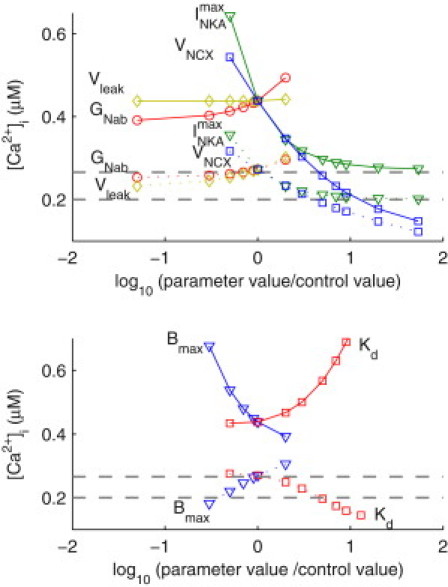
(Top) Changes in peak (solid lines) and diastolic (dotted lines) [Ca2+]i in response to changes in the conductances of the persistent Na+ current (GNab), the SR Ca2+ leak flux (Vleak), the Na+/K+ ATPase current (GNKA) and the NCX (VNCX). Dashed lines indicate the experimentally measured peak and diastolic [Ca2+]i in the KO cardiomyocytes. (Bottom) Changes in peak (solid lines) and diastolic (dotted lines) [Ca2+]i in response to changes total buffer concentration (Bmax) and affinity (Kd).
In Fig. 5, top, the x axis is plotted as the logarithm (base 10) of the ratio between the parameter value and its original value. Zero indicates the parameter value is the same as its original value. Positive values indicate an increase in the parameter value (x = 1 corresponds to a 10-fold increase). Negative values indicate a decrease in the parameter value (x = -1 means the parameter value is 10% of the control value). The gray lines indicate the peak and diastolic [Ca2+]i levels measured experimentally in the KO at 6 Hz, which can be considered as the target values that have been shown to be sufficient for maintaining cardiac function at the 4-week time point.
These results show that decreasing GNab and Vleak had very moderate effects on diastolic [Ca2+]i, which remained significantly higher than the target value even when conductances were decreased to almost zero, whereas, in contrast, increases in the activities of Na+/K+ and NCX sufficiently reduce diastolic [Ca2+]i. Furthermore, the rate of decrease in diastolic [Ca2+]i with increasing VNCX was significantly faster with the target diastolic value being reached when VNCX was between three and five times its control value. The effect of increasing the density of NKA had a similar effect to NCX initially. However, after a fivefold increase in (x = 0.7), the rate of decrease in diastolic [Ca2+]i was reduced and diastolic [Ca2+]i did not decrease to the target value until was 50 times its control value (x = 1.7). It is significant to note that, as VNCX continues to increase, the difference between peak and diastolic [Ca2+]i became smaller, indicating a progressive reduction in Δ[Ca2+]i and thus an anticipated reduction in systolic function that potentially limits the viability of increases in VNCX as a compensatory mechanism for continued loss of SERCA2 function.
A similar analysis was also carried out to test the sensitivity of peak and diastolic [Ca2+]i to the total concentration (Bmax) and affinity (Kd) of the intracellular Ca2+ buffers, which were originally set to 109 and 0.6 μM, respectively, in both the FF and KO models (see Discussion). As shown in Fig. 5, bottom, a decrease in the buffering power by either decreasing Bmax or increasing Kd led to a lower diastolic [Ca2+]i and a higher peak [Ca2+]i. Experimentally observed diastolic [Ca2+]i was reached with a 70% decrease in Bmax or a sevenfold increase in Kd. Further reductions in Bmax led to alternans, whereas Kd could be increased up to 13-fold with sustainable [Ca2+]i transients.
Discussion
The process of SR Ca2+ uptake through SERCA plays a key role in determining the rate of decay of the [Ca2+]i transient and hence relaxation, which in turn is crucial for diastolic filling. Ca2+ uptake into the SR also replenishes SR Ca2+ stores so that Ca2+ can be released again during the next cardiac cycle. The current simulation study demonstrated that given the observed decrease in SERCA activity, sustained [Ca2+]i transients could be maintained in the KO model. Simulated [Ca2+]i transient characteristics in the FF and KO models were consistent with experimental observations in the FF and KO cardiomyocytes. In the following sections, we discuss the relationship between protein expression and functional activity based on our analysis of the [Ca2+]i transients in the KO cardiomyocytes, the changes in Ca2+ dynamics in the KO model and the roles of the compensatory mechanisms. A critical assessment of the assumptions that the model was based on and their implications is provided in the Supporting Material.
Functional activities of the Ca2+ handling proteins in the KO cardiomyocytes
Previously, it has been shown that <5% SERCA2 protein was expressed in myocardial tissue from the KO, compared to the FF (6). In addition, the protein levels of NCX, PMCA were found to increase by 31% and 45%, respectively. In the current study, the functional activities of NCX and PMCA in the FF and KO cardiomyocytes were examined based on the rate of decay of the caffeine-induced [Ca2+]i transient, which in turn were used in conjunction with the decay of the field-stimulated transient to examine SERCA activity. Through the application of this method, sarcolemmal Ca2+ extrusion was calculated to have increased over threefold, and SERCA activity was found to have decreased by 67% (Fig. 1 B, inset). Direct quantitative characterization of the relationship between protein expression and function using experimental methods remains challenging. Our previous measurements of thapsigargin-sensitive 32P-incorporation in left ventricular myocardial tissue homogenates, which showed SERCA activity in the KO mice was ∼15% of the FF level (6), support a nonlinear relationship between expression levels and function.
The nonlinear correlation between the expression and activity of the Ca2+ handling proteins has also been reported by other groups. For example, Schwinger et al. (18) found a nonlinear correlation between expression and activity of the SERCA protein in their study of patients with dilated cardiomyopathy. Schmidt et al. (19) also observed reduced SERCA activity in failing human myocardium compared to nonfailing heart without any difference in SERCA protein level. One possible explanation for such differences could be that an excessive amount of SERCA protein may be present in the normal mouse that is not functional under physiological conditions (13).
Rate of decay of [Ca2+]i transient in the KO cardiomyocytes
Another interesting finding from the model is although the RT50 of the simulated [Ca2+]i transient at 1 Hz in the KO model was more than double of that in the FF model, it was only moderately increased at 6 Hz indicating a much less profound lusitropic effect of SERCA knockout at more physiological pacing frequencies. To find out whether this observation was caused by CaMKII-regulated frequency-dependent increase in Vup, we ran the model at 1 and 3 Hz with Vup kept constant at its 1 Hz level. Simulation results suggest that the increase in Vup at higher pacing frequencies may be important for facilitating the decay of the transient in both FF and KO models. However, when this factor was removed from both models, the resulting percentage increases in RT50 did not differ significantly between the FF and KO models (not shown).
The reason for the aforementioned reduced lusitropic effect of SERCA knockout at 6 Hz can, however, be explained using a simplified theoretical analysis of Ca2+ dynamics during the decay phase of the transient, proposed by Bers and Berlin (20). This method is based on the fact that the rate of [Ca2+]i decline does not only depend on the rate of Ca2+ removal from the cytosol but also on the properties of intracellular Ca2+ buffering. Assuming that Ca2+ transport by SERCA is the main contributor to [Ca2+]i decline in the wild-type mouse, the time taken for [Ca2+]i to decay from its value at time t0 to its value at time t can be expressed as:
| (1) |
where the right hand side is evaluated for [Ca2+]i at times t and t0 (see the Supporting Material for derivation of Eq. 1). Therefore, the RT50 of a [Ca2+]i transient can be obtained by evaluating Eq. 1 for [Ca2+]i at the peak of the transient ([Ca2+]p) and [Ca2+]i at 1/2. ([Ca2+]p + diastolic [Ca2+]i).
Substitution of the parameter values for the FF and KO models (Bmax = 109 μM, Kd = 0.6 μM, Vup = 0.19 (FF 1 Hz), 0.521 (FF 6 Hz), 0.067 (KO 1 Hz), 0.202 (KO 6 Hz) μM/ms, and Km,up = 0.412 μM) and the diastolic [Ca2+]i values (0.125 μM at 1 Hz, 0.2 μM at 6 Hz) into Eq. 1 gives the estimated RT50 of the [Ca2+]i transient as a function of peak [Ca2+]i, as shown in Fig. 6. According to the above theoretical analysis, the shift in RT50 between FF and KO models is much greater at 1 Hz than at 6 Hz, which qualitatively agrees with what is observed experimentally (6). Furthermore, when FDAR was removed from both the FF and KO models, the resulting plot of RT50 against [Ca2+]i still shows a relatively greater shift in RT50 between FF and KO models at 1 Hz, compared to 6 Hz (see details in Supplementary Material). This indicates that the reduced lusitropic impact of SERCA KO at a higher pacing frequency may be independent of FDAR.
Figure 6.
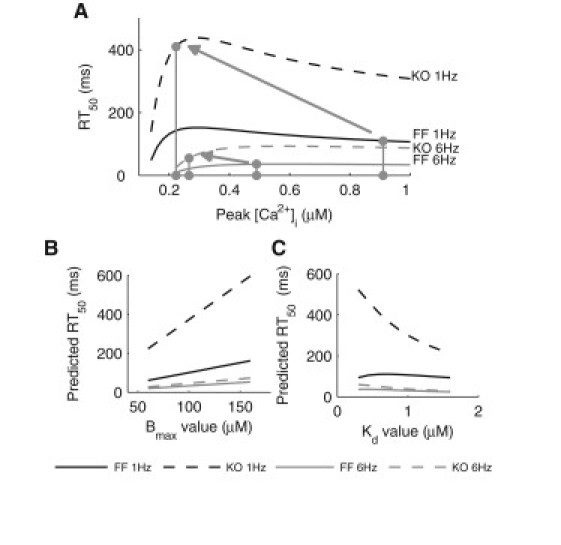
(A) Theoretical analysis of the decay kinetics of the [Ca2+]i transient, showing the RT50 of the transient as a function of peak [Ca2+]i for the FF and KO models. The circles correspond to the experimentally measured peak [Ca2+]i and the predicted RT50 values. (B) Sensitivity of rate of [Ca2+]i decay to the total buffer concentration (Bmax). Predicted RT50 was obtained by substituting the appropriate parameter values into Eq. 1. (C) Sensitivity of rate of [Ca2+]i decay to the affinity of the buffers (Kd).
The sensitivity of RT50 to changes in the buffering parameters was also analyzed using the above approach. Experimental data on buffering properties in mouse ventricular myocytes are sparse, but rat data are available. In rat cardiac myocytes, Diaz et al. (21) measured Bmax to be 160 μM with a Kd of 0.3 μM, whereas Berlin et al. (22) reported an average Bmax of 123 μM with cell-to-cell variations from 68 to 158 μM and an average Kd of 0.96 μM with variations from 0.55 to 1.57 μM. We therefore independently varied Bmax from 60 to 160 μM and Kd from 0.3 to 1.6 μM. The resulting changes in RT50 predicted from substituting the appropriate parameter values into Eq. 1 are shown in Fig. 6, B and C. It can be seen that an increase in the buffering power, by either increasing Bmax or decreasing Kd, led to a slower rate of decay (longer RT50). Nevertheless, within the range of Bmax and Kd examined, the qualitative trend of a smaller shift in RT50 at 6 Hz than at 1 Hz is maintained regardless of the choice of the buffering parameter values. Additional results of RT50 as a function of peak [Ca2+]i for a range of Bmax and Kd values are provided in the Supporting Material.
Finally, it is important to note that although a useful analysis tool, the approach of Bers and Berlin (20) does not take into account the effect of NCX and PMCA on relaxation time that is likely to play an important role in the KO model.
Contributions of the compensatory mechanisms
Simulations with graded reductions in the maximum uptake rate of SERCA showed that at a physiologically relevant pacing frequency for the mouse, diastolic [Ca2+]i rose sharply with decreasing Vup, whereas systolic [Ca2+]i did not change significantly. This observation points to the potential role of the compensatory mechanisms in lowering diastolic [Ca2+]i. Indeed, results from the three perturbation studies (KO-ICaL, KO-NCX, and KO-ICaL-NCX) support this hypothesis. We found that separately increasing ICaL and NCX increased systolic [Ca2+]i and decreased diastolic [Ca2+]i, respectively as one would expect, but the combined effects of the two compensatory mechanisms in simulations appeared to be a decrease in diastolic [Ca2+]i at the expense of a decrease in systolic [Ca2+]i. These results suggest that maintaining a physiological diastolic [Ca2+]i may dominate over the need to maintain systolic [Ca2+]i in the KO mice. In fact, elevated diastolic [Ca2+]i has been reported previously in failing heart preparations (19,23–26). Schmidt et al. (19) found that in failing human myocardium where SERCA function was impaired, diastolic [Ca2+]i was ∼40% higher compared to the control at low pacing frequencies, and increased to almost 100% higher after an increase in stimulation frequency. The elevated diastolic [Ca2+]i was in turn associated with increased generation of diastolic tension in the same preparations, an effect that also increased significantly with increasing stimulation frequency, as well as a decreased systolic tension and a negative force-frequency relationship (19). The level of increase in diastolic [Ca2+]i observed in the study of Schmidt et al. (19) is in the same range as that in the KO-NCX model (50%), suggesting the higher diastolic [Ca2+]i could be of significant physiological relevance to cardiac function, if it had indeed occurred in the KO mice. It is worth noting the differences between the FF model with reduced SERCA activity and the KO-ICaL-NCX model. PMCA activity was kept at the FF level in the former but at the KO level in the latter. In the former, ICaL parameters were kept unchanged with the same values as in the FF model, whereas in the latter, ICaL was clamped to have the same time course as that seen in the original FF model.
We have further demonstrated that, of the many cellular systems that can lower diastolic [Ca2+]i in the presence of impaired SERCA activity, upregulations of NCX and NKA as well as lowering Ca2+ buffering power are the three compensatory mechanisms capable of sufficiently reducing diastolic [Ca2+]i to match its level in the FF controls. For both NCX and NKA, the decrease in diastolic [Ca2+]i with increasing transporter activity occurs at the expense of a reduced systolic [Ca2+]i. On the other hand, a decrease in the buffering power lowered diastolic [Ca2+]i while also increasing systolic [Ca2+]i. Experimentally, diastolic [Ca2+]i was seen to be maintained in the KO cardiomyocytes whereas systolic[Ca2+]i was significantly decreased, compared to the FF values. These observations are consistent with the effects of upregulation of NCX or NKA, but not with lowering buffering power.
Simulation results in Fig. 5 suggest that in the KO model, a physiological diastolic [Ca2+]i can be reached, independently, with: 1), a 50-fold increase in NKA activity; 2), a 70% reduction in total buffer concentration (or a sevenfold decrease in the affinity of the buffer); or 3), a threefold increase in NCX activity. The limitation of upregulating NKA is that the rate of decrease in diastolic [Ca2+]i was progressively slowed after increases in over its control value. Although the reduction in buffering power may be a feasible solution in this model, experimental evidence suggests troponin C contributes to ∼50% of total fast Ca2+ buffering (27,28). Therefore, this compensatory mechanism may necessitate a decrease in troponin concentration or its Ca2+-binding affinity. These effects are likely to have further detrimental consequences on myocardial contraction in the KO cardiomyocytes where the size of the [Ca2+]i transient is already severely reduced. Compared to upregulation of NKA and decreasing buffering power, a threefold increase in NCX activity thus may be the most effective compensatory mechanism. However, this mechanism is not without constraint, as the reduction in [Ca2+]i transient amplitude becomes progressively more severe with further increases in VNCX, suggesting there may exist a balance point that optimizes both diastolic and systolic [Ca2+]i levels. Therefore, with continued reduction in SERCA activity beyond the 4-week time point, as confirmed by our SERCA2 KO study at the 7-week time point (13), cardiac pump function may be compromised without additional compensatory mechanisms such as upregulation of NKA and reduction of buffering power.
In human models of heart failure, the important role of NCX in maintaining diastolic function has been confirmed clinically by Hassenfuss et al. (29). In their study in which failing hearts were grouped according to their diastolic function, it was found that the group with preserved diastolic function also had increased protein levels of NCX. In contrast, in hearts where NCX protein level was unchanged, diastolic function was severely compromised. Therefore, results from our in silico study are consistent with these previous findings and suggest that in the presence of impaired SERCA function, the upregulation of NCX in the KO mice is important for maintaining diastolic [Ca2+]i level and diastolic function. Maintaining diastolic [Ca2+]i levels may additionally have important implications in regards to signaling. For example, CaMKII that is activated by [Ca2+]i, has been implicated in triggering hypertrophy and heart failure (30,31). Thus, the maintenance of diastolic [Ca2+]i levels by NCX upregulation possibly may also underlie the surprising absence of hypertrophy in KO animals (6,13).
Conclusions
We have developed a mathematical model of Ca2+ dynamics in cardiomyocytes of the SERCA2 knockout mice, based on temperature- and species-consistent experimental data. The model confirmed the previous experimental observation that sustained [Ca2+]i transients can be maintained despite severely impaired SERCA function. The importance of the compensatory mechanisms observed in the KO cardiomyocytes was investigated using the model, which demonstrated that the combined effect of the increased L-type Ca2+ current and NCX activity was to lower diastolic [Ca2+]i that was highly sensitive to changes in SERCA activity at the more physiological relevant pacing frequency. In silico investigation into alternative possible compensatory mechanisms identified NCX as the most effective mechanism for lowering diastolic [Ca2+]i although at the cost of a lower systolic [Ca2+]i. Further reductions in SERCA activity beyond the 4-week time point would require additional compensatory mechanisms such as a higher NKA activity or a lower buffering power.
Acknowledgments
The authors also gratefully acknowledge the skilled assistance of Ulla Enger, Morten Eriksen, and Lisbeth Winer.
N.P.S. acknowledges the support of MRC (Medical Research Council). S.A.N. acknowledges the support of EPSRC (Engineering and Physical Sciences Research Council). L.L. thanks the Clarendon Fund for providing funding for this research.
Supporting Material
References
- 1.Feldman A.M., Weinberg E.O., Lorell B.H. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ. Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. [DOI] [PubMed] [Google Scholar]
- 2.Qi M., Shannon T.R., Samarel A.M. Downregulation of sarcoplasmic reticulum Ca(2+)-ATPase during progression of left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 1997;272:H2416–H2424. doi: 10.1152/ajpheart.1997.272.5.H2416. [DOI] [PubMed] [Google Scholar]
- 3.Hasenfuss G., Reinecke H., Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 4.Limas C.J., Olivari M.-T., Simon A. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc. Res. 1987;21:601–605. doi: 10.1093/cvr/21.8.601. [DOI] [PubMed] [Google Scholar]
- 5.Pieske B., Maier L.S., Hasenfuss G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ. Res. 1999;85:38–46. doi: 10.1161/01.res.85.1.38. [DOI] [PubMed] [Google Scholar]
- 6.Andersson K.B., Birkeland J.A.K., Christensen G. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J. Mol. Cell. Cardiol. 2009;47:180–187. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Li L., Chu G., Bers D.M. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am. J. Physiol. 1998;274:H1335–H1347. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- 8.Fink M., Niederer S.A., Smith N.P. Cardiac cell modelling: observations from the heart of the cardiac physiome project. Prog. Biophys. Mol. Biol. 2010 doi: 10.1016/j.pbiomolbio.2010.03.002. Mar 18. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 9.Trafford A.W., Lederer W.J., Sobie E.A. Keeping the beat: life without SERCA—is it possible? J. Mol. Cell. Cardiol. 2009;47:171–173. doi: 10.1016/j.yjmcc.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Bondarenko V.E., Szigeti G.P., Rasmusson R.L. Computer model of action potential of mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H1378–H1403. doi: 10.1152/ajpheart.00185.2003. [DOI] [PubMed] [Google Scholar]
- 11.Jafri M.S., Rice J.J., Winslow R.L. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 1998;74:1149–1168. doi: 10.1016/S0006-3495(98)77832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Louch W.E., Mørk H.K., Sejersted O.M. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J. Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louch W.E., Hougen K., Sejersted O.M. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J. Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng H., Lederer W.J., Cannell M.B. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 15.Li L., Niederer S.A., Smith N.P. A mathematical model of the murine ventricular myocyte: a data-driven biophysically based approach applied to mice overexpressing the canine NCX isoform. Am. J. Physiol. Heart Circ. Physiol. 2010;299:H1045–H1063. doi: 10.1152/ajpheart.00219.2010. [DOI] [PubMed] [Google Scholar]
- 16.Stokke M.K., Hougen K., Trafford A.W. Reduced SERCA2 abundance decreases the propensity for Ca2+ wave development in ventricular myocytes. Cardiovasc. Res. 2010;86:63–71. doi: 10.1093/cvr/cvp401. [DOI] [PubMed] [Google Scholar]
- 17.Berry R.G., Despa S., Shattock M.J. Differential distribution and regulation of mouse cardiac Na+/K+-ATPase alpha1 and alpha2 subunits in T-tubule and surface sarcolemmal membranes.α. Cardiovasc. Res. 2007;73:92–100. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Schwinger R.H., Böhm M., Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca(2+)-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt U., Hajjar R.J., Gwathmey J.K. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J. Mol. Cell. Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 20.Bers D.M., Berlin J.R. Kinetics of [Ca]i decline in cardiac myocytes depend on peak [Ca]i. Am. J. Physiol. 1995;268:C271–C277. doi: 10.1152/ajpcell.1995.268.1.C271. [DOI] [PubMed] [Google Scholar]
- 21.Díaz M.E., Trafford A.W., Eisner D.A. The role of intracellular Ca buffers in determining the shape of the systolic Ca transient in cardiac ventricular myocytes. Pflugers Arch. 2001;442:96–100. doi: 10.1007/s004240000509. [DOI] [PubMed] [Google Scholar]
- 22.Berlin J.R., Bassani J.W., Bers D.M. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 1994;67:1775–1787. doi: 10.1016/S0006-3495(94)80652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gwathmey J.K., Copelas L., Morgan J.P. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ. Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 24.Gwathmey J.K., Hajjar R.J. Relation between steady-state force and intracellular [Ca2+] in intact human myocardium. Index of myofibrillar responsiveness to Ca2+ Circulation. 1990;82:1266–1278. doi: 10.1161/01.cir.82.4.1266. [DOI] [PubMed] [Google Scholar]
- 25.Beuckelmann D.J., Näbauer M., Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 26.Beuckelmann D.J., Näbauer M., Erdmann E. Altered diastolic [Ca2+]i handling in human ventricular myocytes from patients with terminal heart failure. Am. Heart J. 1995;129:684–689. doi: 10.1016/0002-8703(95)90316-x. [DOI] [PubMed] [Google Scholar]
- 27.Bers D.M. 2nd ed. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. [Google Scholar]
- 28.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983;245:C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 29.Hasenfuss G., Schillinger W., Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 30.Wu Y., Temple J., Anderson M.E. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. doi: 10.1161/01.cir.0000027583.73268.e7. [DOI] [PubMed] [Google Scholar]
- 31.Zhang T., Maier L.S., Brown J.H. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.