

Abstract
Both type 2B and type 2M von Willebrand disease result in bleeding disorders; however, whereas type 2B has increased binding affinity between platelet glycoprotein Ibα and von Willebrand factor (vWF), type 2M has decreased binding affinity between these two molecules. We used R687E type 2B and G561S type 2M vWF-A1 mutations to study binding between flowing platelets and insolubilized vWF mutants. We measured rolling velocities, mean stop times, and mean go times at 37°C using high-speed video microscopy. The rolling velocities for wt-wt interactions first decrease, reach a minimum, and then increase with increasing shear stress, indicating a catch-slip transition. By changing the viscosity, we were able to quantify the effects of force versus shear rate for rolling velocities and mean stop times. Platelet interactions with loss-of-function vWF-A1 retain the catch-slip bond transition seen in wt-wt interactions, but at a higher shear stress compared with the wt-wt transition. The mean stop time for all vWF-A1 molecules reveals catch-slip transitions at different shear stresses (gain-of-function vWF-A1 < wt vWF-A1< loss-of-function vWF-A1). The shift in the catch-slip transition may indicate changes in how the different mutants become conformationally active, indicating different mechanisms leading to similar bleeding characteristics.
Introduction
Platelets mediate the homeostatic response to vascular injuries. In arteries, this response is initiated specifically by platelet receptor glycoprotein Ibα (GPIbα) tethering to von Willebrand factor (vWF) on exposed subendothelium. Platelet GPIbα interacts with vWF to initiate rolling interactions that are the first step in the formation of a hemostatic plug at a site of vascular injury. After this initial step, platelets activate, firmly adhere via the GPIIb/IIIa integrin, and aggregate to form a plug and stop bleeding (1). Mutations in either vWF or GPIbα can disrupt this first step in hemostasis, resulting in the bleeding disorders von Willebrand disease (VWD) or platelet type VWD (pt-VWD), respectively (2). Disruption of the normal interactions between these molecules can also lead to thrombosis, as seen in thrombotic thrombocytopenic purpura (3).
vWF is the largest soluble plasma protein and is found both in the bulk fluid in its soluble form and in the subendothelial matrix. At high shear stress, vWF binds to exposed subendothelial matrix components and functions to bridge exposed subendothelial collagens and circulating platelets (4). vWF is a multimeric glycoprotein found in the plasma as a globular protein consisting of a repeating vWF dimer with a total molecular mass ranging from 500 to 20,000 kDa (5). A single vWF monomer contains 2050 amino acids and consists of 11 domains (D′-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2). The A1 domain specifically binds platelet GPIbα (6), collagens I and III (7,8), and collagen IV (9), whereas the A3 domain participates in collagen I and III binding (9–12), and the A2 domain contains the site for cleavage by the ADAMTS-13 enzyme (13). Crystal structures suggest that the β-switch (cystine loop) and β-finger (N-terminus) strands of GPIbα bind directly to vWF-A1, whereupon the region between residues 227 and 241 at the C-terminus undergoes a conformational change (14). Most gain-of-function (GOF) vWF-A1 mutations are located on the A1 surface of the molecule containing its N- and C-termini, whereas loss-of-function (LOF) vWF-A1 mutations are either located on the A1 surface that is the upper face of the domain or buried inside the A1 structure. These mutations do not directly interact with GPIbα, so it is thought that the mutations result in changes that are propagated through the vWF-A1 structure to the binding site with GPIbα (15).
Evidence suggests that mutations in either vWF or GPIbα may lead to abnormal interactions between platelet GPIbα and soluble vWF. It is believed that in type 2B VWD, increased aggregation of platelets with soluble vWF leads to bleeding due to clearance of these aggregates or to a lack of unbound GPIbα receptors to interact at sites of vascular injury. We recently showed that a catch bond governs binding between platelet GPIbα and vWF-A1 at low shear stresses for interactions at room temperature (16). Under normal conditions, wt-wt catch-bond interactions prevent platelet aggregate formation, whereas type 2B VWD GOF mutations form platelet aggregates with soluble vWF in bulk flow at lower shear stresses.
VWD type 2M LOF mutations in vWF show decreased binding affinity resulting in increased rolling velocities and off-rates. These types of mutations are characterized by defective ristocetin binding and normal botrocetin binding. Type 2M mutant G561S shows decreased ristocetin binding and a normal multimerization pattern (17). It is thought that these mutations result in a defect that prevents the vWF molecule from undergoing a conformational change that allows GPIbα binding (7). Atomic force microscopy (AFM) experiments using type 2M mutations in vWF-A1 showed that LOF G561S vWF-A1 binding to GPIbα results in a catch bond, with a transition to a slip bond at higher force (18). It should be noted that the G1324S mutation referred to by some authors (16,18) is the same as the G561S mutation used in this work. However, the tether-bond behavior of type 2M VWD-causing mutations under conditions of flow remains largely unknown.
In this study, we measured rolling interactions to gain insight into bonding behavior. It has been established that cell rolling is predominantly mediated by the dissociation rate of the bonds that are formed and broken during rolling motions (19–21). Bond dissociation under flow is used to determine the bond lifetime, which is inversely proportional to the dissociation rate. We observed a decrease in rolling velocity with increasing wall shear stress, suggesting that the bond lifetime increases (or that the dissociation rate decreases) with increasing force, indicative of a catch bond. Conversely, the observation that the cell rolling velocity increases with increasing shear stress likely indicates that the bond lifetime is decreasing, or that the dissociation rate is increasing, which is indicative of a slip bond. We use the term “catch-slip” to describe these indirect bonding measurements of rolling interactions, where it is likely that catch and slip bonds are present, rather than to indicate that catch and slip bonds were directly measured. These types of catch-slip biphasic transitions have previously been observed in selectins and E. coli (20,22,23), as well as in mathematical models of adhesive behavior (24,25).
To investigate the mechanics of the tether bond, we captured interactions at 250 frames per second and conducted a frame-by-frame analysis of these interactions. The steps in binding of a single tether were analyzed and characterized in stages as either a “stop” or a “go”, as described previously for selectin rolling interactions (20). We analyzed the binding behavior between platelet GPIbα and vWF wt and mutant variations to better understand how the changes seen in mutants associated with VWD occur at the tether-bond level. We show that the rolling velocity and bonding behavior provide insight into how GPIbα and vWF regulate rolling, and demonstrate for the first time (to our knowledge) that platelets rolling on GOF R687E vWF-A1 or LOF G561S vWF-A1 molecules retain the catch-slip bond transition we previously observed for wt-wt interactions (16). Based on the mean stop times obtained under physiological fluid shear stress, we conclude that the catch-slip transition for platelets interacting with GOF R687E vWF-A1 occurs at a lower shear stress than for wt-wt interactions, which occur at a still lower shear stress than for LOF G561S vWF-A1 interactions (GOF vWF-A1 < wt vWF-A1 < LOF vWF-A1). Taken together, these data provide information about the GPIbα-vWF tether bond and the differences that exist between LOF and GOF mutations in vWF-A1, and hence between type 2M and type 2B VWD.
Materials and Methods
Proteins and antibodies
Recombinant vWF-A1 molecules (wt, R687E, G561S) and wt vWF-A1A2A3 molecules were produced as previously described (7,26,27). Whole vWF was purchased from Calbiochem (San Diego, CA).
Platelet isolation
Whole blood was drawn from healthy volunteers according to an approved institutional review board procedure, and all volunteers gave written, informed consent. Whole blood was drawn into citrate buffer (25 g sodium citrate, 8 g citric acid, 500 mL H2O) containing 2 μM prostaglandin E-1. The blood was centrifuged at 150 g for 15 min at room temperature. The supernatant (platelet-rich plasma) was removed and mixed with acid citrate dextrose buffer (6.25 g sodium citrate, 2 H2O, 3.1 g citric acid anhydrous, 3.4 g D-glucose in 250 mL H2O), 10% by volume, and centrifuged at 900 g for 5 min at room temperature. The pellet was suspended in Hepes-Tyrode buffer, and platelets were counted with a hemocytometer. The platelet suspension was then diluted to 1 × 108 platelets per milliliter in Hepes-Tyrode buffer for flow assays.
Measurement of viscosity
The viscosity of the platelet suspension buffer was measured at 37°C with the use of an Ostwald dripping viscometer as previously described (20). In some experiments, 6% Ficoll (MW 400, w/v; Sigma, St. Louis, MO) was added to increase the viscosity 1.8-fold compared with buffer alone.
Preparation of vWF molecule-coated surfaces
The flow-chamber surface was a 35 mm tissue culture dish coated with vWF-A1 molecules (wt at 100 μg/mL, R687E at 100 μg/mL, or G561S at 200 μg/mL), wt vWF-A1A2A3 (at 100 μg/mL), or whole vWF molecules (at 50 μg/mL). The dish was coated with 20 μL of vWF molecule and incubated overnight at 4°C. The surface was rinsed with phosphate-buffered saline and blocked with 20 μL of 1% human serum albumin for 2 h at room temperature, and then rinsed again with phosphate-buffered saline. The prepared dish was brought to 37°C and used as the lower surface for the flow chamber.
Flow-chamber assay
A parallel platelet flow chamber (GlycoTech, Gaithersburg, MD) was used to apply shear stress to platelets interacting with the vWF-coated surface. Platelets were perfused through the flow chamber with a syringe pump (Harvard Apparatus, Holliston, MA). Interactions were visualized using an inverted-stage phase-contrast microscope (DIAPHOT-TMD, X-20 or X-10 phase objective and X-5 projection lens; Nikon, Garden City, NY). The flow chamber was maintained at 37°C by means of an air curtain surrounding the microscope. Images were captured at 250 fps directly to a computer using a high-speed video camera and Phantom 640 software (Phantom V4.2; Vision Research, Wayne, NJ). Images were analyzed post experiment using the Track Object module in Metamorph Offline software (Molecular Devices, Sunnyvale, CA).
Measurement of rolling velocities and rolling step parameters
The rolling velocities (μm/s) were calculated as the total distance traveled in 1 s. Each velocity represents an average of tens to hundreds of cells. The freestream velocities were measured from platelets flowing over surfaces coated with 1% HSA. The rolling step parameters were determined as described previously (20) from video taken at 250 fps. These interactions were classified as stop or go events based on whether a cell was accelerating or decelerating in each frame relative to the previous frame. A cell that was classified as accelerating was considered to be in a go phase, whereas a cell with no acceleration or deceleration was considered to be stopped. Rolling parameters were evaluated on an event basis rather than on a cell basis, as described previously (20). In these cases, data points represent hundreds and in some cases thousands of events, which in turn may result in error bars that are not visible in the figures presented.
Statistical analysis
We analyzed each data set using a one-way analysis of variance followed by a t-test to determine whether neighboring data points were significantly different from each other. To determine which pairs were significantly different for neighboring values of shear stress, we performed t-tests for adjacent data points. All of the catch-slip transition values are statistically significantly different from the data points for one shear stress higher and one shear stress lower than the shear stress at which the catch-slip transition occurs.
Results
Rolling velocities for platelets interacting with vWF molecules
Platelet rolling velocities were calculated for interactions of platelets with wt vWF-A1 (Fig. 1 A), GOF R687E vWF-A1 (Fig. 1 B), LOF G561S vWF-A1 (Fig. 1 C), wt vWF-A1A2A3 (Fig. 1 D), or whole vWF (Fig. 1 E). Fig. 1 B includes the freestream velocities of platelets flowing over 1% HSA, which are higher than the velocities of rolling platelets. Platelets interacting with all vWF molecules display a catch-slip transition, with the rolling velocity first decreasing at lower shear stresses and then transitioning to increasing velocities at higher shear stresses. The minimum rolling velocities were similar for all wt vWF molecules and LOF G561S vWF-A1 (Fig. 1, A and C–E). The catch-slip transition occurs for wt vWF-A1 (Fig. 1 A), whole vWF (Fig. 1 E), and LOF G561S vWF-A1 (Fig. 1 C) at 16 dyn/cm2, whereas the transition for wt vWF A1A2A3 (Fig. 1 D) occurs at 8 dyn/cm2 and the transition for GOF R687E vWF-A1 (Fig. 1 B) occurs at 4 dyn/cm2.
Figure 1.
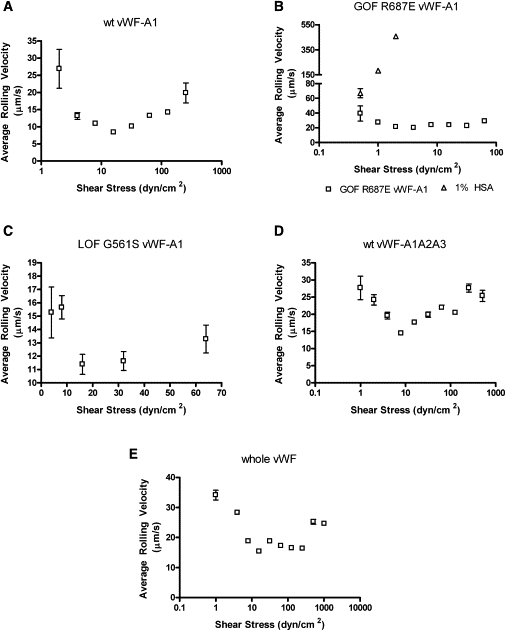
Rolling velocities of platelets on various forms of vWF molecule: (A) wt vWF-A1 (n = 14–256), (B) GOF R687E vWF-A1 (n = 12–353), (C) LOF G561S vWF-A1 (n = 23–98), (D) wt vWF-A1A2A3 (n = 10–36), and (E) wt whole vWF (n = 44–277). Because there is decreased association between platelets and LOF G561S vWF-A1 (C), fewer interactions occur at all shear stresses, and none occur at the highest shear stresses measured for other vWF molecules. Measured freestream velocities of platelets perfused across 1% HSA (Δ) are plotted in B to show that the rolling velocity of platelets rolling on GOF R687E vWF-A1 is less than the freestream values. Data represent the mean ± SE of rolling platelets. Each catch-slip transition is statistically significant with p < 0.05.
Effects of fluid viscosity on rolling velocities in the wt vWF-A1 domain
Purified platelet-rich plasma with or without 6% Ficoll was perfused across a wt vWF-A1-coated coverslip (100 μg/mL) at 37°C, as shown in Fig. 2. The results are plotted against both shear rate (Fig. 2 A) and shear stress (Fig. 2 B). As can be seen in the figure, there is an increase in shear stress by a factor of 1.8 at a given shear rate for samples containing 6% Ficoll. When the rolling velocities are plotted as a function of shear rate (Fig. 2 A), the curves do not align. However, when they are plotted as a function of shear stress (Fig. 2 B), the curves align. These results show that rolling interactions depend on the shear stress applied to the bond. Therefore, these data suggest that the bonding mechanism depends on force rather than transport.
Figure 2.

Effects of viscosity on rolling velocity for (A) shear rate (n = 14–256) and (B) shear stress (n = 10–112) for platelets rolling on wt vWF-A1. Data represent the mean ± SE. Each catch-slip transition is statistically significant with p < 0.05.
Catch-slip transition shear stress varies depending on the vWF-A1 molecule
Interactions of platelets rolling on vWF-A1 molecules were characterized frame by frame as stop or go motions, and rolling interactions were measured at 250 fps. Fig. 3 shows the mean stop time data, i.e., the average time a cell spends in the stop phase during a 1-s interval. For all vWF-A1 molecules, the mean stop time first increases and then decreases as the shear stress and hence the force on the bond increases. The catch-slip transition for GOF R687E vWF-A1 (2 dyn/cm2; Fig. 3 B) is decreased compared with that of wt vWF-A1 (16 dyn/cm2; Fig. 3 A). However, this transition for LOF G561S vWF-A1 (32 dyn/cm2; Fig. 3 C) occurs at an increased shear stress compared with that of wt vWF-A1 molecule (Fig. 3 A).
Figure 3.
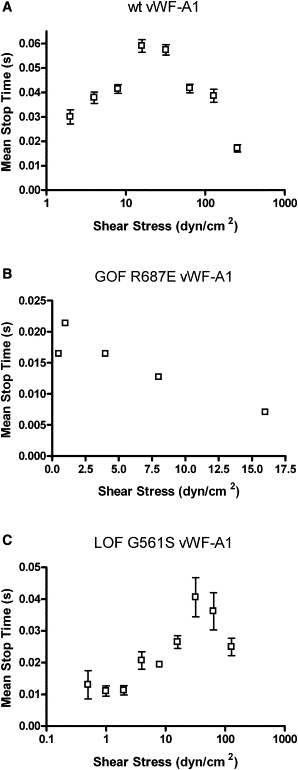
Mean stop times of platelets rolling on vWF-A1 molecules: (A) wt vWF-A1 (n = 14–256), (B) GOF R687E vWF-A1 (n = 5–104), and (C) LOF G561S vWF-A1 (n = 11–382). Data represent the mean ± SE. Each catch-slip transition is statistically significant with p < 0.05.
Effects of viscosity on the mean stop times for platelets rolling on wt vWF-A1
Purified platelet-rich plasma with or without 6% Ficoll was perfused across a wt vWF-A1-coated coverslip (100 μg/mL), as shown in Fig. 4. The results are plotted against both shear rate (Fig. 4 A) and shear stress (Fig. 4 B). The viscosity at a given shear rate was increased by a factor of 1.8 by the addition of 6% Ficoll. The curves do not align when plotted against shear rate (Fig. 4 A), but do align when plotted as a function of shear stress (Fig. 4 B). This alignment with shear stress suggests that bond force governs mean stop time interactions, as opposed to a transport-dependent bonding mechanism.
Figure 4.
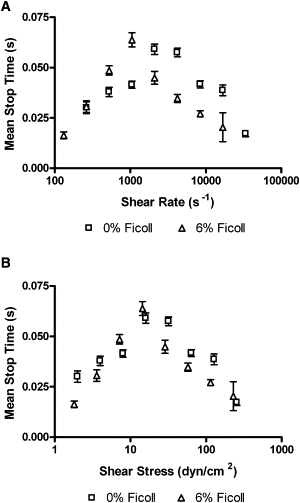
Effects of viscosity on the mean stop times of platelets on wt vWF-A1 molecules: (A) shear rate (n = 14–256) and (B) shear stress (n = 10–112). Data represent the mean ± SE. Each catch-slip transition is statistically significant with p < 0.05.
Mean stop time regulates platelets rolling on wt vWF-A1 molecules
Purified platelet-rich plasma with or without 6% Ficoll was perfused across a wt vWF-A1 coated coverslip (100 μg/mL) at 37°C. The average amount of time a cell spent in either the stop or the go phase over a 1-s interval was calculated from rolling data. Fig. 5 shows that the mean go time is constant when compared with the mean stop time. Furthermore, the mean go time is constant and less than the mean stop time for all shear stresses investigated. These data indicate that the mean go time is unaffected by the shear stress. Further, because the mean stop time displays biphasic behavior with longer durations, the data suggest that the mean stop time is a controlling parameter for rolling interactions.
Figure 5.

Effect of shear stress on the mean stop time and mean go time for platelet rolling interactions with wt vWF-A1 molecules: (A) 0% Ficoll (n = 14–256) and (B) 6% Ficoll (n = 10–112). Data represent the mean ± SE. Each catch-slip transition is statistically significant with p < 0.05.
Discussion
Differences are observed in the bonding interactions between GPIbα and vWF molecules when we compare wt vWF molecules with those with mutations causing type 2M and 2B VWD. The type 2M mutation used in these studies, G561S, occurs naturally and is characterized by decreased platelet binding in the presence of ristocetin (LOF), as well as by a normal multimer pattern and normal number of vWF antigens (17). The type 2B mutation used in these studies, R687E, is characterized by enhanced platelet binding with ristocetin (GOF) (28,29).
We previously showed that biphasic catch-slip bonds govern wt-wt interactions, demonstrating that this bond is force-dependent, and therefore that force plays an important role in the bonding behavior between GPIbα and vWF molecules. However, the same mechanism was not seen for wt GPIb-type 2B VWD interactions, in which the catch bond behavior was absent (16). In this work we obtained functional evidence for catch-slip bonds by observing GPIbα-vWF rolling interactions. We show that platelet wt vWF-A1 interactions have a transition from a catch to a slip bond (Fig. 1 A) at physiological temperatures. Further, we report the novel (to our knowledge) observation that platelet-GOF and platelet-LOF interactions under fluid forces retain the catch-slip transition (Fig. 3, B and C), but these transitions occur at lower and higher shear stresses, respectively, than seen in wt-wt interactions (Fig. 3, A–C). For the GOF R687E vWF-A1 mutation, our data differ from previously reported results that showed a pure slip bond (16). It may be that these differences can be attributed to the difficulty of obtaining data at the lowest shear stresses studied. It is also possible that the differences in temperatures at which the experiments were conducted (37°C herein and room temperature in work presented by Yago and colleagues (16)) are responsible for the differences observed, given that bond kinetics can be affected by temperature.
Our data show that at physiological temperatures, GOF and LOF mutations each differ from wt vWF-A1, but in unique ways (Figs. 1, A–C, and 3). These data show that rolling velocity interactions with each wt, GOF, and LOF vWF-A1 molecules have a catch-slip bond (Fig. 1, A–C).
Further differences were seen in the mean stop time data (Fig. 3), in that the catch-slip transition was seen for all three types of vWF-A1 studied, but at different shear stresses. Together, the rolling velocity and mean stop time data suggest that although type 2M and type 2B VWD have similar clinical manifestations, they employ different mechanisms for tether-bond dissociation.
To study differences in the tether bond, we first observed the rolling velocities for three wt vWF molecules: vWF-A1 (Fig. 1 A), vWF-A1A2A3 (Fig. 1 D), and whole vWF (Fig. 1 E). After completing the wt vWF molecule studies, we compared changes using GOF R687E vWF-A1 (Fig. 1 B) and LOF G561S vWF-A1 (Fig. 1 C). First, we measured the rolling velocities for wt vWF-A1A2A3 and whole vWF (Fig. 1, D and E, respectively). In both cases the rolling velocity of platelets interacting with the vWF molecule first decreased and then increased with increasing shear stress. For whole vWF, the rolling velocity did not increase as dramatically as was seen for both wt vWF-A1 and wt vWF-A1A2A3, which may be due to the presence of the RGD sequence that is found in the whole vWF molecule but not in the other molecules studied. It is likely that the RGD sequence binds integrins that are present on the platelet surface. During the experiments, the platelets were treated with prostaglandin E-1 and citrate buffer to prevent significant activation and thus decrease the likelihood that high-affinity integrins would be present on the platelet surface, since platelet activation is necessary for exposure of these integrins. However, it is possible that some integrins were activated at higher shear stresses, which would result in bonds being formed for interactions with whole vWF that were not present for the other vWF molecules used. Because these interactions likely involve integrin bonds, the rolling interactions result in the formation of some longer-lifetime bonds, causing the platelets to move more slowly. However, because the velocities do not drop to zero, there are not enough integrin bonds to result in complete platelet arrest. The minimum rolling velocity for platelets interacting with whole vWF molecules occurred at 16 dyn/cm2, which is the same minimum observed for both wt vWF-A1 and LOF G561S vWF-A1 surfaces (Fig. 1, A and B, respectively). Of interest, the minimum rolling velocity observed for platelet interactions with wt vWF-A1A2A3 occurred at a shear stress of 8 dyn/cm2 (Fig. 1 D). It was previously suggested that a decrease in the catch-slip transition shear stress indicates a destabilization of the vWF molecule used (18). The term “destabilization” refers to the idea that less shear stress is required to result in adhesion, suggesting that the vWF molecule has undergone a conformational change to become active at a lower shear stress than it would have undergone in its native wt form. Therefore, the fact that the transition occurs at a lower shear stress for the wt-A1A2A3 vWF molecule may suggest that this configuration of the vWF molecule is less stable than wt vWF-A1 and whole vWF. The catch-slip transition for platelets interacting with GOF vWF-A1 occurred at an even lower shear stress: 4 dyn/cm2. This lower transition suggests that the configuration of the GOF vWF-A1 molecule may be less stable than the other vWF molecules we studied.
We extended the rolling interactions to observe more detailed motions of the platelets rolling on each vWF molecule (Figs. 3–5). To quantify these interactions, each frame was characterized as a stop or a go event based on whether the cell was accelerating or decelerating in each frame relative to the previous frame. These values are reported here as mean stop times and mean go times. Previous studies have used mean stop and go times to describe selectin rolling interactions (20). Although such data do not directly measure single-bond behavior, they can be used to infer information about bond lifetimes. For platelet-wt vWF-A1 interactions, we show that the mean stop time first increases and then decreases with increasing shear stress, which is similar to the rolling velocity transition shear stress shown here in Fig. 1 A and reported previously (16). For both rolling velocity and mean stop time data, the catch-slip transition occurs at 16 dyn/cm2 (Figs. 1 A and 3 A). Further, we show that increasing the viscosity (and thus the force) at a given shear rate results in curves that align when plotted versus shear stress, but do not align when plotted versus shear rate (Figs. 2 and 4). These data suggest that, overall, rolling interactions depend on force rather than transport. The mean stop time indicates the average amount of time during a 1-s interval that the cell spends stopped. The mean stop time data suggest how the off-rate or bond lifetime for each interaction is modulated, because this type of measurement indicates how long the bond is stopped before the platelet moves again. Our data, therefore, suggest that the bond lifetime of wt-wt interactions increases and then decreases with increasing applied force. Alternatively, these data suggest that the off-rate decreases (longer lifetime) and then increases (shorter lifetime) as force is increased. Further, the data show that both the overall rolling velocities and more-detailed interactions (in this case, the mean stop time) are governed by force.
To further study the changes introduced by mutations in vWF, we calculated the mean stop times for each vWF-A1 mutant molecule in comparison with wt vWF-A1. Our data show that the transition force for LOF G561S vWF-A1 is greater than that for wt vWF-A1, which is greater than that for GOF R687E vWF-A1 (Fig. 3). These data suggest that greater force is required for the maximum bond lifetime in G561S vWF-A1 compared with wt vWF-A1, whereas less force is required for GOF R687E vWF-A1 compared with wt vWF-A1. These data show that the catch-slip transition seen for rolling velocity is carried over to a parameter that describes a more specific property of the bonding mechanism.
Recently, Auton et al. (18) studied the thermodynamic stability of vWF-A1 wt and mutant molecules. They produced single-molecule AFM data showing that the catch-slip transition for bond lifetimes also exists for both type 2M and type 2B mutants and wt vWF-A1. Further, their AFM and thermodynamic data show that the transition for GOF mutations occurs at lower forces in vWF-A1 compared with wt vWF-A1, which occurs at lower forces than observed for LOF mutations in vWF-A1. Our mean stop time results (Fig. 4) are consistent with their data, suggesting that the stability observed in AFM and thermodynamic measurements can also be seen in the flow-chamber experiments, in which the molecule pairs are subjected to a more physiological environment.
We also studied the mean go times for wt-wt rolling interactions, which represent the average amount of time the platelets spent moving forward after a stop. Based on the model used (20), the mean go time is the combination of the cell first pivoting to break the previous bond and the cell stretching the new trailing bond as it moves forward to form the next bond. Thus, the observations made here can be used to infer whether the rate of association plays a role in the tether bond, but do not directly measure the on-rate. The rolling step interactions show that the mean go time for wt-wt interactions is constant when compared with the mean stop time for wt-wt interactions (Fig. 5). Further, the data show that the mean go time is less than the mean stop time for all shear stresses. Together, these data suggest that the mean stop time is likely the controlling parameter in GPIbα-vWF rolling interactions. Because the stop time suggests information about how long a bond lasts (i.e., the bond lifetime) and the go time suggests information about how bonds are formed, the combined data suggest that the bond lifetime regulates platelet interactions with the wt vWF-A1 molecule when subjected to fluid shear stress. Because the mean go time is constant relative to the mean stop time, these data suggest that the time spent by the cell moving forward will be unaffected by changes in fluid shear stress when compared with changes in the time the cell spends stopped. Because the mean stop time suggests changes in the bond lifetime (regulated by force), these observations can be extended to suggest that force mechanisms, rather than transport mechanisms, regulate the GPIbα-vWF tether bond when subjected to fluid shear stress.
These observations, made over a range of shear stresses, reveal the presence of catch-slip transitions at shear stresses that may reflect changes in vWF structure not only for wt-wt interactions but also for wt-LOF vWF-A1 (rolling and mean stop time data) and wt-GOF vWF-A1 (mean stop time data) interactions. Additional studies will lead to a greater understanding of how naturally occurring mutations in vWF result in each type of VWD. The data shown here provide details about the GPIbα-vWF tether bond and show differences among different types of mutations in the vWF molecule, and hence between type 2M and type 2B VWDs. These data may also explain why bleeding is seen for both GOF and LOF mutations in vWF. For GOF type 2B VWD, there are likely prolonged lifetimes for platelet interactions with GOF R687E vWF-A1 at the lowest shear stresses. This would result in platelet binding to soluble vWF, effectively clearing platelets from the bulk fluid, and consequently there would not be enough to interact at sites of vascular injury. For LOF type 2M VWD mutations, there is decreased adhesion between platelets and LOF vWF mutants when they have an opportunity to bind, and the catch-slip transition (i.e., the shear stress at which bonds last for the longest time) is at a higher shear stress.
Kumar et al. (30) measured the tether frequency for LOF and GOF mutations in the GPIbα molecule, which may display trends similar to those of LOF and GOF mutations in the vWF-A1 molecule. In their work, the tether frequencies for LOF mutations were less than those for wt molecules, which were less than those for GOF mutations, indicating that fewer tethering events were occurring for the LOF mutation and more were occurring for the GOF mutation. Further, they observed that as shear stress increased, the tether frequency for all molecules decreased, indicating fewer tethering events. If similar behavior is occurring for mutations in the vWF-A1 molecule, this, in addition to the data shown here, may help explain bleeding in type 2M patients.
By becoming more stabilized at higher shear stresses compared with wt vWF-A1 molecules, LOF vWF-A1 molecules likely become conformationally active at shear stresses where they are not able to bind platelets effectively because of the decreased number of successful interactions at these higher shear stresses. Further, the data shown in Fig. 4 indicate that the magnitude of the mean stop time at its maximum value is less for LOF vWF-A1 than for wt vWF-A1. Together with the data of Kumar et al. (30), our findings data suggest the occurrence of fewer events that last for a shorter period of time, providing a possible explanation for the decreased binding that is characteristic of type 2M VWD.
Because GOF mutations are less stable at a lower shear stress than wt vWF-A1 molecules (i.e., more susceptible to force than wt vWF-A1 at its normal transition shear stress), it is likely that GOF vWF-A1 molecules become conformationally active at these low shear stresses and are able to participate in the enhanced binding that is characteristic of type 2B VWD. Here we also see that the magnitude of the maximum mean stop time for GOF vWF-A1 molecules is less than that of wt vWF-A1. If the GOF vWF-A1 molecules behave similarly to the GOF GPIbα molecules studied by Kumar et al. (30), it is possible that an increased number of interactions occur at the GOF transition shear stress (i.e., increased tethering frequency), but these interactions are more rapid, which could result an increased number of adhesive events.
Together, these observations may explain why both type 2B and type 2M VWDs result in patient bleeding but have different underlying mechanisms. Our determination of the biomechanical and kinetic properties of wt and mutant molecules elucidates the possible physiological mechanisms inherent in interactions between GPIbα and vWF, and may lead to a better understanding of both type 2B and type 2M VWD bleeding disorders.
Acknowledgments
This work was supported by National Institutes of Health grants HL70537, HL18672, and HL91020.
References
- 1.Reininger A.J. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14(Suppl 5):11–26. doi: 10.1111/j.1365-2516.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- 2.Sadler J.E., Budde U., Working Party on von Willebrand Disease Classification Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J. Thromb. Haemost. 2006;4:2103–2114. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 3.Moake J.L. Thrombotic thrombocytopenic purpura: the systemic clumping “plague”. Annu. Rev. Med. 2002;53:75–88. doi: 10.1146/annurev.med.53.082901.103948. [DOI] [PubMed] [Google Scholar]
- 4.Savage B., Almus-Jacobs F., Ruggeri Z.M. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 5.Andrews R.K., López J.A., Berndt M.C. Molecular mechanisms of platelet adhesion and activation. Int. J. Biochem. Cell Biol. 1997;29:91–105. doi: 10.1016/s1357-2725(96)00122-7. [DOI] [PubMed] [Google Scholar]
- 6.Cruz M.A., Handin R.I., Wise R.J. The interaction of the von Willebrand factor-A1 domain with platelet glycoprotein Ib/IX. The role of glycosylation and disulfide bonding in a monomeric recombinant A1 domain protein. J. Biol. Chem. 1993;268:21238–21245. [PubMed] [Google Scholar]
- 7.Morales L.D., Martin C., Cruz M.A. The interaction of von Willebrand factor-A1 domain with collagen: mutation G1324S (type 2M von Willebrand disease) impairs the conformational change in A1 domain induced by collagen. J. Thromb. Haemost. 2006;4:417–425. doi: 10.1111/j.1538-7836.2006.01742.x. [DOI] [PubMed] [Google Scholar]
- 8.Bonnefoy A., Romijn R.A., Hoylaerts M.F. von Willebrand factor A1 domain can adequately substitute for A3 domain in recruitment of flowing platelets to collagen. J. Thromb. Haemost. 2006;4:2151–2161. doi: 10.1111/j.1538-7836.2006.02111.x. [DOI] [PubMed] [Google Scholar]
- 9.Hoylaerts M.F., Yamamoto H., Vermylen J. von Willebrand factor binds to native collagen VI primarily via its A1 domain. Biochem. J. 1997;324:185–191. doi: 10.1042/bj3240185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalafatis M., Takahashi Y., Meyer D. Localization of a collagen-interactive domain of human von Willebrand factor between amino acid residues Gly 911 and Glu 1,365. Blood. 1987;70:1577–1583. [PubMed] [Google Scholar]
- 11.Cruz M.A., Yuan H., Handin R.I. Interaction of the von Willebrand factor (vWF) with collagen. Localization of the primary collagen-binding site by analysis of recombinant vWF a domain polypeptides. J. Biol. Chem. 1995;270:10822–10827. doi: 10.1074/jbc.270.18.10822. [DOI] [PubMed] [Google Scholar]
- 12.Lankhof H., van Hoeij M., Sixma J.J. A3 domain is essential for interaction of von Willebrand factor with collagen type III. Thromb. Haemost. 1996;75:950–958. [PubMed] [Google Scholar]
- 13.Dong J.F., Moake J.L., López J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 14.Huizinga E.G., Tsuji S., Gros P. Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science. 2002;297:1176–1179. doi: 10.1126/science.107355. [DOI] [PubMed] [Google Scholar]
- 15.Emsley J., Cruz M., Liddington R. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J. Biol. Chem. 1998;273:10396–10401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
- 16.Yago T., Lou J., Zhu C. Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 2008;118:3195–3207. doi: 10.1172/JCI35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabinowitz I., Tuley E.A., Sadler J.E. von Willebrand disease type B: a missense mutation selectively abolishes ristocetin-induced von Willebrand factor binding to platelet glycoprotein Ib. Proc. Natl. Acad. Sci. USA. 1992;89:9846–9849. doi: 10.1073/pnas.89.20.9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Auton M., Sedlák E., Cruz M.A. Changes in thermodynamic stability of von Willebrand factor differentially affect the force-dependent binding to platelet GPIbα. Biophys. J. 2009;97:618–627. doi: 10.1016/j.bpj.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alon R., Chen S., Springer T.A. The kinetics of L-selectin tethers and the mechanics of selectin-mediated rolling. J. Cell Biol. 1997;138:1169–1180. doi: 10.1083/jcb.138.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yago T., Wu J., McEver R.P. Catch bonds govern adhesion through L-selectin at threshold shear. J. Cell Biol. 2004;166:913–923. doi: 10.1083/jcb.200403144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doggett T.A., Girdhar G., Diacovo T.G. Selectin-like kinetics and biomechanics promote rapid platelet adhesion in flow: the GPIb(α)-vWF tether bond. Biophys. J. 2002;83:194–205. doi: 10.1016/S0006-3495(02)75161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marshall B.T., Long M., Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 23.Sarangapani K.K., Yago T., Zhu C. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 2004;279:2291–2298. doi: 10.1074/jbc.M310396200. [DOI] [PubMed] [Google Scholar]
- 24.Dembo M., Torney D.C., Hammer D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B Biol. Sci. 1988;234:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]
- 25.Caputo K.E., Lee D., Hammer D.A. Adhesive dynamics simulations of the shear threshold effect for leukocytes. Biophys. J. 2007;92:787–797. doi: 10.1529/biophysj.106.082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cruz M.A., Diacovo T.G., Handin R.I. Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J. Biol. Chem. 2000;275:19098–19105. doi: 10.1074/jbc.M002292200. [DOI] [PubMed] [Google Scholar]
- 27.Auton M., Cruz M.A., Moake J. Conformational stability and domain unfolding of the Von Willebrand factor A domains. J. Mol. Biol. 2007;366:986–1000. doi: 10.1016/j.jmb.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 28.Matsushita T., Sadler J.E. Identification of amino acid residues essential for von Willebrand factor binding to platelet glycoprotein Ib. Charged-to-alanine scanning mutagenesis of the A1 domain of human von Willebrand factor. J. Biol. Chem. 1995;270:13406–13414. doi: 10.1074/jbc.270.22.13406. [DOI] [PubMed] [Google Scholar]
- 29.de Romeuf C., Hilbert L., Mazurier C. Platelet activation and aggregation induced by recombinant von Willebrand factors reproducing four type 2B von Willebrand disease missense mutations. Thromb. Haemost. 1998;79:211–216. [PubMed] [Google Scholar]
- 30.Kumar R.A., Dong J.F., McIntire L.V. Kinetics of GPIbα-vWF-A1 tether bond under flow: effect of GPIbα mutations on the association and dissociation rates. Biophys. J. 2003;85:4099–4109. doi: 10.1016/S0006-3495(03)74822-X. [DOI] [PMC free article] [PubMed] [Google Scholar]