

Abstract
The ovarian hormone estrogen increases the axospinous synapse density in the hippocampal CA1 region of young female rats but fails to do so in aged rats. This estrogen-mediated alteration of spine synapse structures suggests the coincident requirement for the structural reorganization of the underlying actin cytoskeleton network. Actin reorganization is known to require the deactivation of Cofilin, an actin depolymerization factor. Cofilin is deactivated by LIM Kinase (LIMK), and LIMK activity is modulated by the phosphorylation of specific residues. We have previously demonstrated that estrogen is able to increase phosphorylated LIMK (pLIMK) immunoreactivity (IR) in the hippocampus in vivo and that this estrogen-stimulated pLIMK-IR is decreased in the aged brain. Because Cofilin phosphorylation allows for actin filament elongation and spine synapse growth, we sought to determine if estrogen acts through Cofilin and if such estrogen action requires the observed LIMK activity. Using both hippocampal neurons and the NG108-15 neuroblastoma cell line, we demonstrate here that estrogen stimulates the phosphorylation of Cofilin in vitro. Furthermore, this estrogen action on Cofilin requires LIMK. Lastly, while initiating the phosphorylation of LIMK and Cofilin, estrogen can also stimulate the formation of filopodial extensions, an early step in the formation of nascent spines, demonstrating that estrogen can alter the actin-dependent neuronal morphology. This linkage of estrogen communication to Cofilin via LIMK provides the functionality to the age-sensitive pLIMK-IR that we have observed in vivo.
INTRODUCTION
The hippocampal formation remains receptive and responsive to experience and to stimuli throughout adulthood. This responsiveness includes a sensitivity to circulating steroid hormones that results in the morphological remodeling of hippocampal neurons in vivo (McEwen, 2002). Across the estrous cycle of adult female rats, hippocampal CA1 pyramidal neurons respond to cyclical increases in the circulating estrogen levels with corresponding increases in dendritic spine density and axospinous synapse number (McEwen and Milner, 2007; Woolley and McEwen, 1992). The observation of such estrogen-induced spinogenesis in vivo has been reiterated in vitro with early video microscopy studies on cultured primary hippocampal neurons where stimulation with supra-physiological levels of estrogen induces the formation of immature spines, or neurite filopodia (Brinton, 1993). Together, these in vivo and in vitro observations of estrogen-modulated morphologic plasticity suggest that estrogen directly communicates with and affects the structural reorganization of the cytoskeletal network in hippocampal neurons (Dillon and Goda, 2005; Matus, 2000; Schubert and Dotti, 2007). However, this ability of estrogen to modulate dendritic spine plasticity is lost in the aged female brain (Adams and Morrison, 2003; Morrison et al., 2006).
In the aged female brain, estrogen fails to increase CA1 spine synapse density, presumably due to a loss of the required molecular communication from estrogen to the underlying actin cytoskeletal network (Yildirim et al., 2008). Actin filaments, the major cytoskeletal protein structures in the dendritic spine, impact synaptic plasticity by regulating the formation of synaptic structures as well as by affecting dendritic spine size and shape (Cingolani and Goda, 2008; Sekino et al., 2007). Estrogen may be involved in regulating spine synapse formation by regulating the actin cytoskeletal network. In support of this possibility, we have recently observed that estrogen can increase phosphorylated (p) LIM Kinase (LIMK) immunoreactivity (IR) in the hippocampus in vivo (Spencer et al., 2008a), and that this estrogen-stimulated pLIMK-IR in neurons is diminished in the aged brain (Yildirim et al., 2008).
LIMK is a signal transduction intermediate that is essential for spine formation and synapse function (Sarmiere and Bamburg, 2002) and plays a key role in regulating actin polymerization and depolymerization (Bernard, 2007). LIMK functions as a serine/threonine kinase, and one LIMK target substrate of interest is Cofilin (Yang et al., 1998). Cofilin, a member of the actin depolymerization factor (ADF) family, is a key actin binding protein that promotes the turnover and severing of actin filaments (Bernstein and Bamburg, 2010; Maciver and Hussey, 2002). Cofilin plays an important role in neuronal shape (Sarmiere and Bamburg, 2004), and Cofilin is localized to neuronal structures with high actin filament turnover, including the growth cone of axons and dendrites (Fass et al., 2004; Gungabissoon and Bamburg, 2003). Importantly, Cofilin is present in CA1 spines and is enriched near the membrane an in the post-synaptic density (PSD) where it can regulate actin dynamics in the context of synapse activity and plasticity (Racz and Weinberg, 2006). LIMK phosphorylates Cofilin, rendering Cofilin severing inactive and thereby promoting actin filament extension and dendritic spine formation (Arber et al., 1998; Endo et al., 2007; Meng et al., 2003).
To identify a functional role for the estrogen-stimulated pLIMK-IR that is observed in vivo, we asked whether estrogen affects the phosphorylation of the LIMK substrate Cofilin in cultured hippocampal neurons and in NG108-15 neuronal cells in vitro. We determined that estrogen can stimulate the phosphorylation of both Cofilin and LIMK in vitro, and we demonstrate here that the phospho-modulation of Cofilin by estrogen is LIMK-dependent.
RESULTS
Estrogen stimulates the phosphorylation of ADF/Cofilin
Cofilin is an actin binding protein that regulates the actin assembly into microfilaments (Bernstein and Bamburg, 2010; Maciver and Hussey, 2002). Cofilin promotes the severing of filamentous actin (F-actin) that results in an increased total number of shorter actin filaments and actin monomers (Kiuchi et al., 2007). Cofilin activity is regulated by the phosphorylation of the amino-terminal Ser3 residue whereupon Cofilin no longer binds to actin (Meng et al., 2004). Phosphorylation of Cofilin thus deactivates Cofilin and allows for actin polymerization and stabilization denoted by the accumulation of F-actin content in hippocampal neurons in vivo (Fukazawa et al., 2003). When Cofilin is phospho-deactivated in neurons, dendritic spine initiation is rapidly established as the assembly of elongating actin filaments provides a protrusive motor that results in filopodial extension and spine growth (Gungabissoon and Bamburg, 2003; Svitkina et al., 2003).
Because estrogen stimulates the formation and growth of dendritic spines in vivo and the extension of filopodia in hippocampal neurons in vitro (Brinton, 1993; Murphy and Segal, 1996; Segal and Murphy, 2001), we hypothesized that estrogen also regulates the phosphorylation of Cofilin. To measure changes to Cofilin phosphorylation in vitro, we used the NG108-15 cell line, which provides a good model for examining general cellular and molecular mechanisms that occur in neurons (Docherty et al., 1991; Hamprecht, 1977; Klee and Nirenberg, 1974). Unlike primary cultured neurons, the NG108-15 neuron is readily transfectable with high efficiency, rendering these cells suitable for recombinant manipulations. Moreover, these cells, which have been previously used to study the regulation of actin dynamics and the function of actin-binding proteins such as Cofilin (Tojima et al., 2003; Tojima and Ito, 2004), provide a homogeneous population whose uniform responsiveness allows for the in vitro study of subtle changes in signal transduction. In addition, these cells express estrogen receptors and are estrogen-responsive (Akama and McEwen, 2003; Nishio et al., 2004).
NG108-15 cells were treated with 10 nM of the estrogen 17β-Estradiol (E) at staggered time points, and whole cell lysates were examined by Western blot for phosphorylated Cofilin (pCofilin) levels and total Cofilin (tCofilin) levels. The levels of E-stimulated pCofilin increase in minutes, while the levels of tCofilin remain relatively constant following E-treatment (Fig. 1A). This E-stimulated increase in pCofilin is discernable as soon as 30 min (Fig 1C) post-treatment. Quantified E-stimulated pCofilin enhancement is significant by 4 to 6 hr post-treatment, and at 6 hr, densitometric analyses of Western blots demonstrate a 67% increase in pCofilin levels in the E-treated cells compared to the diluent control (con)-treated cells (Fig. 1C, two-tailed Student’s t test; n = 6, p < 0.05).
Figure 1. Estrogen stimulates the phosphorylation of cofilin in NG108-15 cells.
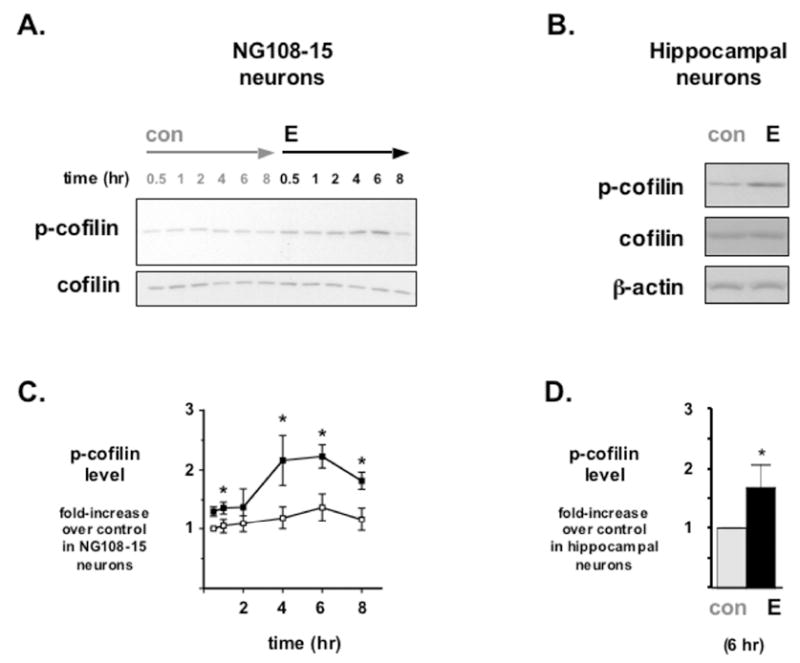
(A) Differentiated NG108-15 neurons were treated with the estrogen 17β-Estradiol (E) or with diluent control (con). Western blot analyses on whole cell lysates revealed a time-dependent increase in phosphorylated Cofilin (p-cofilin) protein levels relative to total Cofilin (cofilin) protein levels. (B) While discernable differences in E-stimulated Cofilin phosphorylation appeared as early as 30 minutes post-treatment, semi-quantitative densitometric analyses of Western blot data indicate a significant increase in p-cofilin protein level (values normalized with total cofilin and expressed as % of control) by 4-8 hr after E treatment. *Statistically significant difference between each paired time point (p<0.05), n=6 each. (C) E also stimulates the phosphorylation of Cofilin in primary cultured rat hippocampal neurons. Cultures were treated with diluent control (con) or with 17β-Estradiol (E) for 6 hr. Western blot analyses on whole cell lysates revealed an increase in p-Cofilin levels in E-treated primary cultures when compared to con-treated neurons. (D) Densitometric analyses of Western blots on hippocampal protein lysates indicated a significant increase in phosphorylated Cofilin protein level (values normalized with total Cofilin and expressed as % of control) after 6 hr exposure to E. *Statistically significant difference over control treated group (p<0.01), n=4 each.
Dissociated cultures of rat hippocampal neurons also were assayed to determine if there is any comparable pCofilin response to E. Primary cultured hippocampal neurons treated with 10 nM E also demonstrate an increase in pCofilin levels (Fig. 1B), and this E-stimulated increase in pCofilin is statistically significant (Fig. 1D, two-tailed Student’s t test; n = 4, p<0.05). Moreover, this 68% increase at 6 hr in primary hippocampal neurons is consistent with the results in NG108-15 cells.
Estrogen stimulates the phosphorylation of LIMK1 in NG108-15 neurons
The upstream kinase to Cofilin is LIMK, which can directly phosphorylate Cofilin at Ser3. LIMK is a key regulator of F-actin-based structures such as filopodia and dendritic spines, and LIMK1 is a neuron-enriched LIMK isoform (Meng et al., 2003; Sarmiere and Bamburg, 2002; Stanyon and Bernard, 1999). LIMK1 is associated with cognitive function (Frangiskakis et al., 1996), and LIMK1 knockout mice demonstrate abnormal dendritic spine morphology and spatial learning disability (Meng et al., 2002).
The activation of LIMK is controlled by phosphorylation at specific residues (Amano et al., 2001; Edwards et al., 1999; Sumi et al., 2001), and in vitro analyses here with LIMK1-positive NG108-15 neuronal cells (Tojima et al., 2003) demonstrated the ability of E to parallel in vivo findings by stimulating the phosphorylation of LIMK (Spencer et al., 2008a; Yildirim et al., 2008). The anti-pLIMK polyclonal antibody used in these experiments requires over-expressed recombinant LIMK according to the manufacturer (also, K.T. Akama unpublished observation), and so for phosphorylated LIMK (pLIMK) analyses, NG108-15 cells were first transfected with a plasmid encoding for the expression of a chimeric enhanced green fluorescent protein (eGFP)-LIMK1 protein (Yang et al., 1998; Yang and Mizuno, 1999). The exogenous expression of this recombinant eGFP-LIMK1 protein is readily distinguishable from the endogenous LIMK protein by the increased apparent molecular weight due to the covalent addition of eGFP. E treatment of transfected NG108-15 cells resulted in significantly increased phosphorylation levels of LIMK1 while the total amount of LIMK and β-actin protein levels were unaffected (Fig. 2A), suggesting a phospho-activation of LIMK1 by E. Densitometric analyses on Western blots demonstrated that E increased pLIMK levels in NG108-15 cells by approximately three-fold (Fig. 2B; two-tailed Student’s t test; n = 5, p<0.01). Additionally, chromogenically enhanced Western blot analyses of whole cell lysates collected from untransfected NG108-15 cells treated with either con or E demonstrated that E treatment also correlated with an elevated level of endogenous LIMK phosphorylation while total LIMK levels remained unchanged (see Supplemental Data).
Figure 2. Estrogen significantly enhances the phosphorylation of exogenous LIMK1.
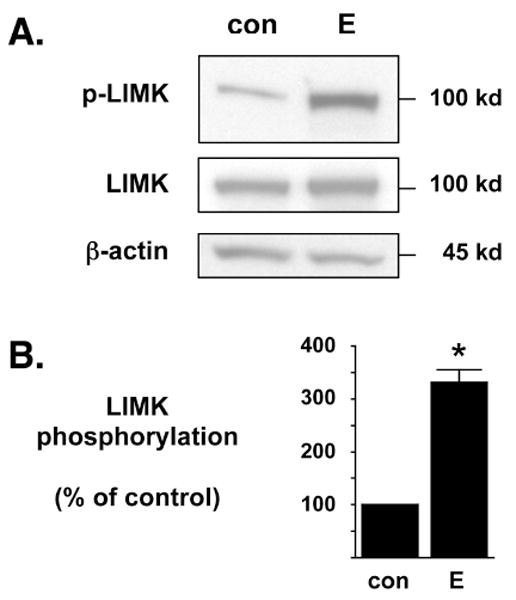
(A) NG108-15 cells were transfected to exogenously express chimeric LIMK1-eGFP and then treated with control (con) or 17β-estradiol (E) for 6 hr. The recombinant LIMK1-eGFP protein (100 kD) is readily discernable from endogenous LIMK protein (72 kD) due to the chimerically attached eGFP marker. In Western blot analyses, the anti-phospho-LIMK antibody is more sensitive to exogenously expressed LIMK protein than to endogenous LIMK protein. Western blot analyses demonstrated that E significantly enhances the phosphorylation of exogenously expressed LIMK1-eGFP (p-LIMK) but does not alter the overall total expression of LIMK1-eGFP (LIMK). As an additional control, the whole cell lysates were also blotted against β-actin protein levels to demonstrate no change in overall endogenous protein levels. (B) Densitometric analyses of immunoblots in (A) demonstrate that estrogen significantly increases the phosphorylation of exogenous LIMK1 when compared to diluent control. The level of phosphorylated LIMK1-eGFP was normalized to the level of total LIMK1-eGFP and densitometry is represented as % of control. *Statistically significant difference between groups (p<0.01), n=5 each.
Estrogen-stimulated Cofilin phosphorylation is LIMK1-dependent
Transfection of NG108-15 cells was subsequently performed with a mutated plasmid encoding for the expression of a recombinant eGFP-dominant negative (dn) LIMK1 chimeric protein (D460A), that is still capable of being phosphorylated but is kinase inactive (Yang et al., 1998; Yang and Mizuno, 1999), to determine the role of LIMK1 in the E-mediated phospho-deactivation of Cofilin. Functionally, this dnLIMK1 chimeric protein blocks any endogenous LIMK1 signaling. As a transfection negative control, NG108-15 cells were transfected instead with eGFP alone. Both eGFP- and eGFP-dnLIMK1-transfected cells were then treated with either diluent control or with E. As demonstrated by Western blot analyses on whole cell lysates, neither construct affected the total protein levels of Cofilin or β-actin, but the over-expression of eGFP-dnLIMK1 dramatically reduced pCofilin levels regardless of treatment (Fig. 3). The reduction of pCofilin levels by eGFP-dnLIMK1 demonstrates that LIMK1 activity is required for Cofilin phosphorylation in NG108-15 cells, and also suggests that the loss of E-mediated Cofilin phosphorylation is not compensated for by another kinase or LIMK isoform.
Figure 3. LIMK-1 is required for estrogen-mediated phosphorylation of cofilin.
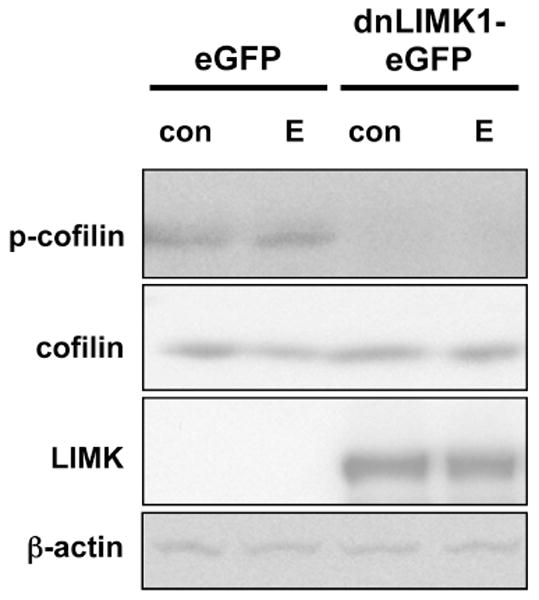
NG108-15 cells were transfected to express either chimeric dominant-negative (dn)LIMK1-eGFP (100 kD) or a corresponding control vector for eGFP expression only. This over-expressed dnLIMK1 protein contains the D460A mutation which renders the protein kinase inactive. Transfected cells were treated with diluent control (con) or 10 nM 17β-estradiol (E). Phosphorylated Cofilin (p-cofilin) and total Cofilin (cofilin) protein levels in whole cell lysates were assayed by Western blot analyses. Dominant-negative LIMK1 interferes with E-stimulated Cofilin phosphorylation but does not interfere with overall Cofilin expression. Trasfection of eGFP alone does not interfere with E-stimulated Cofilin phosphorylation. Lysates were blotted for LIMK expression, and the level of the higher molecular weight exogenous dnLIMK1-eGFP protein (LIMK) is shown. As an additional control, the whole cell lysates were also blotted against β-actin protein levels to demonstrate no change in overall endogenous protein levels across samples. Blots shown are representative of four independent experiments.
Estrogen concomitantly stimulates the formation of filopodia in NG108-15 cells
A morphologic response to estrogen treatment was assessed in NG108-15 cells to determine if E-stimulated Cofilin phosphorylation is also associated with any changes to filopodial outgrowth.
NG108-15 cells were transfected to express eGFP and then treated with either diluent control or E. Fluorescence microscopy on transfected cells revealed the clear presence of several branched dendrite-like structures extended from NG108-15 cell bodies, as well as the presence of numerous thin, elongated projections from these structures (Fig. 4A). The NG108-15 cells were morphologically estrogen-sensitive in the elaboration of filopodia. Fluorescent microscopy imaging of eGFP-expressing NG108-15 cells 6 hr post-treatment revealed an increased number of E-stimulated filopodial projections (Fig. 4B).
Figure 4. NG108-15 elaboration of filopodial projections is estrogen-sensitive.
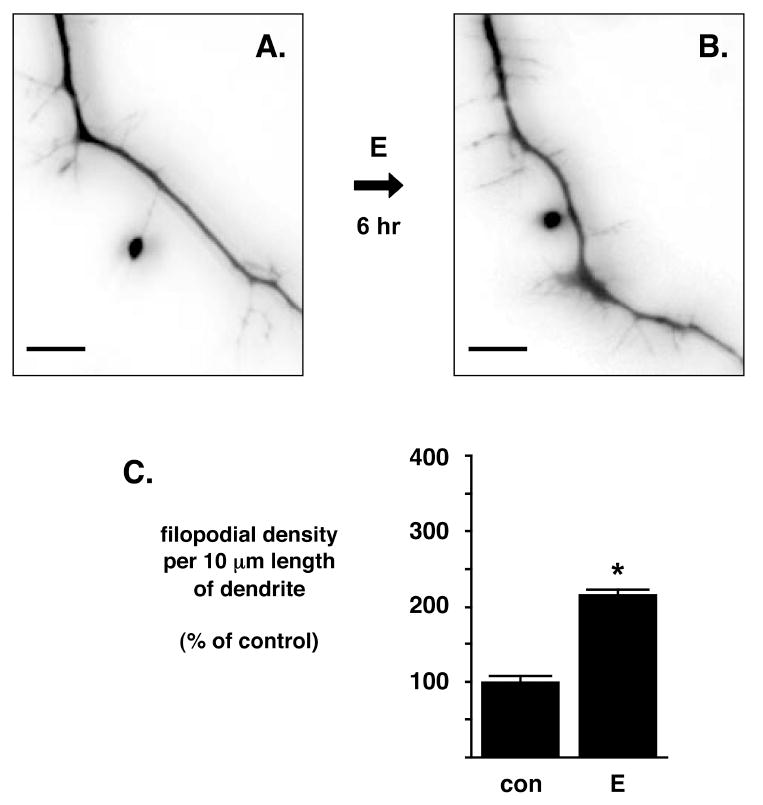
(A and B) Differentiated NG108-15 cells were transfected to express eGFP for inverted fluorescence microscopy imaging. NG108-15 cells show an increased number of filopodial projections 6 hr after the administration of 10 nM 17β-estradiol (E). Scale bar is 5 μm. Imaging of eGFP expression was digitally captured and then white-black inverted in order to facilitate printing and viewing of filopodial extensions. (C) E administration significantly increases the filopodia density in NG108-15 cells. Differentiated NG108-15 cells were treated with E or diluent control (con) for 6 hr and analyzed for number of filopodia/10 μm of dendritic length. Data represent mean ± SEM. *Statistically significant difference between E- and con-treated dendrites (p<0.01), control dendrites, n=80; E dendrites, n=76.
Cells treated with diluent control did not typically exhibit an increase of filopodial projections, and quantification of filopodial projections from cells treated with diluent control or E demonstrated that E increased the number of filopodial projections by approximately two-fold compared to control (Fig. 4C; two-tailed Student’s t test, p<0.01; con group: 1.39±0.11, n = 80; E group: 3.00±0.10, n = 76). This increase in E-stimulated filopodial density (number per 10 μm length of dendrite) is consistent with prior published in vitro results of a two-fold change in primary rat hippocampal neurons (Segal and Murphy, 2001). These data demonstrates that E not only increases the phosphorylation of Cofilin, but also increases the density of filopodial projections in neuronal cells.
In summary, these data indicate that estrogen can stimulate the formation of potential new spines, or filopodia, and at the same time support the actin dynamics required for nascent filopodia via the activation of LIMK1 and the phosphorylated deactivation of Cofilin (Fig 5).
Figure 5. Model of LIMK-dependent phosphorylation of Cofilin.
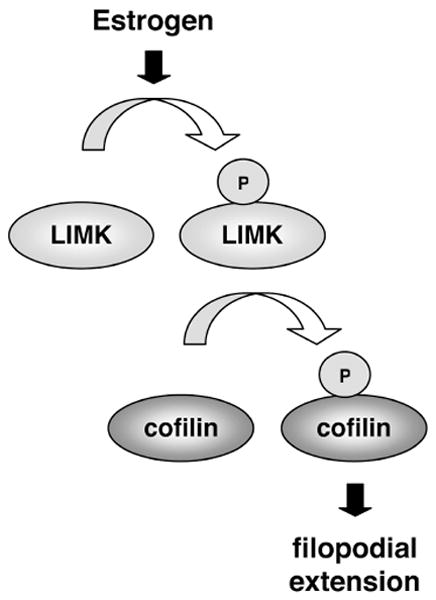
Estrogen stimulates LIMK-dependent phosphorylation of Cofilin. This phosphorylation promotes filopodial extensions along neuronal processes, serving as an initial step in dendritic spine formation. The loss or the reduction of estrogen-stimulated LIMK activity, observed in the aged hippocampus as the reduction of phoshorylated LIMK immunoreactivity, would result in the failure of estrogen to promote new spine formation in the aged brain.
DISCUSSION
The work presented here contributes several important points to our understanding of estrogen effects on spine synapse plasticity. First, these data demonstrate that estrogen controls the phospho-modulation of ADF/Cofilin via LIMK1, thus supporting that estrogen can act on the actin filament network to remodel neuron morphology. Second, the polymerization of actin filaments associated with Cofilin phosphorylation may explain how estrogen concomitantly drives the actin-based projections of filopodia observed in this study. Third, the LIMK-dependent action on Cofilin phosphorylation provides a functional link to the increase in estrogen-stimulated pLIMK-IR observed in the female rat and mouse hippocampus in vivo (Spencer et al., 2008a; Yildirim et al., 2008).
Estrogen modulates Cofilin
The phosphorylation of Cofilin is mediated by LIMK1, which was also phosphorylated and activated following the exposure to estrogen, both in vivo and in vitro. Transduction of the estrogen stimulus to these signaling intermediates may possibly be conveyed by non-genomic signaling mechanisms mediated by non-nuclear and dendritic spine-localized estrogen receptors, such as GPR30, ERα, or ERβ (McEwen et al., 2001; Prossnitz et al., 2008; Raz et al., 2008; Spencer et al., 2008b). A recent report demonstrated that in adult hippocampal slice tissue, estrogen-stimulated Cofilin phosphorylation is blocked pharmacologically with an inhibitor to RhoA kinase (Kramar et al., 2009). Since RhoA kinase feeds into the LIMK pathway, it becomes clear that understanding how estrogen is coupled to LIMK will be critical to understanding why estrogen fails to increase the axospinous synaptic density in the aged brain (Adams and Morrison, 2003; Yildirim et al., 2008).
Cofilin and LIMK are ultimate and penultimate regulators of actin polymerization, respectively (Bamburg, 1999). Both of these molecules have garnered considerable attention for their role in proper dendritic spine formation and synapse function. Neuronal Cofilin plays a key role in synapse-associated learning and memory function by modulating the actin-based dendritic spine architecture. Deficits in either Cofilin or the Cofilin deactivator LIMK1 result in gross morphological abnormalities of spines, neurotransmission, and overall cognitive function (Bamburg and Wiggan, 2002; Meng et al., 2003; Scott and Olson, 2007). Aberrant Cofilin activity is associated with actin rod pathology seen in Alzheimer’s disease, Pick’s disease, and alcoholism thought to cause neurite degeneration and synapse loss (Bamburg and Wiggan, 2002; Bamburg et al., 2010; Maciver and Harrington, 1995; Minamide et al., 2000). LIMK1-/- knockout mice demonstrate abnormal dendritic spine morphology and spatial learning disability (Meng et al., 2002). In addition, LIMK1 deletion is thought to underlie the visuospatial cognitive defects of Williams syndrome (Donnai and Karmiloff-Smith, 2000; Frangiskakis et al., 1996; Morris and Mervis, 2000). Thus, the loss of proper Cofilin regulation results in spine loss or in pathologies associated with aging. Our data point to Cofilin as the critical dysregulated molecular substrate as estrogen-stimulated LIMK activity is diminished in the aged brain (Yildirim et al., 2008).
Estrogen-mediated signal transduction and aging
Estrogen enhances the phosphorylation of Cofilin in NG108-15 cells observable as early as 30 minutes after hormone exposure, which is similar to what is observed in acute hippocampal slice tissue (Kramar et al., 2009). Such a time frame would suggest that estrogen acts on Cofilin at least in part through a non-genomic pathway, independent of a slower, gene expression-dependent process. This effect is sustained through 4 to 6 hours, a point coincident in time with the visible increase in filopodia formation. While other downstream events would determine whether a filopodium is to become a mature dendritic spine, it is noteworthy that through the phosphorylation of Cofilin, estrogen augments the opportunity of neurons to form more eventual spines. Potentially, a brief, peri-menopausal estrogen treatment could therefore provide the brain the necessary opportunity to build or to maintain new synapses (Bohacek and Daniel, 2010; Daniel and Bohacek, 2010; Gibbs, 2010; Sherwin, 2009). Conversely, the diminished LIMK sensitivity to estrogen in the aged brain (Yildirim et al., 2008) would therefore result in the inability of estrogen to stimulate new spine formation in the aged brain.
The effect of estrogen on actin polymerization and assembly is directly relevant to subsequent assembly of synaptic proteins (Kim and Huganir, 1999). Studies on synaptic plasticity point to an obligatory physical interaction between actin and other scaffolding proteins in the dendritic spine in order to position synaptic receptors and to coordinate signal transduction intermediates at the PSD (Allison et al., 1998; Kreienkamp, 2008; Shirao and Sekino, 2001). It is possible then that the loss of estrogen regulation of actin assembly may contribute to the loss of other estrogen-stimulated signal transduction pathways in the aged brain, such as the Akt pathway (Yildirim et al, this issue), since Akt kinase can physically interact with and co-localize with the actin cytoskeleton (Cenni et al., 2003). Prolonged deprivation of estrogen may result in a loss of assembled signal transduction complexes (Lin and Koleske, 2010), rendering the dendritic spine insensitive to future estrogen administration in the aged brain. In order to maintain estrogen sensitivity as the brain ages, it is perhaps possible that estrogen therapy must be administered at the start of ovarian senescence and before a hormone-maintained assembly of actin localized signaling components is lost.
The results presented above support the delineation of estrogen signaling directly to the actin cytoskeleton, and thus this study affords a new way of thinking about the induction of spines by estrogen. Estrogen may not only activate signaling pathways ultimately to regulate and to increase the production of post-synaptic proteins associated with dendritic spine maturation (Akama and McEwen, 2003; Lee et al., 2004), but additionally, estrogen may activate Cofilin-targeting signaling pathways to regulate the mechanical aspects of actin dynamics for nascent spine formation.
Current research on estrogen effects on the aging brain suggests that a discrete “Window of Opportunity” for proper estrogen replacement therapy around the peri-menopause stage is lost beyond the post-menopausal stage. Estrogen signaling that would involve LIMK and Cofilin may play a significant role in maintaining proper synaptic shape and function and should be considered when designing estrogen replacement therapies.
EXPERIMENTAL PROCEDURES
Materials
Rabbit polyclonal antibodies raised against phospho-cofilin (p-cofilin), total cofilin (t-cofilin), phospho-LIMK1/2 (p-LIMK), total LIMK, and β-actin were all purchased from Cell Signaling Technology (Beverly, MA). As stated by the manufacturer, this anti-pLIMK polyclonal antibody requires exogenous, recombinant protein for phospho-detection. The eGFP-chimeric wild-type (wt) LIMK-1 and eGFP-chimeric dnLIMK-1 plasmid constructs were kindly provided by Kensaku Mizuno, PhD (Tohoku University, Sendai, Japan) (Yang et al., 1998; Yang and Mizuno, 1999).
Animals
All methods have been approved by The Rockefeller University Institutional Animal Care and Use Committee and conform to National Institutes of Health guidelines. Animals were housed with food and water available ad libitum under a 12 hr light-dark cycle in a temperature-controlled room. For primary hippocampal neuron preparations, dissections were performed as similarly described (Brinton, 1993; Murphy and Andrews, 2000). Briefly, primary hippocampal cell cultures were derived from brains of E18 rat embryos that were harvested and dissected in ice cold Hank’s Balanced Saline Solution (HBSS, Invitrogen, Carlsbad, CA). All hippocampi were dissected into and triturated in HBSS. Neurons were plated in phenol red-free Neurobasal (Nb) media supplemented with B-27, 2 mM L-glutamine (Invitrogen) and fresh 25 μM glutamate (Sigma, St. Louis, MO). Cells were subsequently maintained in B27-supplemented phenol red-free Nb media without glutamate for 21 days in vitro before the performance of any experimental treatment.
Cell culture and DNA transfection
The NG108-15 neuroblastoma cell line was purchased from American Type Cell Culture (Manassas, VA; cell line number HB-12317). NG108-15 cells were propagated in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% PenStrep (100 U/ml penicillin G sodium and 100 μg/ml streptomycin sulfate; Invitrogen). To differentiate NG108-15 neurons, cells were plated in wells that had been coated with poly-L-lysine (Sigma). Two days after plating, cells were washed with Dulbecco’s PBS (Invitrogen), and media was replaced with phenol red-free DMEM supplemented with 0.5% charcoal-stripped FBS (HyClone, Logan, UT), 1% PenStrep, and 1 mM dibutyryl cAMP (Sigma). NG108-15 cells were differentiated for 10 days prior to all experimental treatments. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. NG108-15 neurons were analyzed 48 hr post-transfection. For estrogen treatments, ethanol-soluble 17β-Estradiol (Sigma) was prepared by dissolving in 100% EtOH first at a 10 mM stock concentration and then diluting this stock 1000 fold further to an intermediate concentration of 10 μM in phenol red-free DMEM. Final treatment concentrations were 10 nM 17β-Estradiol. Control treatments (con) entailed a comparable dilution of the ethanol diluent in phenol red-free DMEM (0.0001% EtOH).
Imaging
NG108-15 cells that had been eGFP-transfected were viewed by inverted fluorescent microscopy using a Nikon Eclipse TE200 station and images were captured with a CoolSnap digital camera. Black and white images were captured via IP Labs software (Scanalytics, Fairfax, VA) and then white-black inverted via Photoshop software (Adobe, San Jose, CA) for ease in filopodial visualization and printing.
Western blot analysis
Experimental samples were harvested in 2x Laemmli sample lysis buffer that had protease and phosphatase inhibitor cocktails (Sigma) added freshly. For time-point assays, cells were treated in staggered fashion and then harvested at the same time. Lysates were sonicated and boiled for 5 minutes before separation by SDS-PAGE. Protein transfer was performed onto polyvinylidene difluoride membrane (Millipore, Bedford, MA). Membranes were blocked and then incubated in primary antibody overnight at 4°C. HRP-conjugated goat anti-rabbit or goat anti-mouse IgG secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used at a dilution of 1:5000 for 1 hr at room temperature. Visualization was achieved by enhanced chemiluminescence (Perkin Elmer, Boston, MA). Semi-quantitative densitometry was performed on scanned Western blot films using Image Quant 5.2 software (Molecular Dynamics, Sunnyvale, CA). Phosphorylated protein levels were normalized to total (phosphorylated and unphosphorylated) protein levels when used in quantification.
Statistical analyses
All numerical data are presented as average ± SEM. Statistical significance was calculated using Student’s two-tailed t-test (paired two-sample for mean values) to determine if groups are distinct. P-values of p < 0.05 was used to determine statistically significant difference between treatment groups.
Supplementary Material
Acknowledgments
This study was supported in part by the NIH NS 07080, P01 AG 16765 (BSMc and KTA). Additional support for this work comes from the NIH MSTP grant GM07739, The Surdna Foundation Medical Scientist Fellowship, and the Barbara and Stephen Friedman Fellowship Endowment (GSY).
Abbreviations
- ADF
actin depolymerization factor
- eGFP
enhanced green fluorescent protein
- IR
immunoreactivity
- LIMK
LIM kinase
- dn
dominant negative
- p
phosphorylated
- PSD
post-synaptic density
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MM, Morrison JH. Estrogen and the aging hippocampal synapse. Cereb Cortex. 2003;13:1271–5. doi: 10.1093/cercor/bhg078. [DOI] [PubMed] [Google Scholar]
- Akama KT, McEwen BS. Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci. 2003;23:2333–9. doi: 10.1523/JNEUROSCI.23-06-02333.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–36. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano T, Tanabe K, Eto T, Narumiya S, Mizuno K. LIM-kinase 2 induces formation of stress fibres, focal adhesions and membrane blebs, dependent on its activation by Rho-associated kinase-catalysed phosphorylation at threonine-505. Biochem J. 2001;354:149–59. doi: 10.1042/0264-6021:3540149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Wiggan OP. ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 2002;12:598–605. doi: 10.1016/s0962-8924(02)02404-2. [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bernstein BW, Davis RC, Flynn KC, Goldsbury C, Jensen JR, Maloney MT, Marsden IT, Minamide LS, Pak CW, Shaw AE, Whiteman I, Wiggan O. ADF/Cofilin-actin rods in neurodegenerative diseases. Curr Alzheimer Res. 2010;7:241–50. doi: 10.2174/156720510791050902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–6. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–95. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohacek J, Daniel JM. The beneficial effects of estradiol on attentional processes are dependent on timing of treatment initiation following ovariectomy in middle-aged rats. Psychoneuroendocrinology. 2010;35:694–705. doi: 10.1016/j.psyneuen.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Brinton RD. 17[beta]-Estradiol Induction of Filopodial Growth in Cultured Hippocampal Neurons within Minutes of Exposure. Mol Cell Neurosci. 1993;4:36–46. doi: 10.1006/mcne.1993.1005. [DOI] [PubMed] [Google Scholar]
- Cenni V, Sirri A, Riccio M, Lattanzi G, Santi S, de Pol A, Maraldi NM, Marmiroli S. Targeting of the Akt/PKB kinase to the actin skeleton. Cell Mol Life Sci. 2003;60:2710–20. doi: 10.1007/s00018-003-3349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–56. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Daniel JM, Bohacek J. The critical period hypothesis of estrogen effects on cognition: Insights from basic research. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbagen.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Robbins J, Brown DA. NG108-15 neuroblastoma x glioma hybrid cell line as a model neuronal system. In: Chad J, Wheal HV, editors. Cellular Neurobiology: a practical approach Vol. Oxford University Press; New York: 1991. pp. 74–95. [Google Scholar]
- Donnai D, Karmiloff-Smith A. Williams syndrome: from genotype through to the cognitive phenotype. Am J Med Genet. 2000;97:164–71. doi: 10.1002/1096-8628(200022)97:2<164::aid-ajmg8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nature Cell Biol. 1999;1:253–9. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Mizuno K. LIM kinase and slingshot are critical for neurite extension. J Biol Chem. 2007;282:13692–702. doi: 10.1074/jbc.M610873200. [DOI] [PubMed] [Google Scholar]
- Fass J, Gehler S, Sarmiere P, Letourneau P, Bamburg JR. Regulating filopodial dynamics through actin-depolymerizing factor/cofilin. Anat Sci Int. 2004;79:173–83. doi: 10.1111/j.1447-073x.2004.00087.x. [DOI] [PubMed] [Google Scholar]
- Frangiskakis JM, Ewart AK, Morris CA, Mervis CB, Bertrand J, Robinson BF, Klein BP, Ensing GJ, Everett LA, Green ED, Proschel C, Gutowski NJ, Noble M, Atkinson DL, Odelberg SJ, Keating MT. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell. 1996;86:59–69. doi: 10.1016/s0092-8674(00)80077-x. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–60. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gibbs RB. Does short-term estrogen therapy produce lasting benefits in brain? Endocrinology. 2010;151:843–5. doi: 10.1210/en.2009-1453. [DOI] [PubMed] [Google Scholar]
- Gungabissoon RA, Bamburg JR. Regulation of growth cone actin dynamics by ADF/cofilin. J Histochem Cytochem. 2003;51:411–20. doi: 10.1177/002215540305100402. [DOI] [PubMed] [Google Scholar]
- Hamprecht B. Structural, electrophysiological, biochemical, and pharmacological properties of neuroblastoma-glioma cell hybrids in cell culture. Int Rev Cytol. 1977;49:99–170. doi: 10.1016/s0074-7696(08)61948-8. [DOI] [PubMed] [Google Scholar]
- Kim JH, Huganir RL. Organization and regulation of proteins at synapses. Curr Opin Cell Biol. 1999;11:248–54. doi: 10.1016/s0955-0674(99)80033-7. [DOI] [PubMed] [Google Scholar]
- Kiuchi T, Ohashi K, Kurita S, Mizuno K. Cofilin promotes stimulus-induced lamellipodium formation by generating an abundant supply of actin monomers. J Cell Biol. 2007;177:465–76. doi: 10.1083/jcb.200610005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee WA, Nirenberg M. A neuroblastoma × glioma hybrid cell line with morphine receptors. Proc Nat’l Acad Sci USA. 1974;71:3474–7. doi: 10.1073/pnas.71.9.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, Lynch G. Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J Neurosci. 2009;29:12982–93. doi: 10.1523/JNEUROSCI.3059-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp HJ. Scaffolding proteins at the postsynaptic density: shank as the architectural framework. Handb Exp Pharmacol. 2008:365–80. doi: 10.1007/978-3-540-72843-6_15. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Romeo RD, Svenningsson P, Campomanes CR, Allen PB, Greengard P, McEwen BS. Estradiol affects spinophilin protein differently in gonadectomized males and females. Neuroscience. 2004;127:983–8. doi: 10.1016/j.neuroscience.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Lin YC, Koleske AJ. Mechanisms of Synapse and Dendrite Maintenance and their Disruption in Psychiatric and Neurodegenerative Disorders. Annu Rev Neurosci. 2010 doi: 10.1146/annurev-neuro-060909-153204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK, Harrington CR. Two actin binding proteins, actin depolymerizing factor and cofilin, are associated with Hirano bodies. Neuroreport. 1995;6:1985–8. doi: 10.1097/00001756-199510010-00008. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Hussey PJ. The ADF/cofilin family: actin-remodeling proteins. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-5-reviews3007. Reviews3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–8. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Akama KT, Alves SE, Brake WG, Bulloch K, Lee S, Li C, Yuen GS, Milner TA. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Nat’l Acad Sci USA. 2001;98:7093–100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Estrogen actions throughout the brain. Recent Prog Horm Res. 2002;57:357–84. doi: 10.1210/rp.57.1.357. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Milner TA. Hippocampal formation: Shedding light on the influence of sex and stress on the brain. Brain Res Rev. 2007 doi: 10.1016/j.brainresrev.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–33. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev Neurosci. 2003;14:233–40. doi: 10.1515/revneuro.2003.14.3.233. [DOI] [PubMed] [Google Scholar]
- Meng Y, Takahashi H, Meng J, Zhang Y, Lu G, Asrar S, Nakamura T, Jia Z. Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacol. 2004;47:746–54. doi: 10.1016/j.neuropharm.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nature Cell Biol. 2000;2:628–36. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- Morris CA, Mervis CB. Williams syndrome and related disorders. Annu Rev Genomics Hum Genet. 2000;1:461–84. doi: 10.1146/annurev.genom.1.1.461. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Brinton RD, Schmidt PJ, Gore AC. Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J Neurosci. 2006;26:10332–48. doi: 10.1523/JNEUROSCI.3369-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–68. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Andrews SB. Culture models for the study of estradiol-induced synaptic plasticity. J Neurocytol. 2000;29:411–7. doi: 10.1023/a:1007121525399. [DOI] [PubMed] [Google Scholar]
- Nishio M, Kuroki Y, Watanabe Y. Subcellular localization of estrogen receptor beta in mouse hippocampus. Neurosci Lett. 2004;355:109–12. doi: 10.1016/j.neulet.2003.10.064. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- Racz B, Weinberg RJ. Spatial organization of cofilin in dendritic spines. Neurosci. 2006;138:447–56. doi: 10.1016/j.neuroscience.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW. Rapid estrogen signaling in the brain. Neurosignals. 2008;16:140–53. doi: 10.1159/000111559. [DOI] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR. Head, neck, and spines: a role for LIMK-1 in the hippocampus. Neuron. 2002;35:3–5. doi: 10.1016/s0896-6273(02)00759-6. [DOI] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR. Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol. 2004;58:103–17. doi: 10.1002/neu.10267. [DOI] [PubMed] [Google Scholar]
- Schubert V, Dotti CG. Transmitting on actin: synaptic control of dendritic architecture. J Cell Sci. 2007;120:205–12. doi: 10.1242/jcs.03337. [DOI] [PubMed] [Google Scholar]
- Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med. 2007;85:555–68. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- Segal M, Murphy DD. Estradiol induces formation of dendritic spines in hippocampal neurons: functional correlates. Horm Behav. 2001;40:156–9. doi: 10.1006/hbeh.2001.1688. [DOI] [PubMed] [Google Scholar]
- Sekino Y, Kojima N, Shirao T. Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem Int. 2007;51:92–104. doi: 10.1016/j.neuint.2007.04.029. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Estrogen therapy: is time of initiation critical for neuroprotection? Nat Rev Endocrinol. 2009;5:620–7. doi: 10.1038/nrendo.2009.193. [DOI] [PubMed] [Google Scholar]
- Shirao T, Sekino Y. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci Res. 2001;40:1–7. doi: 10.1016/s0168-0102(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Milner TA, McEwen BS. Estrous cycle regulates activation of hippocampal Akt, LIM kinase, and neurotrophin receptors in C57BL/6 mice. Neuroscience. 2008a;155:1106–19. doi: 10.1016/j.neuroscience.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008b;29:219–37. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanyon CA, Bernard O. LIM-kinase1. Int J Biochem Cell Biol. 1999;31:389–94. doi: 10.1016/s1357-2725(98)00116-2. [DOI] [PubMed] [Google Scholar]
- Sumi T, Matsumoto K, Nakamura T. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J Biol Chem. 2001;276:670–6. doi: 10.1074/jbc.M007074200. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 2003;160:409–21. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima T, Takahashi M, Ito E. Dual regulation of LIM kinase 1 expression by cyclic AMP and calcium determines cofilin phosphorylation states during neuritogenesis in NG108-15 cells. Brain Res. 2003;985:43–55. doi: 10.1016/s0006-8993(03)03113-5. [DOI] [PubMed] [Google Scholar]
- Tojima T, Ito E. Signal transduction cascades underlying de novo protein synthesis required for neuronal morphogenesis in differentiating neurons. Prog Neurobiol. 2004;72:183–93. doi: 10.1016/j.pneurobio.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–54. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- Yang N, Mizuno K. Nuclear export of LIM-kinase 1, mediated by two leucine-rich nuclear-export signals within the PDZ domain. Biochem J. 1999;338(Pt 3):793–8. [PMC free article] [PubMed] [Google Scholar]
- Yildirim M, Janssen WG, Tabori NE, Adams MM, Yuen GS, Akama KT, McEwen BS, Milner TA, Morrison JH. Estrogen and aging affect synaptic distribution of phosphorylated LIM kinase (pLIMK) in CA1 region of female rat hippocampus. Neuroscience. 2008;152:360–70. doi: 10.1016/j.neuroscience.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.