

Abstract
A simple steady-state kinetic high-throughput assay was developed for the salicylate synthase MbtI from Mycobacterium tuberculosis, which catalyzes the first committed step of mycobactin biosynthesis. The mycobactins are small-molecule iron chelators produced by M. tuberculosis, and their biosynthesis has been identified as a promising target for the development of new antitubercular agents. The assay was miniaturized to a 384-well plate format and high-throughput screening was performed at the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Diseases (NSRB). Three classes of compounds were identified comprising the benzisothiazolones (class I), diarylsulfones (class II), and benzimidazole-2-thiones (class III). Each of these compound series was further pursued to investigate their biochemical mechanism and structure–activity relationships. Benzimidazole-2-thione 4 emerged as the most promising inhibitor owing to its potent reversible inhibition.
Keywords: high-throughput screening, mycobactins, salicylate synthase, siderophores, tuberculosis
Introduction
Tuberculosis (TB) is the leading cause of bacterial infectious disease mortality, and the World Health Organization (WHO) estimates that at least one third of the world’s population is infected with a latent form of Mycobacterium tuberculosis (Mtb), the etiological agent of TB.[1] Based on the emergence of multidrug-resistant tuberculosis (MDR-TB), the high incidence of cases of HIV/TB co-infection, and the lack of new antitubercular drugs, the WHO has declared TB a global health emergency. New TB drugs should be effective against MDR-TB strains and should ideally decrease the treatment duration by targeting latent non-replicating mycobacteria through a new mechanism of action.[2] New TB drugs must also be compatible with antiretroviral agents used for HIV treatment and current second-line antitubercular agents, especially the fluoroquinolones.
M. tuberculosis, like most pathogens, requires iron for its survival, as this metal is a required cofactor for more than 40 proteins involved in numerous biological redox processes including respiration, DNA biosynthesis, and amino acid biosynthesis.[ 3] However, unlike most pathogens, which use multiple redundant pathways for iron acquisition, M. tuberculosis relies on a single iron acquisition system employing mycobactins, which are small-molecule iron chelators also known as siderophores. Targeted genetic inactivation of mbtB, a gene involved in mycobactin synthesis, resulted in a mutant strain incapable of growth under iron-deficient conditions and attenuated for growth in macrophages, serving to validate mycobactin biosynthesis as a potential pathway for the development of a novel class of antitubercular agents.[4]
The biosynthesis of mycobactins begins from salicylic acid and is catalyzed by a non-ribosomal peptide synthetase–polyketide synthase assembly line of one dozen proteins.[5] The salicylic acid starter unit is in turn prepared from chorismate by a magnesium-dependent bifunctional salicylate synthase known as MbtI, which catalyzes the two-step conversion of chorismate to salicylic acid via isochorismate.[6] The conversion of chorismate to isochorismate likely occurs via a concerted SN2″ reaction mechanism, while pyruvate elimination in the second half-reaction is believed to proceed through an intramolecular [3+3]-sigmatropic rearrangement, formerly a retro-Ene reaction, resulting in aromatization to afford salicylic acid (Figure 1).[7] MbtI is an ideal target in the mycobactin biosynthetic pathway because this enzyme catalyzes the first committed biosynthetic step of the mycobactins, has no human orthologues, appears to be essential based on whole-genome saturated transposon mutagenesis studies in M. tuberculosis as well as targeted disruption in M. smegmatis, and has been structurally and functionally characterized.[6d, 8] Abell and co-workers reported the synthesis of analogues of chorismate and isochorismate as inhibitors of chorismate-using enzymes including salicylate synthase, isochorismate synthase, and anthranilate synthase.[ 9] Transition-state inhibitors of the isochorismate synthase EntC from E. coli were synthesized over a decade ago that could represent potential MbtI inhibitors.[10] In another study, a peptide-based inhibitor of chorismate-using enzymes was prepared that exhibited noncompetitive inhibition against the isochorismate synthase domain of EntB from E. coli, with a Ki value of 56 μM.[11] Recently, Payne and co-workers reported the first inhibitors of MbtI including both substrate and intermediate mimics, with the most potent compound possessing a Ki value of 11 μM.[12] Herein we report a complementary approach using high-throughput screening (HTS) to identify novel MbtI inhibitors and also disclose limited structure–activity relationships for the most promising scaffolds.
Figure 1.
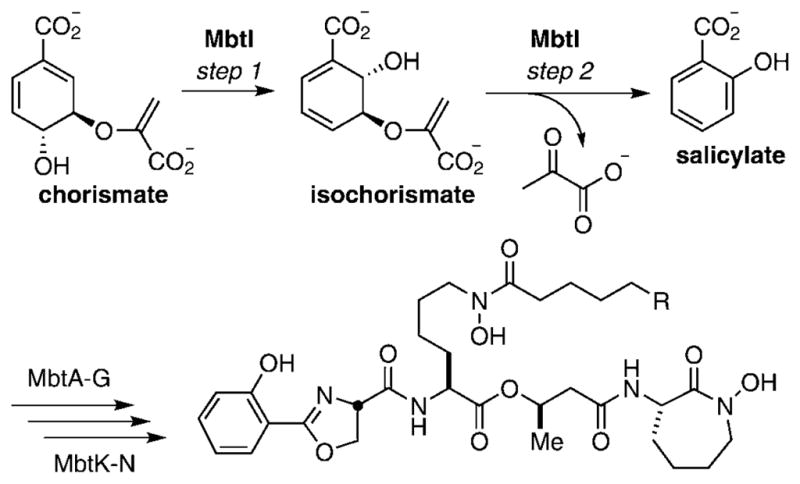
MbtI-catalyzed conversion of chorismate to salicylate.
Results and Discussion
High-throughput screening
To identify small-molecule inhibitors of MbtI, we developed a steady-state kinetic high-throughput assay that exploits the intrinsic fluorescence of salicylic acid based on the assay reported by Lott and co-workers.[6d, 12] The assay measures the formation of salicylic acid from chorismate, catalyzed by MbtI in the presence of the essential divalent Mg2+ cofactor. Fluorescence interference was expected to be a major source of false positives, as the fluorescence excitation and emission wavelengths are respectively in the UV and violet spectral regions; however, this concern was mitigated by the ability to directly detect the reaction product salicylic acid. The relatively low catalytic activity of MbtI (kcat~3 min−1) precluded the use of a coupled assay involving indirect detection of pyruvate via consumption of NADH employing lactate dehydrogenase. To perform HTS, the assay was miniaturized to a 30 μL 384-well plate format, and the protein and chorismate concentrations were decreased to reduce reagent consumption while maintaining initial velocity conditions. The impact of incubation temperature, time, and DMSO concentration were individually evaluated. Given the lack of any reported MbtI inhibitors at the time of the assay, a positive control for the HTS that mimicked full inhibition of the enzyme activity was obtained by excluding MgCl2 from the assay mixture. The final optimized conditions involved 100 mM Tris-HCl pH 8.0, 65 μM chorismate (KM=6.0 μM), 415 nM MbtI, Igepal CA-630 at 0.0025% (w/v), 0.5% (v/v) DMSO and an incubation time of 3 h at 24 °C with excitation and emission wavelengths of 305 and 420 nm, respectively. The nonionic detergent Igepal CA-630 was included to minimize nonspecific inhibition caused by aggregation.[13] Under the optimal conditions the Z′ score (see Equation (1), Experimental Section), which is a measure of assay robustness, was calculated at 0.85. Z′ scores in the range of 0.5–1.0 are considered excellent, thus the assay was deemed suitable for HTS.
HTS was performed at the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Diseases (NSRB) at Harvard Medical School. A total of 104 802 compounds were screened in duplicate. The normalized percent inhibition (NPI, see Equation (2), Experimental Section), calculated for each experimental well plate-by-plate based on the respective positive and negative controls, was chosen as a scoring function to eliminate plate-to-plate variability. Library compounds that exhibited NPI values greater than 50% in both assay replicates and that did not exhibit significant fluorescence background (less than 130% of the fluorescence background of the control wells) were initially selected for further analysis, which resulted in a total of 931 hits representing a 0.89% hit rate. Based on chemical tractability for subsequent medicinal chemistry efforts and presence of potentially reactive functionality, 293 compounds (~0.3% of the library) were manually selected for further analysis, and 1 μL DMSO stock solutions of the compounds were provided by the NSRB. All compounds were rescreened at a single concentration of 30 μgmL−1 (~100 μM for a nominal Mr of 300 Da) using a continuous assay format as detailed in the Experimental Section in order to re-confirm inhibition and rank order compounds. From this analysis a total of 84 compounds were identified that exhibited greater than 50% enzyme inhibition, and 17 of the most promising inhibitors, based on enzyme inhibition and chemical tractability, were purchased from their respective suppliers. All compounds were analyzed by 1H NMR spectroscopy and HRMS to verify structural assignment, and 16 of the compounds were positively identified (see Table 1), while one compound could not be authenticated and appeared to be a fragment of the reported structure. Fresh DMSO stock solutions from dry powders of the 16 confirmed compounds were prepared, and each compound was screened in duplicate using a steady-state continuous assay measuring fluorescence intensity and by an HPLC end-point assay using a threefold serial dilution from 0.5 to 400 μM as described in the Experimental Section. The IC50 values and Hill slopes were measured for each compound from the resulting concentration–response plots, and in general, both assays were in excellent agreement except for a few of the weakest inhibitors such as 13 and 16 (Table 1). Notably, the benzisothiazolone compounds 1 and 2 exhibited extremely potent activity, with IC50 values approaching the enzyme concentration, suggesting tight binding or irreversible inhibition as further evidenced by the high Hill slopes. The next four most active compounds possessed moderate potency, with IC50 values in the range of 2–12 μM. The potency of the remaining hits 6–16 rapidly declined from 28 μM for 7 to 109 μM for 16. Based on the observed potencies and ease of preparing analogues, we selected three chemical series for further chemical and biochemical evaluation: class I compounds are characterized by the benzisothiazolone core of 1 and 2, class II compounds contain the diarylsulfone core found in 8, and class III compounds possess the benzimidazole-2-thione core as found in 4 and 16.
Table 1.
Structures of confirmed hits against MbtI.
| Compd | Structure | Fluorescence Assay[a] | HPLC Assay[b] | Compd | Structure | Fluorescence Assay[a] | HPLC Assay[b] | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 [μM] | Hill | IC50 [μM] | Hill | IC50 [μM] | Hill | IC50 [μM] | Hill | ||||
| 1 | 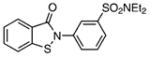 |
0.86±0.04 | 2.28 | 0.96±0.04 | 2.19 | 9 | 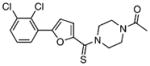 |
51.09±1.87 | 1.47 | 80.81±3.39 | 1.36 |
| 2 | 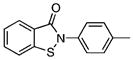 |
1.59±0.11 | 2.12 | n.d.[c] | n.d. | 10 | 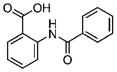 |
52.68±2.92 | 1.37 | 81.72±4.87 | 1.51 |
| 3 | 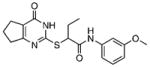 |
2.19±0.04 | 1.33 | 2.91±0.31 | 1.45 | 11 | 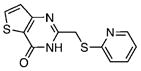 |
54.19±2.03 | 1.59 | 80.81±4.16 | 1.79 |
| 4 | 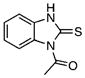 |
5.89±0.27 | 1.71 | 5.16±0.49 | 1.23 | 12 | 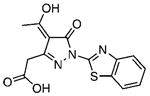 |
56.11±4.14 | 1.60 | 123.6±6.14 | 1.36 |
| 5 | 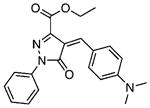 |
6.80±0.62 | 1.17 | 9.51±0.52 | 1.54 | 13 | 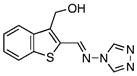 |
69.80±6.90 | 1.38 | 304.8±26.0 | 1.24 |
| 6 | 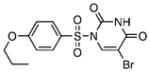 |
11.50±0.51 | 1.41 | 11.72±0.57 | 1.43 | 14 | 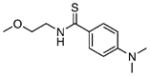 |
79.61±8.91 | 1.40 | 159.6±18.8 | 1.16 |
| 7 | 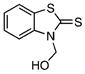 |
27.88±1.59 | 1.59 | 42.25±2.78 | 1.00 | 15 | 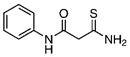 |
102.8±9.6 | 1.04 | 129.3±7.0 | 1.19 |
| 8 | 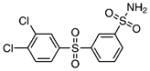 |
35.13±5.55 | 0.85 | 49.17±11.63 | 0.7 | 16 | 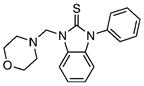 |
109.3±6.8 | 1.11 | 484.6±17.2 | 1.55 |
IC50 values and the corresponding Hill slopes were determined by the continuous fluorescence assay as described in the Experimental Section.
IC50 values and the corresponding Hill slopes were determined by the end-point HPLC assay as described in the Experimental Section.
Not determined. All assays were performed under initial velocity conditions at 37 °C and pH 8.0 with 1.0 μM MbtI, 100 μM chorismate, 0.5 mM MgCl2, in 100 mM Tris-HCl with 1% DMSO and 0.0025% Igepal CA-630.
Class I: benzisothiazolones
The potent activity of benzisothiazolones 1 and 2 was initially very encouraging, and there is an abundance of reports of the antiviral, antitumor, and antioxidant properties of this class of molecules.[14] However, the activity is likely due to the electrophilic isothiazolone moiety that can react with thiol nucleophiles to form mixed disulfide linkages.[15] The MbtI crystal structure reveals four surface-exposed cysteine residues (residues 50, 211, 380, and 436).[6d] Incubation of 2 with MbtI resulted in rapid enzyme inactivation (<30 s), which precluded assessment of the kinact/Ki ratio that defines irreversible inhibitor efficiency. To confirm the irreversible nature of inhibition, MbtI was incubated with 2, and the protein was subsequently analyzed by ESIMS. The deconvoluted masses of MbtI treated with 2 corresponded precisely to the expected mass of the quadruply labeled protein (51751 Da; see figure S1, Supporting Information). Because none of the four cysteines directly interact in the active site, we hypothesized that modification of the cysteine residues must introduce a conformational change in the enzyme, thereby inhibiting enzyme function. To investigate this further, we conducted circular dichroism (CD) experiments to study the change in the secondary structure of MbtI induced by the binding of benzisothiazolone 2. Indeed, compound 2 induced a significant change in the α-helical content of MbtI, from 20.3% in the native protein to 6.8% in the modified form (see figure S2, Supporting Information). Although none of the cysteines in MbtI are involved in the catalytic action of the enzyme, Cys211 and Cys380 pack against Arg417 and Arg382, respectively, which in turn make contact with Tyr385. This tyrosine residue interacts directly with the substrate in the active site, and any perturbation with the Cys211 and Cys380 could indirectly affect the binding of substrate to the active site. While the activity of 2 is mechanistically interesting, we believe that benzisothiazolones are pan-assay-interference compounds (PAINS) as defined by Baell and Holloway, due to the nonspecific labeling of protein cysteines and the findings that the benzisothiazolone substructure is found as a hit in a large percentage of high-throughput assays deposited in the PubChem database (http://pubchem.ncbi.nlm.nih.gov/).[16]
Class II: diarylsulfones
Next, we investigated the chemical synthesis of a small series of class II diarylsulfones, as the parent hit 8 bears a close structural similarity to the known anti-mycobacterial agent, dapsone (4,4′-diaminodiphenylsulfone (33); see Table 2). Analogues of 8 were synthesized to understand the significance of the two chlorine atoms and to study the tolerance of the sulfonamide moiety to N substitution. Friedel–Crafts sulfonylation of appropriately substituted aromatic compounds 17–19 using phenylsulfonyl chloride provided diarylsulfones 20–22 (Scheme 1).[17] Regioselective chlorosulfonylation of 20–22 furnished 23–25 in moderate yields ranging from 50 to 55% over two steps. Subsequent reaction of 23–25 with various amines afforded the final desired sulfonamides 3 and 26–31. Acetylation of 3 with acetic anhydride provided 32.
Table 2.
SAR of class II diarylsulfones.
| Compd | Structure | IC50 [μM] | Hill Slope |
|---|---|---|---|
| 8 | 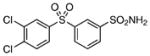 |
35 | 0.85 |
| 26 | 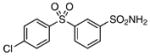 |
>200 | – |
| 27 | 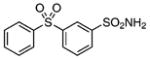 |
>200 | – |
| 28 | 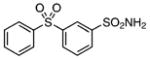 |
>200 | – |
| 29 | 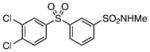 |
>200 | – |
| 30 | 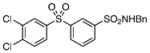 |
>200 | – |
| 31 | 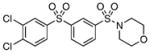 |
>200 | – |
| 32 | 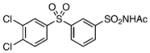 |
>200 | – |
| 33 |  |
118±19 | 0.81 |
Scheme 1.
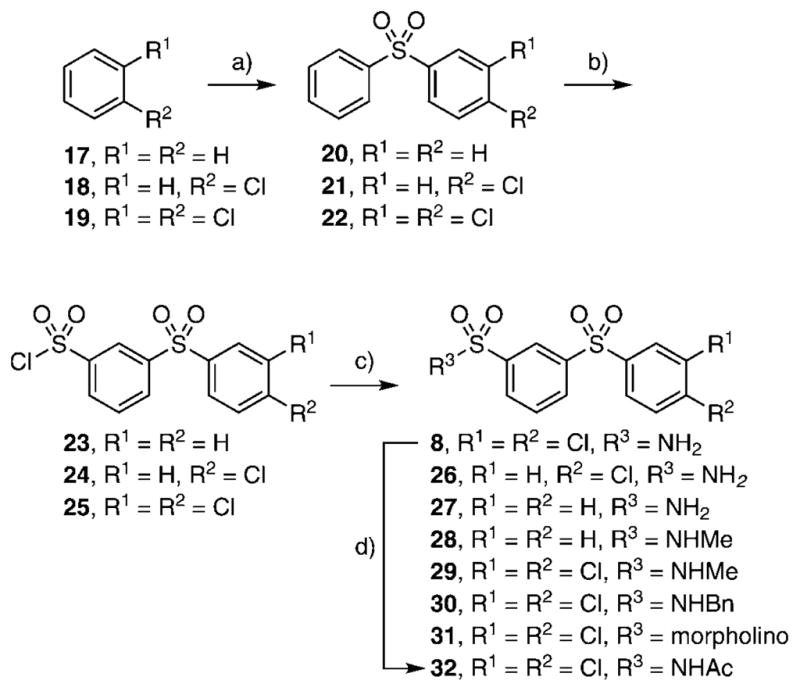
Reagents and conditions: a) PhSO2Cl, AlCl3, RT, 16 h; b) ClSO3H, 50°C, 6 h, 50–55 %; c) RNH2, CH2Cl2, DIPEA/NH3-CH3OH, RT, 16 h, 60–65%; d) Ac2O, ZnCl2, RT, 50°C, 30 min, 87%.
Class II compounds were analyzed for their activity, and it was found that deletion of either of the meta- and para-chloro substituents in 26–27 resulted in complete loss of activity. Similarly, N-alkyl sulfonamide derivatives 28–31 and N-acetyl derivative 32 were completely inactive, demonstrating a remarkably tight structure–activity relationship at this position. Finally, we evaluated the anti-mycobacterial agent dapsone (33), which showed modest MbtI inhibition, with an IC50 value of 118 μM.
Class III: benzimidazole-2-thiones
The synthesis of the class III benzimidazole-2-thiones was performed because this series had two members in the top-16 compounds from the HTS campaign. Additionally, the simple heterocyclic scaffold has a low molecular weight and hence a fairly high ligand efficiency (binding energy per heavy atom). A systematic series of analogues were prepared to examine the importance of the imidazole and benzene rings in the parent compound 4. Compounds 5, 41–45, and 47 were synthesized by acetylation of benzimidazole-2-thione 34, benzimidazoline 35, 5-methylbenzimidazole-2-thione 36, benzoxazole-2-thione 37, 2-oxindole 38, benzimidazol-2-one 39, and imidazole-2-thione 48, respectively, and in yields ranging from 93 to 96% (Scheme 2).[18] Benzimidazoline 35 was prepared by nickel boride reduction of 34,[19] while the analogues N-methylbenzimidazole-2-thione 40 and 2-thiomethylbenzimidazole 46 were commercially available.
Scheme 2.
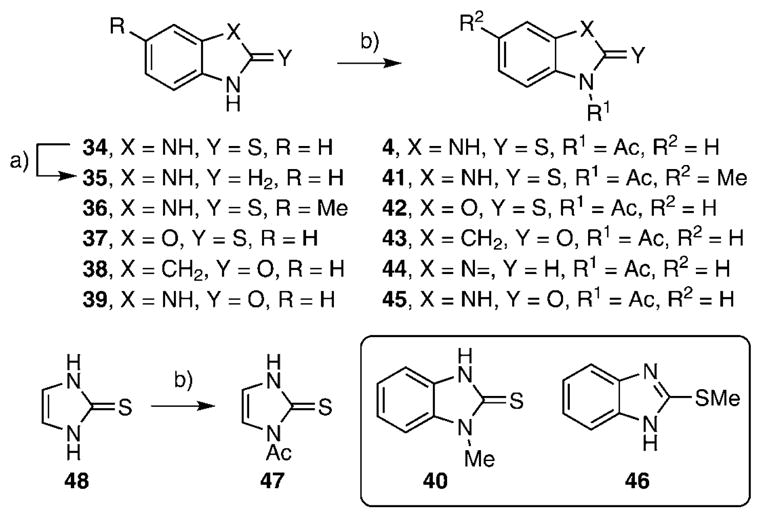
Reagents and conditions: a) NiCl2, NaBH4, CH2Cl2, 0°C, 60 min, 83%; b) Ac2O, 93–96% for substrates 34–38 and 1. Ac2O, 2. BnNH2, RT, 60 min, CH2Cl2, 96% for substrate 39.
Class III benzimidazole-2-thiones were next evaluated for enzyme inhibition. The synthetically prepared compound 4 provided an IC50 value of 9.2 μM, which agrees closely with the value of 5.9 μM obtained for the purchased compound (Table 3). Deletion of the N-acetyl group or replacement with an N-methyl group in analogues 34 and 40 led to six- to nine-fold decreases in potency, demonstrating the importance of the acetyl substituent at N1. Substitution of the aryl ring was explored in a single example with 5-methylbenzimidazole-2-thione 41, and no impact on potency was observed. Benzoxazole 42 was prepared to examine the importance of the NH group of benzimidazole-2-thione 5, and this compound was about twofold less potent, showing that the H-bond donor capacity of the NH group in 4 is dispensable for activity. While the aforementioned small set of analogues defined positions amenable to modification, we also observed that the 2-thione group in 4 is essential for activity, as 2-oxindole 43, benzimidazoline 44, benzimidazol-2-one 45, and 2-methylthiobenzimidazole 46 were completely inactive. Finally, the benzene ring in benzimidazole-2-thione 4 was shown to be absolutely essential, as deletion of this group in imidazole-2-thione 47 abolished all activity. Further characterization of 4 was performed to determine its mode of inhibition by measuring the reaction velocity at various inhibitor concentrations at different fixed concentrations of the substrate chorismate (see figure S3, Supporting Information). Compound 4 exhibited noncompetitive inhibition (α=1) with respect to chorismate, providing a Ki value of 9.7 μM, which agrees closely with the IC50 value, as expected for a noncompetitive inhibitor with an α value equal to unity. Overall, this small series of analogues suggests further opportunities for improving potency and defines the minimal structural features required for biochemical activity.
Table 3.
SAR of class III benzimidazole-2-thiones.
| Compd | Structure | IC50 [μM] | Hill Slope |
|---|---|---|---|
| 4 | 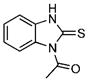 |
9.2±0.9 | 1.00 |
| 34 | 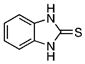 |
58.8±3.6 | 0.90 |
| 40 | 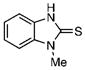 |
83.9±4.7 | 1.20 |
| 41 | 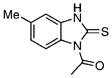 |
7.6±0.4 | 1.20 |
| 42 | 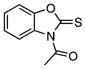 |
21.8±1.4 | 1.10 |
| 43 | 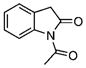 |
>200 | – |
| 44 | 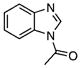 |
>200 | – |
| 45 | 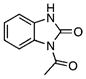 |
>200 | – |
| 46 | 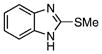 |
>200 | – |
| 47 | 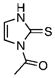 |
>200 | – |
Conclusions
High-throughput screening for inhibitors of MbtI using the reported direct continuous assay that measures salicylate fluorescence was successful, and led to the identification of several novel MbtI inhibitors with modest low-micromolar IC50 values. Three of the most promising hits were pursued to investigate the biochemical mechanism of inhibition and to establish structure–activity relationships. The benzisothiazolones or class I compounds were shown to be irreversible inhibitors that covalently label all four cysteine residues in MbtI, as determined by ESIMS. Based on the affinity labeling of cysteine residues along with findings that this compound class is found as a hit in a large number of assays, the benzisothiazolones were deemed unsuitable for further study, and we nominate this substructure as a pan-assay-interference compound (PAINS) according to the nomenclature defined by Baell and Holloway.[16] The diarylsulfone or class II compounds were shown to possess extremely tight SAR, as all modifications to the parent compound 8 abolished biochemical activity. Interestingly, the structurally related drug dapsone, used to treat leprosy, was found to have very modest activity, with an IC50 value of 118 μM. Based on the remarkably tight SAR, this chemical series also does not merit further investigation. The class III benzimidazole-2-thiones were observed to be well-behaved reversible noncompetitive inhibitors, and the SAR pinpoints opportunities to further augment potency and defines the essential features of this new inhibitor scaffold.
Experimental Section
High-throughput screening
HTS of libraries of commercially available compounds was performed against MbtI at the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Diseases (NSRB), Harvard Medical School. Library descriptions can be found at http://iccb.med.harvard.edu/screening/compound_libraries/index.htm. The assay was performed with 384-well plates (Corning catalogue number 3575) in a final volume of 30 μL, in duplicate. The master mix (25 μL) containing all components of the assay excluding MgCl2 (100 mM Tris-HCl pH 8.0, 0.0025% (w/v) Igepal CA-630, 415 nM MbtI, 65 μM chorismate) was prepared freshly before the assay and was dispensed into 384-well plates using a Matrix Welmate liquid dispenser. Compound libraries (5 mgmL−1 DMSO stock solutions) were transferred to the assay plates using a 100 nL pin array transfer robot. Fluorescence intensity was measured in a PerkinElmer Envision plate reader, to account for possible autofluorescence of test compounds, using a Photometric λex 330 nm filter (band pass 80 nm) and a Photometric λem 420 nm filter (band pass 8 nm), and a LANCE/DELFIA dichroic mirror for a 400 nm cutoff. The plates were shaken for 30 s, and 5 μL of a 6mM MgCl2 solution in 100 mM Tris-HCl pH 8.0, 0.0025% (w/v) Igepal CA-630 were dispensed to all wells in columns 1–23 of the 384-well plates, and 5 μL of 100 mM Tris-HCl pH 8.0, 0.0025% (w/v) Igepal CA-630 were dispensed to column 24. This resulted in final assay concentrations of 345 nM MbtI, 54.2 μM chorismate, and library compound concentrations of 25 μgmL−1. Each plate had one column of negative controls (no test compounds) and one column for positive controls (no test compounds and no MgCl2). The plates were incubated for 3 h at room temperature, and the reaction was quenched by adding 5 μL of 0.5M EDTA (pH 8.0) to all wells of the plate, using a Matrix Welmate liquid dispenser. Fluorescence intensity was then measured in a PerkinElmer Envision plate reader. A total of 104802 compounds were screened in duplicate over seven days: Biomol ICCB known bioactives 2, NINDS custom collection 2, Prestwick 1 collection, Asinex 1 (plates 1671–1705), ChemBridge 3, ChemDiv 4, ChemDiv 3, Enamine 2, Life Chemicals 1 (plates 1649–1659), Maybridge 5 (plates 1661–1669), Maybridge 4, ChemDiv 2 (plates 1369–1379), and Enamine 1 (plates 1394–1410). Library descriptions can be found at http://iccb.med.harvard.edu/screening/compound_libraries/index.htm.
HTS data analysis
The fluorescence intensity measured before starting the enzymatic reaction was subtracted from the final fluorescence reading, after EDTA quenching, for each well of all plates. Outliers within the positive and negative controls, i.e., wells that exhibited ≥ 10% deviation for the respective fluorescence average in each plate, were discarded. Normalized percentage inhibition (NPI) was then calculated for each well, plate by plate, based on the respective controls. A hit list was extracted by selecting compounds that exhibited NPI >50% in both assay replicates and the intrinsic fluorescence intensities of which (measured before the enzymatic reaction) did not differ by >30% of that of the controls. Compounds that showed NPI >80% and moderate intrinsic fluorescence (between 130 and 250% of the controls FI) were also considered as hits at this stage.
| (1) |
| (2) |
Secondary screening
Selected hit compounds were purchased from Enamine (Kiev, Ukraine), ChemDiv (San Diego, CA, USA) and ChemBridge (San Diego, CA, USA). Reactions were performed under initial velocity conditions in a total volume of 50 μL at 37°C for 20 min, and the production of salicylic acid was monitored continuously by following changes in fluorescence at λem 420 nm with λex 305 nm on a Molecular Devices M5e multimode plate reader. Assays were set up in duplicate and contained 1.0 μM MbtI in reaction buffer (100 mM Tris-HCl pH 8.0, 0.5 mM MgCl2, 100 μM chorismate, and 0.0025% Igepal CA-630). A threefold serial dilution of inhibitors was added to black 384-well plates coated with a nonbinding surface (Greiner) to give a final DMSO concentration of 1%. Additionally, a positive control (DMSO only) and negative control (10 mM EDTA) were also included. MbtI was pre-incubated with inhibitors for 10 min before the reactions were initiated by the addition of MgCl2. For the HPLC end-point assay, the reactions (50 μL) were quenched at the end of 20 min by the addition of 10 mM EDTA and CH3OH (50 μL), then centrifuged at 13000×g for 2 min. Calibration curves were prepared in triplicate by the addition of salicylic acid to blank cell lysate to afford final concentrations of 0.125, 0.25, 0.5, 1.0, and 2.0ngmL−1. Standard curve R2 values were 0.999, and the percent coefficient of variance (CV) at all concentrations was <10%. HPLC was performed with an Agilent 1100 instrument (Agilent Technologies, Santa Clara, CA, USA), a Phenomenex Luna C18(2) 50 mm × 3.0 mm, 5 μm reversed-phase column (Phenomenex, Torrance, CA, USA), and a Jasco FP-920 fluorescence detector (Jasco Inc., Easton, MD, USA) with excitation and emission wavelengths set at 300 and 400 nm, respectively. The mobile phase consisted of an ammonium acetate buffer pH 4.6/CH3OH (90:10, v/v) at a flow rate of 1.0 mLmin−1. Under these conditions, salicylic acid eluted at 2.16 min. The IC50 values and Hill slopes were calculated using both continuous fluorescence and HPLC data from the Hill equation [Eq. (3)]. In this equation, the fractional activity (vi/v0) versus inhibitor concentration was fit by nonlinear regression analysis using GraphPad prism version 4.0; vi is the reaction velocity at a given [I], v0 is the reaction velocity of the DMSO control, and h is the Hill slope.
| (3) |
Cloning, expression, and purification of MbtI
Primers CC CATATG TCC GAG CTC AGC GTC G and GG CTCGAG TTA CTG GCG TGC AAC CAG ATA AG were used to amplify mbtI from H37Rv genomic DNA and cloned into the PCR capture vector pCR2.1 TOPO (Invitrogen) using standard procedures. After the gene was sequenced and found to be free of PCR errors, the plasmid was digested at the restriction sites NdeI and XhoI (underlined in the primers above) and cloned into the expression vector pET15b (Novagen) to create the MbtI expression vector pCDD138. Small-scale expression of pCDD138 transformed into the E. coli expression strain BL21(DE3) yielded 2.2 mgL−1 MbtI. Large-scale fermentation was performed with a 75 L bioreactor at the Biotechnology Resource Center at the University of Minnesota (http://www.bti.umn.edu/brc/index.html). Several colonies were used to inoculate 50 mL LB containing 50 μgmL−1 carbenicillin. The seed was grown for 14.5 h at 30°C with rotary agitation at 200 rpm. The primary seed (10 mL) was used to inoculate a secondary culture in 1 L LB with 50 μgmL−1 carbenicillin. The secondary seed was grown for 2.5 h at 37 °C and 200 rpm. Upon reaching an OD600 of 0.69, the secondary seed was added to 55 L production media (10 gL−1 tryptone, 5 gL−1 yeast extract, 5 gL−1 NaCl, 0.1 gL−1 Mazu 204, 50 μgmL−1 carbenicillin) in a 75 L NBS bioreactor. The cells were grown at 37°C, with an airflow of 25 Lmin−1, a pressure of 4.0 psi, and agitation at 300 rpm. The dissolved oxygen content and pH were monitored, but not controlled. At 2.5 h post-inoculation, the OD600 reached 0.67, and the reactor was cooled to 30°C. Upon reaching 30 °C, IPTG was added to a final concentration of 0.4 mM, and the cultures were allowed to grow for an additional 4 h. The reactor was then cooled to 24°C, and the cells were harvested using a Sharples T1-P semi-continuous flow centrifuge yielding a wet weight of 162 g. The pellet was frozen in liquid N2 and resuspended in 244 mL cracking buffer (11.9 gL−1 HEPES, 17.5 gL−1 NaCl, 0.681 gL−1 imidazole, pH 8.0) at 4°C. The resulting slurry was divided into 30–35 mL ali- quots and passed twice through a French press at 15000 psi. The lysate was cleared at 10000×g for 20 min to yield 184 mL supernatant. To isolate MbtI, 10 mL 50% Ni-NTA was added to the cleared lysate and incubated at 4 °C for 1 h. The resin was loaded into a column and washed with 50 mL wash buffer (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, pH 8.0) and eluted with 13 mL elution buffer (50 mM HEPES, 300 mM NaCl, 250 mM imidazole, pH 8.0). The binding, wash, and elution steps were repeated, and the purified protein was desalted into storage buffer (20 mM HEPES pH 8.0, 1% glycerol) using PD-10 columns (GE Healthcare) to yield 200 mg protein that was greater than 95% pure by SDS-PAGE.
Mass spectrometric analysis of MbtI
Protein solutions for ESIMS measurement were prepared at a concentration of 10 μM MbtI in 100 mM sodium phosphate buffer, pH 7.4. Inhibitor-treated MbtI was prepared by incubating 10 μM MbtI with 100 μM benzisothiazolone 2 in 100 mM sodium phosphate buffer, pH 7.4 containing 2% DMSO in a final volume of 50 μL for 20 min. The solution was adjusted to pH 2 with 0.1% aqueous TFA. To desalt the protein for subsequent MS analysis, the protein solution (10 μL) was applied to a C4 ZipTip (Millipore) and eluted with 75:25 CH3CN/H2O (10× 5 μL). The sample was diluted tenfold in 1:1 CH3CN/H2O containing 0.1% formic acid, and 10 μL of this desalted sample was directly injected into a QSTAR pulsar ESIMS instrument.
Determination of secondary structure content of MbtI
CD spectroscopy was performed on native MbtI and MbtI pre-incubated with benzisothiazolone 2 in order to determine the effect of the drug on protein secondary structure. CD spectra were recorded on a JASCO J-810 spectropolarimeter with quartz cuvettes (1 mm path length) at 25°C. CD data were collected between 165 and 260 nm with a scanning speed of 50 nmmin−1 at concentration of 12 μM MbtI in 20 mM phosphate buffer, pH 7.4. Deconvolution of spectra was performed using the algorithms SELCON, CONTIN, and CDSSTR, available at http://dichroweb.cryst.bbk.ac.uk/html/home.shtml. The α helix and β sheet content were calculated at 20.3 and 27.3%, respectively, whereas treatment of MbtI with benzisothiazolone 2 resulted in respective values of 6.8 and 32.5%.
General chemistry procedures
All commercial reagents (Sigma–Aldrich, Acros, Alfa Aesar) were used as provided. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying CH2Cl2. Flash chromatography was performed with an ISCO Combiflash Companion® purification system with prepacked 4 g silica gel cartridges with the indicated solvent system. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual CH3OH (3.31 ppm) or CHCl3 (7.27 ppm), and carbon chemical shifts are reported using an internal standard of residual CH3OH (49.1 ppm) or CHCl3 (77.0 ppm). All reactions were monitored by TLC on silica gel 60 F254 (Merck), and detection was carried out under UV light λ 254 nm. Proton chemical data are reported as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, ovlp=overlapping, br=broad), coupling constant, and integration. FTIR spectra were obtained on a Nicolet Protégé 460 ESP spectrometer. High-resolution mass spectra were acquired on an Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. Melting points were measured on an electrothermal Mel-Temp manual melting point apparatus and are uncorrected.
General procedure for the preparation of compounds 8, 26–31
The appropriate aromatic compound 17–19 (6.0 mmol) and AlCl3 (1.2 mmol) were added to phenylsulfonyl chloride (1.0 mmol). The reaction was stirred at room temperature for 16 h then poured into ice–H2O. The resulting mixture was extracted with EtOAc (10 mL). The organic solution was washed with a solution of 1N NaOH (7 mL) and saturated aqueous NaCl (7 mL). The organic layer was dried (MgSO4) and concentrated to provide the crude sulfones 20–22 in 92–93% yield.
ClSO3H (10 mmol) was added to the crude sulfone (1 mmol) prepared above, and the reaction was heated at 50°C for 6 h, then cooled to room temperature. The reaction mixture was poured slowly onto ice (50 g), resulting in the formation of a white solid. The solid was extracted with EtOAc (2×15 mL), and the combined organic extracts were washed with saturated aqueous NaCl (10 mL), dried (MgSO4), then concentrated to afford the desired product 23–25 in 50–55% yield.
Either 7N methanolic NH3 (3 mL) or CH2Cl2 (5.0 mL) was added to the crude phenylsulfonyl chloride (1.0 mmol) prepared above followed by an appropriate amine (2.0 mmol) and Et3N (2.0 mmol). The reaction mixture was stirred at room temperature for 16 h, then concentrated and purified by silica gel chromatography to afford the desired sulfonamides 8, 26–31 (60–65% yield).
3-(3,4-Dichlorophenylsulfonyl)benzenesulfonamide (8)
The title compound was prepared by following the general procedure from 25 (0.38 g) to afford a white amorphous solid (0.23 g, 63 %): Rf= 0.20 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CD3OD + [D6]DMSO): δ=8.45 (s, 1H), 8.21 (d, J=8.4 Hz, 1 H), 8.19–8.16 (m, 2 H), 7.92 (dd, J=8.4, 2.4 Hz, 1H), 7.79 (ovlp t, J=8.4 Hz, 1H), 7.77 (ovlp d, J=9.0 Hz, 1H); 13C NMR (150 MHz, CD3OD + [D6]DMSO): δ=155.9, 145.9, 142.0, 141.2, 133.8, 132.0, 131.2, 131.0, 130.9, 129.6, 127.5, 125.2; HRMS (ESI−) calcd for C12H8Cl2NO4S2: [M−H]− 363.9277, found: 363.9281 (error 1.1 ppm).
3-(4-Chlorophenylsulfonyl)benzenesulfonamide (26)
The title compound was prepared by following the general procedure from 24 (0.35 g) to afford a white amorphous solid (0.19 g, 60 %): Rf= 0.2 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CDCl3): δ=8.43 (s, 1 H), 8.16 (d, J=8.4 Hz, 1H), 8.14 (d, J=8.4 Hz, 1 H), 7.97 (d, J= 9.0 Hz, 2H), 7.76 (t, J=8.4 Hz, 1 H), 7.62 (d, J=9.0 Hz, 2H); 13C NMR (150 MHz, CD3OD): δ=145.5, 142.4, 140.2, 139.5, 130.63, 130.57, 130.44, 129.67, 129.27, 124.9; HRMS (ESI−) calcd for C12H9ClNO4S2: [M−H]− 329.9667, found: 329.9662 (error 1.5 ppm).
3-(Phenylsulfonyl)benzenesulfonamide (27)
The title compound was prepared by following the general procedure from 23 (0.31 g) to afford a white amorphous solid (0.20 g, 65%): Rf=0.30 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CD3OD): δ=8.45 (s, 1H), 8.13 (t, J=8.7 Hz, 2H), 7.97 (d, J=7.8 Hz, 2 H), 7.73 (td, J=7.8, 2.4 Hz, 1 H), 7.64–7.62 (m, 1 H), 7.59–7.55 (m, 2H); 13C (150 MHz, CD3OD): δ= 145.7, 143.1, 141.0, 133.9, 130.8, 130.7, 130.6, 129.6, 127.7, 125.0; HRMS (ESI−) calcd for C12H10NO4S2: [M−H]− 296.0057, found: 296.0066 (error 3.0 ppm).
N-Methyl-3-(phenylsulfonyl)benzenesulfonamide (28)
The title compound was prepared by following the general procedure from 23 (0.31 g) to afford a white amorphous solid (0.20 g, 62 %): Rf= 0.35 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CD3OD): δ=8.34 (s, 1 H), 8.19 (d, J=7.8 Hz, 1H), 8.06 (d, J=7.8 Hz, 1 H), 8.00 (d, J= 7.2 Hz, 2H), 7.78 (t, J=7.8 Hz, 1 H), 7.67 (t, J=7.2 Hz, 1 H), 7.60 (t, J=7.2 Hz, 2 H), 2.50 (s, 3H); 13C (150 MHz, CD3OD): δ=143.3, 141.5, 140.9, 134.0, 131.6, 131.2, 130.7, 129.7, 127.8, 125.8, 28.0; HRMS (ESI−) calcd for C13H12NO4S2: [M−H]− 310.0213, found: 310.0207 (error 1.9 ppm).
3-(3,4-Dichlorophenylsulfonyl)-N-methylbenzenesulfonamide (29)
The title compound was prepared by following the general procedure from 25 (0.38 g) to afford a white amorphous solid (0.23 g 62%): Rf=0.40 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CDCl3): δ=8.32 (s, 1 H), 8.03 (d, J=7.8 Hz, 1 H), 8.00 (d, J=7.8 Hz, 1 H), 7.96 (d, J=2.4 Hz, 1H), 7.71 (dd, J=8.4, 2.4 Hz, 1H), 7.65 (t, J=7.8 Hz, 1H), 7.55 (d, J=8.4 Hz, 1H), 2.52 (s, 3H); 13C NMR (150 MHz, CDCl3): δ=142.2, 141.5, 140.2, 139.2, 134.4, 132.2, 131.9, 131.4, 130.8, 129.8, 127.1, 126.4, 29.0; HRMS (ESI−) calcd for C13H10Cl2NO4S2: [M−H]− 377.9434, found: 377.9436 (error 0.5 ppm).
N-Benzyl-3-(3,4-dichlorophenylsulfonyl)benzenesulfonamide (30)
The title compound was prepared by following the general procedure from 25 (0.38 g) to afford a white amorphous solid (0.32 g, 60%): Rf=0.40 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CD3Cl3 + CD3OD): δ=8.29 (s, 1H), 7.98 (d, J=7.8 Hz, 1 H), 7.95 (d, J=2.4 Hz, 1 H), 7.93 (d, J=7.8 Hz, 1 H), 7.69 (dd, J=8.4, 2.4 Hz, 1 H), 7.56 (ovlp t, J=7.8 Hz, 1H), 7.55 (ovlp d, J=8.4 Hz, 1H), 7.16–7.13 (m, 3 H), 7.07–7.05 (m, 2 H), 4.08 (s, 2H); 13C NMR (150 MHz, CDCl3+ CD3OD): δ=143.0, 142.0, 140.3, 139.2, 136.0, 134.5, 132.0, 131.9, 131.2, 130.7, 129.8, 128.7, 128.1, 128.0, 127.1, 126.3, 47.2; HRMS (ESI−) calcd for C19H14Cl2NO4S2: [M−H]− 453.9747, found: 453.9756 (error 1.9 ppm).
N-[3-(3,4-Dichlorophenylsulfonyl)phenylsulfonyl]morpholine (31)
The title compound was prepared by following the general procedure from 25 (0.38 g) to afford a white amorphous solid (0.30 g, 61%): Rf=0.40 (hexane/EtOAc 1.5:1); 1H NMR (600 MHz, CDCl3): δ=8.30 (s, 1 H), 8.16 (d, J=7.8 Hz, 1 H), 8.04 (d, J=1.8 Hz, 1 H), 7.97 (d, J=7.8 Hz, 1 H), 7.78 (ovlp dd, J=8.4, 1.8 Hz, 1H), 7.76 (ovlp t, J=7.8 Hz, 1H), 7.63 (d, J=8.4 Hz, 1 H), 3.75 (t, J=4.8 Hz, 4 H), 3.02 (t, J=4.8 Hz, 4H); 13C NMR (150 MHz, CDCl3): δ=142.8, 140.4, 139.4, 137.8, 134.7, 132.6, 132.1, 132.0, 131.0, 130.0, 127.14, 127.11, 66.2, 46.1; HRMS (APCI−) calcd for C16H13Cl2NO4S2: [M−H2O]− 416.9669, found: 416.9670 (error 0.2 ppm).
N-[3-(3,4-Dichlorophenylsulfonyl)phenylsulfonyl]acetamide (32)
Ac2O (0.5 mL) was added to 3 followed by ZnCl2 (1.1 mg, 3 mol%). The reaction mixture was heated at 50°C for 30 min to afford a clear solution. The reaction mixture was then cooled to room temperature, concentrated in vacuo, and purified by silica gel chromatography with 60% EtOAc/hexane to afford the title compound (96 mg, 87%) as a white amorphous solid: Rf=0.30 (hexane/EtOAc 1:1); 1H NMR (600 MHz, CD3OD): δ=8.56 (s, 1 H), 8.26 (ovlp d, J= 7.8 Hz, 1 H), 8.24 (ovlp d, J=7.8 Hz, 1H), 8.15 (d, J=2.4 Hz, 1 H), 7.90 (dd, J=8.4, 2.4 Hz, 1 H), 7.82 (t, J=7.8 Hz, 1H), 7.77 (d, J= 8.4 Hz, 1 H), 1.94 (s, 3H); 13C NMR (150 MHz, CD3OD): δ=169.8, 142.0, 141.3, 141.0, 138.6, 133.9, 132.9, 132.5, 132.0, 130.8, 129.6, 127.6, 127.4, 22.1; HRMS (APCI−) calcd for C14H10Cl2NO5S2: [M−H]− 405.9383, found: 405.9378 (error 1.2 ppm).
General procedure for the acetylation 4, 41–44, and 47
Ac2O (6 mL) was added to compound 34–38, and 48 (1 mmol). The mixture was heated at reflux for 3 h to provide a clear solution. The reaction was cooled to room temperature, whereupon the product precipitated. The resulting white solid was then filtered and washed with cold CH2Cl2 to remove Ac2O to afford the product (93–96% yield).
1-Acetylbenzimidazole-2-thione (5)
The title compound was prepared by following the general procedure from 34 (0.15 g) to afford a white amorphous solid (0.18 g, 94% yield): Rf=0.30 (hexane/EtOAc 4:1); mp: 200–203°C; 1H NMR (600 MHz, [D6]DMSO): δ=13.30 (br s, 1H), 7.98 (d, J=7.8 Hz, 1 H), 7.28 (t, J= 7.8 Hz, 1H), 7.21 (t, J=7.8 Hz, 1 H), 7.17 (d, J=8.4 Hz, 1H), 3.31 (s, 3H); 13C NMR (150 MHz, [D6]DMSO): δ=172.8, 170.7, 131.8, 131.6, 126.1, 124.1, 116.1, 110.2, 28.7; IR: νmax=1725 cm−1 (C=O); HRMS (ESI+) calcd for C9H9N2OS: [M+H]+ 193.0430, found: 193.0423 (error 3.6 ppm).
1-Acetyl-5/6-methylbenzimidazole-2-thione (41)
The title compound was prepared by following the general procedure from 36 (0.16 g) to afford a white amorphous solid (0.19 g, 93%) as an inseparable 1:1 mixture of 5-methyl and 6-methyl regioisomers: Rf= 0.30 (hexane/EtOAc 4:1); 1H NMR (600 MHz, CD3OD + [D6]DMSO): δ=7.94 (d, J=8.4 Hz, 1 H), 7.92 (s, 1 H), 7.10 (d, J=7.8 Hz, 1H), 7.02 (d, J=8.4 Hz, 2 H), 6.96 (s, 1 H), 3.04 (s, 3 H), 3.02 (s, 3 H), 2.39 (s, 3 H), 2.38 (s, 3H); 13C NMR (150 MHz, CD3OD + [D6]DMSO): δ= 172.3, 172.1, 170.9, 170.7, 135.4, 133.3, 131.2, 129.2, 125.8, 124.0, 115.8, 115.2, 109.6, 109.1, 109.0, 108.5, 27.4, 27.3, 20.5, 20.2; IR: νmax=1688 cm−1 (C=O); HRMS (ESI+) calcd for C10H11N2OS: [M+H]+ 207.0587, found: 207.0579 (error 3.9 ppm).
1-Acetylbenzoxazole-2-thione (42)
The title compound was prepared by following the general procedure from 37 (0.15 g) to afford a white amorphous solid (0.18 g, 94%): Rf=0.30 (hexane/EtOAc 4:1); 1H NMR (600 MHz, CDCl3): δ=8.06 (d, J=8.4 Hz, 1 H), 7.33–7.29 (m, 3 H), 3.05 (s, 3H); 13C NMR (150 MHz, CDCl3): δ= 179.4, 171.1, 146.7, 130.0, 126.4, 125.7, 116.7, 109.8, 27.9; IR: νmax= 1738 cm−1 (C=O); HRMS (ESI+) calcd for C9H8NO2S: [M+H]+ 194.0270, found: 194.0263 (error 3.6 ppm).
1-Acetyl-2-oxindole (43)
The title compound was prepared by following the general procedure from 38 (0.13 g) to afford a white amorphous solid (0.18 g, 96%): Rf=0.35 (hexane/EtOAc 4:1); 1H NMR (600 MHz, CDCl3): δ=8.21 (d, J=8.4 Hz, 1 H), 7.31 (t, J= 8.4 Hz, 1H), 7.26 (d, J=7.8 Hz, 1 H), 7.18 (t, J=7.2 Hz, 1H), 3.71 (s, 2 H), 2.67 (s, 3H); 13C NMR (150 MHz, CDCl3): δ=175.3, 170.8, 141.4, 128.2, 124.9, 123.9, 123.4, 116.6, 36.6, 26.6; IR: νmax=1760, 1709 cm−1 (C=O); HRMS (ESI+) calcd for C10H10NO2: [M+H]+ 176.0706, found: 176.0710 (error 2.3 ppm).
1-Acetylbenzimidazole (44)
NaBH4 (2.27 g, 60.0 mmol) was added portionwise (in 10 equal portions) to a solution of benzimidazole-2-thione 34 (1.00 g, 6.65 mmol) and NiCl2 (2.6 g, 20.0 mmol) in CH3OH (50 mL) over 60 min at 0 °C. Once the addition of the final portion of NaBH4 was complete, the reaction was filtered, and the filtrate was concentrated to afford a white solid, which was dissolved in EtOAc (50 mL), washed with H2O (40 mL), dried (MgSO4) and concentrated to afford crude 35 (0.65 g, 83%).
Crude 35 (0.2 g, 1.7 mmol) prepared above was acetylated using the general procedure for acetylation and purified by flash chromatography (hexane/EtOAc 4:1) to afford the title compound 44 (0.25 g, 93% yield) as a white amorphous solid: Rf=0.35 (hexane/EtOAc 4:1); 1H NMR (600 MHz, CD3OD): δ=8.69 (s, 1 H), 8.16 (d, J= 8.4 Hz, 1H), 7.66 (d, J=8.4 Hz, 1H), 7.37–7.35 (m, 2 H), 2.72 (s, 3H); 13C NMR (150 MHz, CD3OD): δ=168.6, 143.3, 143.2, 131.5, 125.7, 124.9, 119.4, 115.5, 22.5; IR: νmax=1726 cm−1 (C=O); HRMS (ESI+) calcd for C9H9N2O: [M+H]+ 161.0709, found: 161.0718 (error 5.5 ppm).
1-Acetylbenzimidazole-2-one (45)
Benzimidazole-2-one (1.00 g, 7.5 mmol) 39 was acetylated by using the general procedure for acetylation, which afforded the bis-acetylated adduct. Benzylamine (0.25 g, 2.5 mmol) was added to a solution of the crude bis-acetylated product (0.5 g, 2.3 mmol) in CH2Cl2 (5 mL), and the reaction was stirred for 1 h at room temperature. The reaction was filtered to collect the white solid, which was washed with CH2Cl2 to afford the title compound (0.19 g, 96% yield) as a white amorphous solid: Rf=0.25 (hexane/EtOAc 4:1); 1H NMR (600 MHz, [D6]DMSO): δ=8.32 (d, J=7.8, Hz, 1H), 7.40 (t, J=7.8 Hz, 1H), 7.33 (t, J= 7.8 Hz, 1H), 7.25 (d, J=7.8, 1 H), 2.95 (s, 3H); 13C (150 MHz, [D6]DMSO): δ=171.1, 153.1, 130.3, 129.5, 127.7, 125.0, 122.2, 121.1, 25.9; IR: νmax=1724, 1705 cm−1 (C=O); HRMS (ESI+) calcd for C9H9N2O2: [M+H]+ 177.0659, found: 177.0663 (error 2.2 ppm).
1-Acetylimidazole-2-thione (47)
The title compound was prepared by following the general procedure from 48 (0.10 g) to afford a yellow amorphous solid (0.13 g, 96 %): Rf=0.30 (hexane/EtOAc 4:1); 1H NMR (600 MHz, CD3OD): δ=7.46–7.44 (m, 1 H), 6.67– 6.65 (m, 1H), 2.98 (s, 3H); 13C (150 MHz, CD3OD): δ=172.3, 169.9, 115.8, 114.9, 25.4; IR: νmax=1716 cm−1 (C=O); HRMS (ESI+) calcd for C5H7N2OS: [M+H]+ 143.0274, found: 143.0268 (error 4.1 ppm).
Supplementary Material
Acknowledgments
This research was supported by NIH grant R01 AI070219 (to C.C.A.). Facilities and compound libraries for high-throughput screening were made available through the NSRB and the New England Regional Center of Excellence in Biodefense and Emerging Infectious Disease (U54 AI057159). We thank members of the NSRB (Harvard Medical School) for their ongoing advice.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201000275.
Supporting Information: mass spectrometry and circular dichroism analysis of MbtI treated with and without compound 2. Michaelis –Menten and Lineweaver–Burk plot of MbtI and 4.
References
- 1.World Health Organization. Global Tuberculosis Control: Surveillance and Financing, WHO Report 2008. WHO Press; Geneva (Switzerland): 2008. [Google Scholar]
- 2.Laurenzi M, Ginsberg A, Spigelman M. Infect Disord Drug Targets. 2007;7:105–119. doi: 10.2174/187152607781001817. [DOI] [PubMed] [Google Scholar]
- 3.a) De Voss JJ, Rutter K, Schroeder BG, Barry CE., 3rd J Bacteriol. 1999;181:4443–4451. doi: 10.1128/jb.181.15.4443-4451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Monfeli RR, Beeson C. Infect Disord Drug Targets. 2007;7:213–220. doi: 10.2174/187152607782110031. [DOI] [PubMed] [Google Scholar]
- 4.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd Proc Natl Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]; b) Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, Gokhale RS. Proc Natl Acad Sci USA. 2006;103:2069–2074. doi: 10.1073/pnas.0507924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) He Z, Stigers Lavoie KD, Bartlett PA, Toney MD. J Am Chem Soc. 2004;126:2378–2385. doi: 10.1021/ja0389927. [DOI] [PubMed] [Google Scholar]; b) Bulloch EM, Jones MA, Parker EJ, Osborne AP, Stephens E, Davies GM, Coggins JR, Abell C. J Am Chem Soc. 2004;126:9912–9913. doi: 10.1021/ja048312f. [DOI] [PubMed] [Google Scholar]; c) Bulloch EM, Abell C. ChemBioChem. 2005;6:832–834. doi: 10.1002/cbic.200400385. [DOI] [PubMed] [Google Scholar]; d) Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS. J Bacteriol. 2006;188:6081–6091. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. Biochemistry. 2007;46:954–964. doi: 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- 7.a) Wright SK, DeClue MS, Mandal A, Lee L, Wiest O, Cleland WW, Hilvert D. J Am Chem Soc. 2005;127:12957–12964. doi: 10.1021/ja052929v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeClue MS, Baldridge KK, Kunzler DE, Kast P, Hilvert D. J Am Chem Soc. 2005;127:15002–15003. doi: 10.1021/ja055871t. [DOI] [PubMed] [Google Scholar]
- 8.a) Sassetti CM, Boyd DH, Rubin EJ. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]; b) Nagachar N, Ratledge C. FEMS Microbiol Lett. 2010;308:159–165. doi: 10.1111/j.1574-6968.2010.02004.x. [DOI] [PubMed] [Google Scholar]; c) Nagachar N, Ratledge C. FEMS Microbiol Lett. 2010;311:193–199. doi: 10.1111/j.1574-6968.2010.02091.x. [DOI] [PubMed] [Google Scholar]
- 9.a) Payne RJ, Kerbarh O, Miguel RN, Abell AD, Abell C. Org Biomol Chem. 2005;3:1825–1827. doi: 10.1039/b503800f. [DOI] [PubMed] [Google Scholar]; b) Payne RJ, Bulloch EMM, Toscano MM, Jones MA, Kerbarh O, Abell C. Org Biomol Chem. 2009;7:2421–2429. doi: 10.1039/b901694e. [DOI] [PubMed] [Google Scholar]; c) Payne RJ, Bulloch EM, Kerbarh O, Abell C. Org Biomol Chem. 2010;8:3534–3542. doi: 10.1039/c004062b. [DOI] [PubMed] [Google Scholar]
- 10.a) Kozlowski MC, Bartlett PA. J Am Chem Soc. 1991;113:5897–5898. [Google Scholar]; b) Kozlowski MC, Tom NJ, Seto CT, Sefler AM, Bartlett PA. J Am Chem Soc. 1995;117:2128–2140. [Google Scholar]
- 11.Ziebart KT, Dixon SM, Avila B, El-Badri MH, Guggenheim KG, Kurth MJ, Toney MD. J Med Chem. 2010;53:3718–3729. doi: 10.1021/jm100158v. [DOI] [PubMed] [Google Scholar]
- 12.Manos-Turvey A, Bulloch EM, Rutledge PJ, Baker EN, Lott JS, Payne RJ. ChemMedChem. 2010;5:1067–1079. doi: 10.1002/cmdc.201000137. [DOI] [PubMed] [Google Scholar]
- 13.Feng BY, Shoichet BK. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Collier PJ, Ramsey AJ, Austin P, Gilbert P. J Appl Bacteriol. 1990;69:569–577. doi: 10.1111/j.1365-2672.1990.tb01550.x. [DOI] [PubMed] [Google Scholar]; b) Collier PJ, Ramsey A, Waigh RD, Douglas KT, Austin P, Gilbert P. J Appl Bacteriol. 1990;69:578–584. doi: 10.1111/j.1365-2672.1990.tb01551.x. [DOI] [PubMed] [Google Scholar]; c) Combrink KD, Gulgeze HB, Meanwell NA, Pearce BC, Zulan P, Bisacchi GS, Roberts DG, Stanley P, Seiler SM. J Med Chem. 1998;41:4854–4860. doi: 10.1021/jm9804580. [DOI] [PubMed] [Google Scholar]; d) Sharmeen L, McQuade T, Heldsinger A, Gogliotti R, Domagala J, Gracheck S. Antiviral Res. 2001;49:101–114. doi: 10.1016/s0166-3542(00)00143-1. [DOI] [PubMed] [Google Scholar]; e) Morley JO, Oliver Kapur AJ, Charlton MH. Org Biomol Chem. 2005;3:3713–3719. doi: 10.1039/b509529h. [DOI] [PubMed] [Google Scholar]; f) Wiles JA, Hashimoto A, Thanassi JA, Cheng J, Incarvito CD, Deshpande M, Pucci MJ, Bradbury BJ. J Med Chem. 2006;49:39–42. doi: 10.1021/jm051066d. [DOI] [PubMed] [Google Scholar]; g) Gorsuch S, Bavetsias V, Rowlands MG, Aherne GW, Workman P, Jarman M, McDonald E. Bioorg Med Chem. 2009;17:467–474. doi: 10.1016/j.bmc.2008.11.079. [DOI] [PubMed] [Google Scholar]
- 15.a) Terentis AC, Freewan M, Plaza TSS, Raftery MJ, Stocker R, Thomas SR. Biochemistry. 2010;49:591–600. doi: 10.1021/bi901546e. [DOI] [PubMed] [Google Scholar]; b) Loo JA, Holler TP, Sanchez J, Gogliotti R, Maloney L, Reily MD. J Med Chem. 1996;39:4313–4320. doi: 10.1021/jm960253w. [DOI] [PubMed] [Google Scholar]; c) Sarma BK, Mugesh G. Chem Eur J. 2008;14:10603–10614. doi: 10.1002/chem.200801258. [DOI] [PubMed] [Google Scholar]; d) Sarma BK, Mugesh G. J Am Chem Soc. 2007;129:8872–8881. doi: 10.1021/ja070410o. [DOI] [PubMed] [Google Scholar]; e) Sivaramakrishnan S, Keerthi K, Gates KS. J Am Chem Soc. 2005;127:10830–10831. doi: 10.1021/ja052599e. [DOI] [PubMed] [Google Scholar]
- 16.Baell JB, Holloway GA. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 17.Gopalsamy A, Shi MX, Stauffer B, Bahat R, Billiard J, Ponce-De-Leon H, Seestaller-Wehr L, Fukayama S, Mangine A, Moran R, Krishnamurthy G, Bodine P. J Med Chem. 2008;51:7670–7672. doi: 10.1021/jm801069w. [DOI] [PubMed] [Google Scholar]
- 18.a) Dyer E, Minnier CE. J Heterocycl Chem. 1969;6:23–28. [Google Scholar]; b) Chung IH, Cha KS, Seo JH, Kim JH, Chung BY, Kim CS. Heterocycles. 2000;53:529–533. [Google Scholar]; c) Saxena DB, Khajuria RK, Suri OP. J Heterocycl Chem. 1982;19:681–683. [Google Scholar]; d) Nishio T, Shiwa K. Heterocycles. 2004;62:313–324. [Google Scholar]
- 19.Khurana JM, Kukreja G, Bansal G. J Chem Soc Perkin Trans. 2002;1:2520–2524. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.