

Inherited mutations and polymorphisms that alter the polypeptide sequence can affect the stability of the protein fold, triggering onset of many different diseases at birth and during aging. A major challenge is to understand the underlying mechanisms responsible for such diseases and to develop effective therapeutic approaches. A central pathway responsible for the generation and maintenance of the protein fold is the protein homeostasis or proteostasis network (PN) (1). The PN has both folding and ubiquitin-mediated degradation branches serving as the Yin and the Yang of proteostasis (Fig. 1). Our understanding of the role of the PN in generating and maintaining a functional protein (the Yang), or directing its removal from the cell (the Yin) during normal protein turnover or in cases of misfolding, is limited. This balance is critical for normal cellular, tissue and organismal physiology. In the current issue of Science, Okiyoneda et al (REF) provide an important clue to the operation of the PN in their analysis of a mutant of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) that is responsible for inherited disease Cystic Fibrosis (CF). Their work highlights the importance of continuous surveillance of the mutant CFTR protein fold by the PN throughout its cellular itinerary- from birth by co-translational insertion into the endoplasmic reticulum (ER), to removal as an unstable protein at the plasma membrane (PM) by the degradative branch of the PN. Their work support a view that it is the globally acting Yin and Yang properties of the PN that set an energetic standard for protein stability that directs cellular, tissue and organismal function (Fig. 1) (2). The ramifications of their observations impact our understanding and treatment of a broad range of human misfolding diseases.
Figure 1. The Yin and Yang of proteostasis.
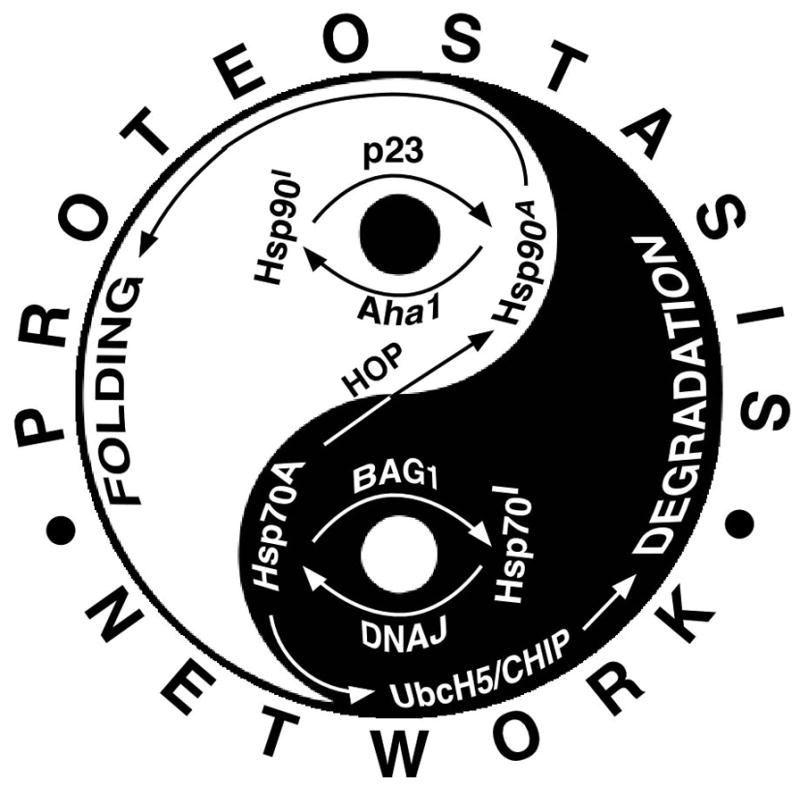
Schematic representation of the balance between the folding (Yang) and degradation (Yin) branches of the cellular proteostasis network (PN) that globally manage protein folding in the cell in health and disease. Hsp70 and Hsp90, core components of the PN, are cycled between active and inactive forms by regulatory co-chaperones. The ATP bound, client binding competent state of Hsp90 (Hsp90A) is stabilized by p23, a step involved in protein folding. The release of clients is accomplished by inactivation of the Hsp90 (Hsp90I) through activation of its ATPase activity, which is regulated by Aha1. The active, client binding competent state of Hsp70 (Hsp70A) is the ADP bound state, which is generated by DNAJ family of (Hsp40) proteins, which are ATPase activating co-chaperones. Client release is achieved either by transfer of the client to Hsp90 via the activity of Hsp70/90 organizing protein (HOP) or by BAG/TRP-domain containing proteins. BAG proteins mediate the ADP/ATP exchange of Hsp70. Prolonged binding of clients to Hsp70 results in the recruitment of E2 ubiquitin conjugating and E3 ubiquitin ligating enzymes leading to degradative pathways. UbcH5 and CHIP are the respective enzymes mediating the ubiquitination and ER-associated degradation or lysosomal delivery of CFTR (Okiyoneda et al. REF).
The most prominent disease causing mutation in CF is the deletion of Phe508 (ΔF508) in the cytosol-oriented Nucleotide Binding Domain 1 (NBD1) of CFTR. Loss of Phe508 leads to energetic destabilization that alters folding, targeting ΔF508 for ER-associated degradation (ERAD) (3). While much work in the past has focused on components that dictate folding and degradation in the ER (4), Okiyoneda et al. (REF) have now identified a number of factors regulating the PM stability of ΔF508- referred to as peripheral quality control. It is well known that ΔF508 is a temperature sensitive mutant as reduced temperature (26°C) will partially rescue the mutant from degradation in the ER and promote its delivery to the cell surface. Although stable at the PM at the reduced temperature, a return to physiological temperature (37°C) triggers its rapid degradation (3), a feature uncharacteristic of the more energetically stable WT CFTR.
To understand the underlying mechanisms of ΔF508 destabilization at 37°C, the authors took advantage of the temperature sensitivity of the mutant to achieve a PM pool of the protein followed by a return to 37°C, and examined the role of various PN components in the turnover of PM localized ΔF508. In so doing, they identified UbcH5c and CHIP as the critical E2 ubiquitin-conjugating and E3 ubiquitin-ligase, respectively, as key factors initiating ubiquitination, internalization and lysosomal degradation. In addition, they elucidated a critical role for both heat shock proteins 70 and 90 (Hsp70/90) as well as numerous regulatory co-chaperones including specific Hsp40 family proteins and BCL2-associated anthanogene (BAG) to be involved in the PM stability of ΔF508. While the UbcH5 family, CHIP and the Hsp70/90 machinery have been shown to facilitate folding, export and/or degradation of ΔF508 at the ER membrane during nascent synthesis (4, 5), it was not anticipated that the same proteins would play similar roles at the PM. In general, the data provide important support for the emerging principle that the proteostasis environment, as defined by the PN, dictates the fate of proteins throughout their functional life cycle. Thus, the old adage that simply getting ΔF508-CFTR out of the ER would be sufficient to correct disease may not hold.
These observations have far reaching implications for both basic research and therapeutic approaches to human disease. Proteostasis encompasses a vast network of folding chaperones/co-chaperones, redox enzymes, degradative machineries and their regulatory signaling pathways that mediate protein folding and degradation. The Yin and Yang of PN function is largely centered around core Hsp70 and 90 chaperone systems (6), which have cytosolic and luminal homologues in all organelles (2), suggesting common principles have evolved to generate and maintain the protein fold to ensure proper cell, tissue and organismal function. The balance between the folding Yang and degradative Yin pathways are tightly linked to the Hsp70/90 ATPase cycles which are regulated through ATP loading and ATPase activating co-chaperones (ATP to ADP) (Fig. 1). In the case of Hsp70, BAGs act as nucleotide exchange factors (ADP to ATP) whereas Hsp40 family members serve as ATPase activators (7). Hsp90 ATPase activity is regulated by the co-chaperones Aha1 (ATPase activator) and p23 (ATPase inhibitor), and many ancillary co-chaperones including a large family TRP-domain containing cyclophilin/immunophilin proteins that likely facilitate client delivery (8). Moreover, the composition of the PN varies remarkably between different cell types (2, 9). PN function is, in turn, tightly linked to the folding energy landscape, a pathway of folding intermediates the nascent protein must navigate to achieve the native state (10). Recent modeling (2, 11) suggests that the rates of folding and misfolding as well as thermodynamic stability collectively influence the success or failure of the protein fold along the landscape, a process heavily managed by the Yin and Yang components of the PN machinery (10).
The observations by Okiyoneda et al. (REF) that now suggest that PM-localized proteins are subject to the same surveillance system used by the ER raises the specter that protein stability and function is continuously sensitive to the local cellular PN following export. Moreover, because the pathways dictated by the degradative Yin side of the PN differ for the various compartments in which the protein client resides (e.g., chaperone-mediated retro-translocation to the proteasome from the ER opposed to vesicular-based lysosomal delivery at the PM), these results suggest that vesicular trafficking does not make the choice of life and death. Rather, it is a standard provided by the PN (2) that decides protein fate in health and disease. Thus, the PN can provide global management to overcome loss- and/or gain-of-function mutations seen in numerous misfolding diseases. Intriguingly, this standard can be a highly variable between cell types and even within a given cell in response to a broad range of signaling pathways including the unfolded protein response (UPR), heat shock response (HSR), oxidative stress pathways, diet restriction, inflammatory and insulin growth factor 1 receptor (IGF1R) signaling pathways (1), all of which protect us from the environment (12) and during aging (13, 14). The underlying operating principles of the PN (2) highlighted by Okiyoneda et al. (REF) suggests that while various chemical, biochemical and physiologic approaches could solve the folding and trafficking defects in one compartment, we must ultimately solve the folding and function problem for the cell as a whole, perhaps through the malleable PN (Fig. 1). This view at the moment is an underappreciated feature of organismal physiology. It is now apparent that diverse human diseases including, for example, CF, Alzheimer’s, type II diabetes, and bacterial and viral pathologies (15), would benefit from a better understanding how the PN functions globally as a folding management program in order to develop better tools to control the network therapeutically to restore human health. That is, we need to learn to use the biology of the protective Yang to correct the biology oftentimes mis-managed by the Yin (Fig. 1).
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Science. 2008;319:916. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Annu Rev Biochem. 2009;78:959. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 3.Riordan JR. Annu Rev Biochem. 2008;77:701. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 4.Turnbull EL, Rosser MF, Cyr DM. BMC Biochem. 2007;8(Suppl 1):S11. doi: 10.1186/1471-2091-8-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, et al. Cell. 2006;127:803. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 6.Broadley SA, Hartl FU. FEBS Lett. 2009;583:2647. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 7.Vos MJ, Hageman J, Carra S, Kampinga HH. Biochemistry. 2008;47:7001. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- 8.Wandinger SK, Richter K, Buchner J. J Biol Chem. 2008;283:18473. doi: 10.1074/jbc.R800007200. [DOI] [PubMed] [Google Scholar]
- 9.Hutt DM, Powers ET, Balch WE. FEBS Lett. 2009;583:2639. doi: 10.1016/j.febslet.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartl FU, Hayer-Hartl M. Nat Struct Mol Biol. 2009;16:574. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 11.Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. Cell. 2007;131:809. doi: 10.1016/j.cell.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 12.Morimoto RI. Genes Dev. 2008;22:1427. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panowski SH, Dillin A. Trends Endocrinol Metab. 2009;20:259. doi: 10.1016/j.tem.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Morimoto RI, Cuervo AM. J Gerontol A Biol Sci Med Sci. 2009;64:167. doi: 10.1093/gerona/gln071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geller R, Vignuzzi M, Andino R, Frydman J. Genes Dev. 2007;21:195. doi: 10.1101/gad.1505307. [DOI] [PMC free article] [PubMed] [Google Scholar]