

Abstract
Here, we have investigated mitochondrial biology and energy metabolism in human embryonic stem cells (hESCs) and hESC-derived neural stem cells (NSCs). Although stem cells collectively in vivo might be expected to rely primarily on anaerobic glycolysis for ATP supply, to minimise production of reactive oxygen species, we show that in vitro this is not so: hESCs generate an estimated 77% of their ATP through oxidative phosphorylation. Upon differentiation of hESCs into NSCs, oxidative phosphorylation declines both in absolute rate and in importance relative to glycolysis. A bias towards ATP supply from oxidative phosphorylation in hESCs is consistent with the expression levels of the mitochondrial gene regulators peroxisome-proliferator-activated receptor γ coactivator (PGC)-1α, PGC-1β and receptor-interacting protein 140 (RIP140) in hESCs when compared with a panel of differentiated cell types. Analysis of the ATP demand showed that the slower ATP turnover in NSCs was associated with a slower rate of most energy-demanding processes but occurred without a reduction in the cellular growth rate. This mismatch is probably explained by a higher rate of macromolecule secretion in hESCs, on the basis of evidence from electron microscopy and an analysis of conditioned media. Taken together, our developmental model provides an understanding of the metabolic transition from hESCs to more quiescent somatic cell types, and supports important roles for mitochondria and secretion in hESC biology.
Keywords: Mitochondria, Pluripotency, Embryonic stem cells, Neural stem cells, Differentiation, Secretion
Introduction
Human embryonic stem cells (hESCs) are long-term self-renewing cells that are able to differentiate into any cell type in the body (Thomson et al., 1998). The potential of hESCs to act as an unlimited source for somatic cell generation is of major interest for downstream applications, including drug testing, disease modelling and cell replacement therapy (Braam et al., 2009; Desbordes et al., 2008; Han et al., 2009; Kiskinis and Eggan, 2010; Passier et al., 2008; Zeng and Rao, 2008). Additionally, these cells might yield insights into the processes underlying cellular senescence, cancer and ageing (Zeng, 2007). Obtaining a more complete understanding of the biology of pluripotent cells is important for maximising the benefits from these areas.
The cells of the inner cell mass of the blastocyst, from which hESCs are derived, divide rapidly. This property of aggressive growth is reflected in cultured ESCs by their ability to form teratomas and their short-term growth-factor-autonomous cell cycling (Becker et al., 2010; Blum and Benvenisty, 2009), although there are differences in the division rate of different pluripotent cell types (Hanna et al., 2010). Progenitor cells in the adult organism, such as multipotent neural stem cells (NSCs), must possess the unique properties of long-term viability and self-maintenance, while remaining in what is typically a quiescent state. Multipotent NSCs can be derived in pure populations from cultured hESCs (Reubinoff et al., 2001; Swistowski et al., 2009; Zhang et al., 2001), but little is known about their bioenergetic requirements or how closely their energy metabolism is governed by the constraints of the genetic developmental programme. The transition from a pluripotent stem cell through progressive stages of differentiation probably involves dynamic changes in the energy demand for cellular processes, depending on the needs of the individual cell types. ATP generation in aerobic cells is divided between two main pathways: anaerobic glycolysis and mitochondrial oxidative phosphorylation. On the basis of changing energy demands, and the need to limit the production of mitochondrial reactive oxygen species, it might be expected that the relative contribution of these two pathways would change during differentiation.
Some models suggest that an expansion of the mitochondrial population regardless of the differentiation lineage might be a universal phenomenon from the onset of ESC differentiation (Facucho-Oliveira and St John, 2009). However, many signalling and energy-consuming processes might be decreased in the early transition to a differentiated state, and it might not be expected that this would be accompanied by a mitochondrial expansion (Armstrong et al., 2006; Becker et al., 2006; Efroni et al., 2008; Meshorer and Misteli, 2006). By contrast, the evidence that mitochondrial biogenesis occurs in some instances of somatic stem-cell differentiation could be consistent with the acquisition of specialised energy-requiring processes, as reported for mesenchymal stem-cell differentiation (Chen et al., 2008; Uldry et al., 2006). Changes in mitochondrial regulation might also be driven by genetic developmental programmes, reflecting the typical energy requirements of these cell types in vivo as opposed to the energy requirements in culture. This provides a rationale for following the genetic regulators of energy metabolism in concert with the bioenergetics to illuminate the different forces driving changes in mitochondrial function.
In the present study, we characterise mitochondrial function in hESCs and in several stages of neuronal differentiation using advanced bioenergetic technologies. We conclude that hESCs have a high mitochondrial activity that declines disproportionately, alongside several related parameters of mitochondrial biogenesis, with differentiation into NSCs and that this is coupled to a decrease in overall ATP turnover, a decrease in macromolecule biosynthesis and a decrease in protein secretion.
Results
hESCs have a high rate of energy turnover
To study the bioenergetics of hESC neural differentiation, we examined hESCs (Fig. 1Ai), hESC-derived NSCs (Fig. 1Aii) and NSCs differentiated for 35 days, which consist of >80% β-III-tubulin-positive neurons (termed D-35; Fig. 1Aiii), as shown previously (Swistowska et al., 2010; Swistowski et al., 2009). It has been demonstrated previously that these NSCs are nestin- and Sox1-positive and have a wide differentiation capacity (Swistowski et al., 2009). For comparisons with another somatic cell type, we chose to examine a well-characterised primary human fetal-foreskin-derived fibroblast line (BJ; termed HDF).
Fig. 1.

hESCs show high levels of mitochondrial activity and a bias for aerobic ATP production. (A) The differentiation scheme used for measurements. Pluripotent I6 hESCs (i), multipotent I6 neural stem cells (NSCs) (ii) and differentiated I6 NSCs (D-35) (iii), stained for neuronal Tuj-β-tubulin. BJ fibroblasts (HDF) were also analysed (image not shown). (B) Oxygen consumption rate (OCR) measured on adherent cultures normalised to cell protein levels. Basal, endogenous rate; Oligomycin, ATP-synthase-inhibited rate; FCCP, maximal uncoupled rate; and Rot + Ant A, rotenone- and antimycin-A-inhibited rate. (C) Proton production rate from measurements taken in tandem with respiration rates. (D) ATP production rate from oxidative phosphorylation and glycolysis. Results shown are the means+s.e.m. from independent experiments (hESC, n=10; NSC, n=11; D-35, n=3; and HDF, n=4). The statistical significance was calculated from a one-way ANOVA using Dunnett's correction. *P<0.05, **P<0.01.
The oxygen consumption rate and extracellular acidification rate were determined in adherent cultures using a Seahorse XF-24 extracellular flux analyzer, as described previously (Gerencser et al., 2009). Basal respiration rate in hESC colonies was 1.8-fold higher than in NSCs and 2.8-fold higher than in HDFs, when normalised to the cell protein level (Fig. 1B). The coupled rate (the total basal rate minus the rate with oligomycin, where oligomycin is an inhibitor of ATP synthesis) was 111 pmol of O2 per minute per 10 μg of cell protein in hESCs and dropped to 54 pmol of O2 per minute per 10 μg of cell protein in NSCs, indicating that the mitochondrial ATP output decreases in the transition from hESCs to NSCs. Data were combined from the I6 and H9 lines for hESCs and NSCs to increase the reliability of our final determinations (no significant difference was observed between these lines). Differentiation of NSCs into D-35 cells caused little change in respiration or oxidative phosphorylation compared with the parental NSCs. Furthermore, the maximal uncoupler-evoked oxygen consumption rate (indicating maximal respiratory capacity) was significantly decreased in NSCs when compared with hESCs. The extracellular acidification rate is mainly due to lactate and bicarbonate production and, when calibrated as the proton production rate, indicates glycolytic rate (Wu et al., 2007). In contrast with respiration, the glycolytic rate was slightly increased in NSCs but declined significantly in D-35 cells (Fig. 1C). An absolute quantification of both the oligomycin-sensitive oxygen consumption rate and glycolytic rate can be converted into the ATP production rate, to assess the relative contribution in each cell type. The oligomycin-sensitive oxygen consumption was converted into the ATP production rate using a P/O ratio of 2.3 (Brand, 2005), and there is a one-to-one relationship between the lactate production rate and the ATP production rate from glycolysis. Remarkably, hESCs made at least 77% of their ATP using oxidative phosphorylation, whereas this value was significantly lower in NSCs (55%) and in HDFs (59%) (Fig. 1D, both P<0.01 compared with hESCs), but was similar to that of hESCs in D-35 cells (76%). The results in Fig. 1D show that hESCs had a faster turnover of ATP than NSCs, D-35 cells or HDFs and that this demand is met primarily by higher oxidative phosphorylation in hESCs.
Mitochondrial content during hESC neural differentiation
To address the apparent bias hESCs show towards high oxidative phosphorylation for ATP production, we assessed various parameters of mitochondrial content in each cell type. Western blots showed that although the levels of voltage-dependent anion-selective channel (VDAC) remained unchanged upon neural differentiation, cytochrome c levels were markedly decreased in NSCs but were upregulated again upon differentiation into D-35 cells (Fig. 2A,B). The important antioxidant proteins manganese superoxide dismutase (MnSOD) (mitochondrial) and copper/zinc superoxide dismutase (CuZnSOD) (cytosolic) showed little change (MnSOD was slightly reduced in NSCs), indicating that the cellular antioxidant system remains fairly stable between these states.
Fig. 2.

Mitochondrial content is altered with hESC differentiation. (A) Western blots for mitochondrial [cytochrome c (Cyt C) and VDAC] and antioxidant proteins (MnSOD and CuZnSOD) in whole-cell lysates from hESC cultures with corresponding NSCs and differentiated NSCs (D-35), compared with BJ fibroblasts (HDF). (B) Protein band quantification for cytochrome c and VDAC from three independent lysates for each sample normalised to the hESC protein level. Results represent the means+s.d. of combined data from the I6 and H9 lines. (C) mtDNA copy number per cell in hESCs and differentiated cells. Results shown are the means+s.e.m. for the H9 and I6 stem cell lines and the HDFs. (D) mtDNA copy number changes during hESC differentiation as embryoid bodies or as a monolayer. Results shown are the means+s.e.m. for independent experiments in the H9 and I6 stem cell lines. (E) Confocal images for mitochondrial volume ratio determination. Calcein-AM labels the cytoplasm and MitoTracker Red (MTR) labels the mitochondria. The upper two panels show the raw images for each channel and the lower two panels show the binary processed images, which were used as input for the calculation. (F) Mitochondrial volume ratio quantification from confocal imaging. Data represent means±s.e.m. for at least four independent experiments with both H9 and I6 stem cell lines. The statistical significance was calculated from a one-way ANOVA using Dunnett's correction. **P<0.01.
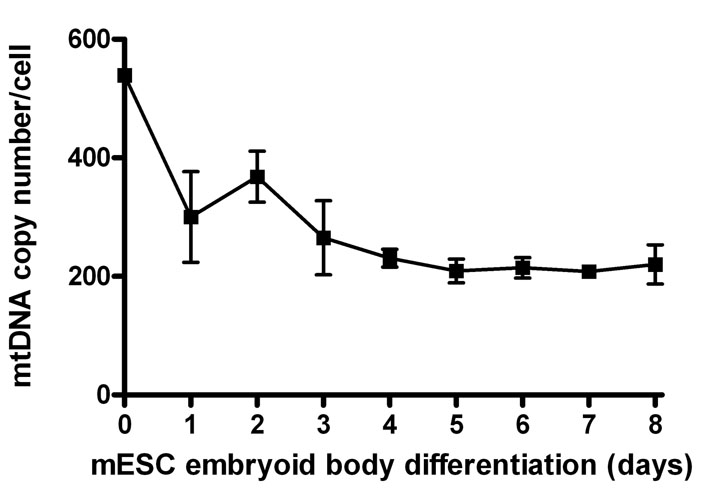
Quantification of absolute mitochondrial DNA (mtDNA) copy number revealed that hESCs are rich in mtDNA, with around 1200–1500 copies per cell, whereas NSCs, D-35 cells and HDFs contain 600–1000 copies per cell (Fig. 2C; values are the means of the H9 and I6 cell lines at each differentiation stage). A third hESC line BG01 was found to have approximately 1200 copies per cell. No significant difference was found in mtDNA content between hESCs maintained on feeder cells and those maintained on matrix in mouse embryonic fibroblast (MEF)-conditioned medium (data not shown). When normalised for cell size (see supplementary material Fig. S1), these values equate to an approximately 1.5- to 2-fold greater mtDNA density in hESCs. Differentiation of hESCs, as embryoid bodies or as a monolayer in inactivated serum-replacement-containing medium, showed that this progressive transition in mtDNA copy number occurred during the differentiation process itself and that by day 12 the amount was close to the content of the NSCs (Fig. 2D). This pattern was mimicked in B6 mouse ESC differentiation (supplementary material Fig. S2).
We also measured mitochondrial organelle content by employing live-cell confocal imaging stereology in cells labelled with MitoTracker Red and calcein-AM. In agreement with differentiation-based changes in respiration and mtDNA content, stereology revealed a small but significant reduction in mitochondrial volume density from 5.4±0.3% in hESCs to 4.0±0.3% in NSCs (Fig. 2E,F; P<0.01), which was reversed upon further differentiation (D-35: 5.9±0.4%). The mitochondrial volume density of the HDFs was similar to that of the NSCs (HDF: 4.0±0.2%; P<0.01 compared with hESCs).
Pluripotent stem cells express mitochondrial gene regulators in a pattern indicative of high reliance on oxidative phosphorylation
To investigate whether the high levels of mitochondrial activity and changes in mitochondrial biogenesis upon differentiation had underlying nuclear genetic regulation, we examined the expression of the ‘master regulators’ of mitochondrial gene expression – that is transcriptional coactivators and corepressors thought to control the expression of multiple downstream targets (Feige and Auwerx, 2007; Handschin and Spiegelman, 2006). We measured the expression levels of the genes encoding the two transcriptional coactivators peroxisome-proliferator-activated receptor γ coactivator (PGC)-1α and PGC-1β and the transcriptional corepressor receptor-interacting protein 140 (RIP140). We first compared hESCs with several somatically derived primary cell types and two cancer cell lines. Consistent with our functional observations, the mRNAs encoding PGC-1α and PGC-1β were both expressed to a greater extent in hESCs than in most of the somatic cell types (Fig. 3A,B). The cell types most similar to hESCs were melanocytes (HEMN-LP) and the cancer cell line MCF7, which showed high expression of the mRNAs encoding both PGC-1α and PGC-1β. RIP140, a repressor of genes involved in oxidative metabolism, was weakly expressed in hESCs compared with all the other cell types (Fig. 3C). PGC-1α and PGC-1β expression decreased and RIP140 expression increased during hESC differentiation into NSCs (Fig. 3D,E). Similar changes in PGC-1α and PGC-1β expression with hESC embryoid body differentiation have also been reported recently (Prigione et al., 2010). A large induction in PGC-1α upon differentiation into D-35 cells is consistent with our observation of increased mitochondrial biogenesis and a return to a reliance on mitochondrial ATP production in these cells.
Fig. 3.

Gene expression profiles for three metabolic master regulators highlight similarities between pluripotent stem cells and other energy demanding cell types. Real-time reverse transcription–PCR data for expression of the mRNA encoding PGC-1α (A), PGC-1β (B) and RIP140 (C) in I6 and BG01 hESC lines maintained on feeder cells (MEF) or on Geltrex in conditioned medium (GTX) compared with seven other primary cell types and two cancer cell lines. HDF, human diploid fibroblast; HEK, human epidermal keratinocyte; HEMN-LP, human epidermal melanocyte-low pigmented; HMVEC, human mammary vein endothelial cells; HUVEC, human umbilical vein endothelial cells; HPASMC, human pulmonary artery smooth muscle cells; HMEC, human mammary epithelial cells, MCF7, breast cancer cell line; and HeLa, cervical cancer cell line. Results represent means+s.e.m. for 2–4 independent RNA preparations. (D) Real-time reverse transcription–PCR data during differentiation of hESCs as embryoid bodies or as a monolayer. Data represent the mean values for two hESC lines (H9 and I6). (E) mRNA expression for NSCs and D-35 cells relative to their parental hESC lines. (F) mRNA expression changes during reprogramming of IMR31 fibroblasts into iPSCs and differentiation into iPSC-NSCs and iPSC-D35 cells.
To confirm further the expression pattern of these genes in human pluripotent cells, we assessed the expression of these genes in an induced pluripotent stem cell (iPSC) line derived from IMR90 fibroblasts (Mali et al., 2010). There was a strong induction of the mRNA encoding PGC-1β, whereas that encoding RIP140 was repressed upon reprogramming (Fig. 3F). PGC-1α was not induced but already had elevated expression in these fibroblasts compared with the other fibroblast strains we measured. iPSCs that were induced to differentiate into NSCs and D-35 cells, as described previously (Swistowski et al., 2010), showed similar mRNA expression changes to the hESC differentiations (Fig. 3F).
Finally, we attempted to control the energetic supply in differentiated cells by stable overexpression of PGC-1β or short hairpin RNA (shRNA) depletion of RIP140 in I6 NSCs or HDFs. Surprisingly, neither manipulation substantially boosted mitochondrial respiration or respiratory capacity (supplementary material Fig. S3). RIP140 knockdown did subtly increase maximal respiratory capacity in both cell types and stimulated glycolysis in NSCs. These findings suggest that manipulation of these genes individually is insufficient to alter mitochondrial bioenergetics in these cell types.
Plasma and mitochondrial membrane potential measurements in hESCs, NSCs and HDFs
The higher basal ATP turnover and maximal ATP-generating capacity of hESCs cannot be explained solely by the abundance of mitochondria in these cells. Mitochondrial membrane potential (Δψm) accounts for the majority of the proton-motive force used for driving ATP synthesis and therefore has a significant effect on the maximal ATP-generating capacity (Nicholls, 2006). The resting Δψm reflects the balance between supply (substrate and terminal oxidation) and demand (ATP consumption). A high ATP demand in hESCs might outstrip supply and thus result in a lower Δψm than in differentiated cells such as NSCs and HDFs, where ATP demand is smaller. Conversely, a high ATP supply originating from Δψm might keep ATP consumption running faster in hESCs.
Δψm in adherent cultures of hESCs and related lines was determined by fluorescence microscopy. All known Δψm measurement techniques (fluorescence, voltammetric or isotopic) rely on the potential-dependent distribution of lipophilic cations, such as the fluorescent indicator tetramethylrhodamine methyl ester (TMRM). The mitochondrial accumulation of these cations is a function of Δψm, the plasma membrane potential (Δψp) and the mitochondrial-matrix-to-cell-volume ratio (Nicholls, 2006). These parameters are expected to differ between cell types. To unveil Δψm, Δψp was followed with a lipophilic anionic plasma membrane potential indicator (PMPI) (Nicholls, 2006), and volume ratios were determined separately. TMRM and PMPI fluorescence of whole microscopic view fields were monitored in time, and Δψp and Δψm were determined in millivolts by temporally deconvoluting the fluorescence intensity time lapses, taking into account the slow potential-dependent redistribution and nernstian behaviour of these probes (A.A.G. and M.D.B., unpublished).
Δψp was calibrated by establishing a K+-equilibrium potential {a potential governed by the intra- and extra-cellular (EC) [K+]} using a cocktail of K+-channel openers and making stepwise increments in [K+]EC (Fig. 4A–C). The resting Δψp pooled from the two hESC lines (I6 and H9), as well as from HDFs is shown in Fig. 4D. The mitochondrial-matrix-to-cell-volume ratio was obtained as the product of the mitochondria-to-cell-volume ratio (see above) and the matrix-to-mitochondria-volume ratio. The latter was measured as the volume density of matrix by transmission electron microscopic stereology and was 0.84 for hESCs and 0.76 for NSCs. For HDFs, we assumed the same ratio as for NSCs. Interestingly, mitochondria in hESCs were mostly in the ‘orthodox’ conformation (Hackenbrock, 1966), whereas NSCs showed a ‘condensed’ conformation, with higher electron density in the matrix and more dilated cristae (Fig. 4G). The resting Δψm was calculated from the efflux kinetics of TMRM following an acute and complete mitochondrial depolarisation (Fig. 4E,F). The decay of TMRM fluorescence under these conditions was determined by the previous resting Δψm, volume ratios and the present Δψp (see the supplementary methods).
Fig. 4.

Resting plasma and mitochondrial membrane potential determination in hESCs and differentiated cells. (A) Time-lapse images of a hESC colony labelled with TMRM and PMPI potentiometric probes, under basal conditions, following addition of the K+ equilibrium cocktail (KEC) and the complete depolarisation cocktail (CDC). (B) Fluorescence intensity traces corresponding to A. Following the KEC addition, sequential replacement steps with KCl-based medium were performed as indicated. Finally the plasma membrane was completely depolarised with the addition of the CDC. (C) Mean normalised PMPI fluorescence values obtained for the three indicated cell types at the indicated extracellular K+ concentrations. (D) Mean (+s.e.m.) resting plasma membrane potentials for hESCs, NSCs and HDFs (hESC and NSC, n=8; HDF, n=3). Potentials were derived from C as given in the supplementary methods. (E) Time-lapse images of a hESC colony labelled with TMRM and PMPI, under basal conditions, 40 seconds after addition of the mitochondrial depolarisation cocktail (MDC) and after addition of the CDC. (F) Fluorescence intensity traces corresponding to E. Mitochondrial depolarisation was triggered with the MDC as indicated and finally complete depolarisation was achieved with the CDC. Parameters derived from the fluorescence traces are shown in supplementary material Fig. S3. (G) Electron micrographs of typical mitochondria in hESCs and NSCs (viewed at 105,000×). (H) Mean (+s.e.m.) resting mitochondrial membrane potential values for hESCs, NSCs and HDFs (hESC, n=28; NSC, n=16; and HDF, n=13). Potentials were derived from experiments similar to those shown in F and as given in the supplementary methods. The statistical significance was calculated from a one-way ANOVA using Dunnett's correction. **P<0.01.
The mean determinations are shown in Fig. 4H. We conclude that there is no significant change in Δψm upon differentiation of hESCs into NSCs. Therefore the decrease in respiration in NSCs when compared with hESCs is not caused by a large decrease in ATP supply or demand; instead supply and demand are kept in balance by concerted regulation.
The higher ATP turnover in hESCs compared with NSCs at slower growth rates is probably explained by macromolecule secretion in hESCs
Our data suggest that hESCs turnover ATP more rapidly than differentiated cells. To pinpoint the process(es) responsible for the increased ATP demand, we compiled a complete budget of the ATP demand in hESCs and NSCs. Individual ATP-consuming processes were acutely inhibited, causing a consequential drop in ATP demand and triggering a proportional drop in respiration. Dose–response curves for the protein translation inhibitor cycloheximide, the DNA and/or RNA synthesis inhibitor actinomycin D and the proteasome inhibitor MG132 are shown in supplementary material Fig. S4. In addition, the tubulin and actin turnover and the activities of Na+/K+-ATPase, the endoplasmic reticulum (ER) and plasma membrane Ca2+-ATPase and xanthine oxidase were investigated. Using the minimum saturating concentrations of each inhibitor, the relative contributions to the total respiration of these cells were assigned cumulatively for each of these processes (Table 1; Fig. 5). Inhibition of these processes caused a measurable drop in the oxygen consumption rate for both cell types, with the exception of the Ca2+-ATPases and xanthine oxidase. The relative demand from each of these processes was largely maintained, with protein synthesis and DNA and/or RNA synthesis still accounting for the majority of mitochondrial respiration in both cell types. However, the absolute oxygen consumption rates that were sensitive to inhibition of protein synthesis, DNA and/or RNA synthesis and Na+/K+-ATPase were significantly higher in hESCs, indicating that these processes consume more ATP in hESCs than in NSCs.
Table 1.
Rates of energy-consuming reactions in hESCs and NSCs

Fig. 5.

The relative division of energy-consuming reactions is largely maintained between hESCs and NSCs. The relative division of total respiration is shown, corresponding to the values in Table 1.
We considered the possibility that differences in cell division rates between hESCs and NSCs could account for some of the observed differences in ATP turnover. Hence, generation times were determined through time-lapse imaging. Surprisingly, the generation time of NSCs was significantly shorter than that of hESCs (H9 hESCs at 20.3 hours compared with NSCs at 14.2 hours; I6 hESCs at 24.2 hours compared with NSCs at 14.1 hours) (Fig. 6A–C; supplementary material Movie 1). No significant differences in cell death rates were observed between hESCs and NSCs (Fig. 6D). In conclusion, the higher ATP turnover rate in hESCs cannot be explained by a higher growth rate. Instead, processes that consume ATP in the same proportion as at lower growth rates must be increased; these might include autophagy or secretion of macromolecules.
Fig. 6.

Cell generation times for hESCs and NSCs. (A) hESC colony labelled with Hoechst 33342 under time-lapse imaging. Cells undergoing mitosis or cell death are labelled. (B) hESC mean cell generation times deduced from individual colonies from the I6 and H9 hESC lines. (C) Estimated mean (+s.e.m.) cell generation times for I6 and H9 hESCs (n=12 each) and NSCs (n=4 each). (D) Mean (+s.e.m.) frequency of cell death over a 24-hour period. Significance between values was calculated using a Student's t-test. **P<0.01.
Electron microscopy showed that there were hallmarks of macromolecule secretion in hESCs but not in NSCs (Fig. 7A,B). hESCs typically contained clusters of single-membrane-bound vesicles with homogeneous content and a less electron-dense core (Fig. 7A–D). Although no definite exocytotic events were observed, electron micrographs suggest a goblet-cell-like secretion mechanism (Fig. 7C), which is supported by previous results showing the secretion of cytosolic and cytoskeletal proteins in the proteome of hESC-conditioned medium (LaFramboise et al., 2010). By contrast, evidence of autophagy was uncommon, but both lines contained large multivesicular bodies signifying heterophagy. To assess the relative rates of protein secretion directly, we analysed medium conditioned for 10 hours by hESCs and NSCs and found a significantly higher concentration of secreted proteins in medium conditioned by hESCs (Fig. 8).
Fig. 7.

Secretory-like vesicles are uniquely present in hESCs. (A) Transmission electron micrographs of a typical field inside an I6 hESC colony with clusters of secretory vesicles in most cells. (B) Typical I6 NSC; no structures resembling the vesicle clusters of hESCs are seen in NSCs. (C,D) Magnifications of hESC secretory-vesicle-rich areas of H9 hESC and I6 hESCs lines, respectively. The arrow marks the goblet-cell-like secretion mechanism. Panel D shows that secretory vesicles are often surrounded by mitochondria and granules resembling polysomes. v, secretory vesicles appearing as membrane-bound structures with homogeneous content; m, mitochondrion; n, nucleus.
Fig. 8.

hESCs secrete more protein than NSCs. (A) Proteins precipitated from the medium conditioned by I6 hESCs or NSCs for 10 hours were subjected to SDS-PAGE and stained with GelCode Coomassie Blue stain. Loading of precipitated material was corrected for the amount of cell protein in each experiment. Whole-cell lysate lanes are shown for 85 μg of protein. The control lane is from a medium- or Geltrex-only precipitation. The gels are representative of two independent experiments. (B) Quantification of total precipitated protein using densitometry. The intense band at 55 kDa, also visible in the control, was excluded from the quantification. Values are means+s.d. for three independent experiments. Significance between values was calculated using a Student's t-test. **P<0.01.
Thus, the high ATP production rate in hESCs might be required to support RNA synthesis, protein synthesis, ion pumping and cytoskeletal rearrangements during macromolecule secretion by a goblet-cell-like mechanism, in which large parts of the cell, including cytosolic and cytoskeletal components, are secreted.
Discussion
To our knowledge, this is the first report of the bioenergetics of hESC differentiation. Our principal finding is that hESCs maintain a high rate of mitochondrial oxidative phosphorylation, which funds their high ATP demand for processes other than growth. We report a discrepancy wherein decreasing energy turnover is concomitant with increasing cell division rate during the transition from hESCs into NSCs, which might be explained by macromolecule secretion in hESCs.
ESCs have a fundamentally different in vivo biological role compared with that of somatic progenitor cells, which can remain quiescent for long periods of time. It seems sensible that these cell types would be bioenergetically tuned towards their normal developmental roles. Using a novel experimental platform enabling measurement of oxygen consumption rates and glycolytic rates in normal adherent cultures, we discovered that hESCs had an elevated rate of oxidative phosphorylation compared with hESC-derived NSCs, but without increased glycolysis. The hESC lines we investigated also had a relatively high mitochondrial biogenesis compared with NSCs and HDFs (Fig. 2), and overall our data support a model whereby mitochondrial biogenesis is decreased upon the transition from hESCs into early progenitors but increased again on differentiation into more specialised cell types, such as the neuronal population examined in the present study. The changes in mitochondrial biogenesis correlated with the changing contribution of oxidative phosphorylation to ATP output during differentiation. The higher maximal respiratory capacity of hESCs could be caused by the higher matrix-to-mitochondria and mitochondria-to-cell-volume ratios and the greater amounts of respiratory complexes, as indicated by the abundance of mtDNA and cytochrome c. Other studies have presented data suggesting that some indicators of mitochondrial content might increase overall during hESC differentiation in a mixed cell population differentiating in the presence of serum (Armstrong et al., 2010; Saretzki et al., 2008). This could reflect the preferential induction of other cell types, such as cardiomyocytes, in the presence of serum, but conclusions are difficult to draw on the basis of whole-cell fluorescent measurements because changes in cell size, and both mitochondrial and plasma membrane potentials, will affect these readings (see Fig. 4D). We have shown in the present study that, in a pure population of hESC-derived NSCs, mitochondrial content and activity are reduced compared with those of the parent cells. Further study will be needed to assess the mitochondrial and bioenergetic changes in other cell lineages. However, preliminary data from a cardiac differentiation model suggest a unique mitochondrial development for beating compared with non-beating cells (M.J.B., unpublished), highlighting further the importance of studying pure populations of cells.
From these observations, we inferred that hESCs might have a genetic programme that facilitates their high mitochondrial activity and identified expression patterns consistent with this for the ‘master regulators’ PGC-1α, PGC-1β and RIP140. The PGC-1 coactivators have been widely studied in other models and shown to be potent regulators of mitochondrial gene expression (Arany et al., 2007; Fritah et al., 2010; Lelliott et al., 2006; Lin et al., 2003; Meirhaeghe et al., 2003; Powelka et al., 2006; Seth et al., 2007). We observed high levels of expression of the mRNA encoding coactivators PGC-1α and PGC-1β in hESCs and, interestingly, also in the cancer cell line MCF7, pointing to potential commonalities in energy metabolism. MCF7 cells have been reported to obtain 80% of their ATP from oxidative phosphorylation (Guppy et al., 2002). Nearly all the other primary somatic cell types studied showed reduced expression of the mRNA encoding PGC-1α and PGC-1β, together with increased expression of that encoding the corepressor RIP140. Recent publications support these observations by showing that many other genes involved in mitochondrial biogenesis, including those encoding the mtDNA transcription factor TFAM and the mitochondrial RNA polymerase POLRMT, are also downregulated upon hESC and iPSC differentiation (Armstrong et al., 2010; Prigione et al., 2010). Whether these expression changes occur as a programme of differentiation or whether they change in response to reduced energy demand remains an unanswered question. Increasing energy demand in fibroblasts through mitochondrial uncoupling has been reported to induce the PGC-1 coactivators (Rohas et al., 2007). The expression levels of the PGC-1 coactivator could decrease if energy demand fell in the early stages of differentiation. However, as we see a strong induction of PGC-1α upon neuronal differentiation from NSCs, without any increase in ATP demand, and a return towards reliance on oxidative phosphorylation-generated ATP, a developmental genetic programme might also be a driving force in both of these transitions. Manipulation of PGC-1β or RIP140 in NSCs and HDFs was insufficient to drive a change in the bioenergetics, and so other genes might oppose or limit their effects in these cells.
Measurements of mitochondrial membrane potential showed that hESCs and NSCs had a similar Δψm of approximately −130 mV. The higher rate of oligomycin-sensitive respiration in hESCs in the absence of a significantly different mitochondrial membrane potential suggests a parallel increase in the rate of ATP-supplying and -demanding processes. Concerted regulation of demand and supply could be in favour of avoiding increases in Δψm and an associated increase in the production of reactive oxygen species (Brand, 2010; Murphy, 2009). A breakdown of ATP budgets revealed that in both hESCs and NSCs ATP was consumed through largely the same pathways. Protein and nucleic acid synthesis together accounted for the largest portion of the energy budget (hESCs, 41%; NSCs, 52% of mitochondrial respiration), indicating that most of the ATP drives macromolecule synthesis. As the total energy budget was greater in hESCs than in NSCs, this indicated a faster macromolecule synthesis rate in hESCs; however, in the absence of increased cell division, these extra macromolecules must ultimately be degraded or secreted. hESCs have been shown to secrete a diverse range of chemokines, cytokines, growth factors and matrix remodelling enzymes, along with cytosolic and cytoskeletal components (Bendall et al., 2009; LaFramboise et al., 2010). We found that hESCs had morphological indications of goblet-cell-like secretion and secreted significantly more protein into the extracellular environment than did NSCs. Further study is warranted to investigate the importance of this secretory activity. Secretion could be a driving force necessitating an increased rate of biosynthesis; however, an elevated biosynthesis rate in the absence of faster division might also drive secretion instead of increasing cell size. Goblet-cell-like hESCs have also been observed by Sathananthan and colleagues (Sathananthan et al., 2002). hESCs are known to divide relatively slowly – much slower than mouse ESCs – so it is plausible that in hESCs the division rate might not keep pace with the programme driving biosynthesis, but this hypothesis will require further study. The expression of potent oncoproteins such as c-Myc is a characteristic of ESCs and could be an example of such a programme. Myc itself is an important regulator of ribosome biogenesis and protein synthesis, and also a regulator of mitochondrial activity and biogenesis (Fan et al., 2010; Kim et al., 2008; Li et al., 2005; van Riggelen et al., 2010). The importance of biosynthesis and energy metabolism in ESCs has also been highlighted in recent studies (Dejosez et al., 2010; Wang et al., 2009).
In summary, we provide what we believe to be the first evidence that the energy demand for many cell processes in hESCs is elevated relative to their division rate and funded primarily by ATP generated through mitochondrial oxidative phosphorylation. Developing embryos of several species studied in vitro show a large increase in oxygen consumption at the blastocyst stage, during which protein synthesis is at its maximal rate, and it has been estimated that the majority of ATP comes from oxidative phosphorylation during this phase (Houghton et al., 1996; Sturmey and Leese, 2003; Thompson et al., 1996). In this respect, hESCs might mirror the metabolic characteristics of cells of the developing blastocyst during this important phase of expansion.
Materials and Methods
Cell culture and differentiation
hESC lines I6, H9 and BG01 were maintained as described previously (Zeng et al., 2004) or were grown on Geltrex-coated dishes (1:50) (Invitrogen, Carlsbad, CA) in the same medium previously conditioned for 24 hours on inactivated mouse embryonic fibroblasts (MEFs). B6 mouse ESCs were grown in the same unconditioned medium but supplemented with 1000 units/ml leukaemia inhibitory factor (LIF) (Millipore) instead of fibroblast growth factor 2 (FGF2). For hESC differentiation, colonies were detached with collagenase IV (1 mg/ml), triturated and suspended as embryoid bodies for up to 16 days or re-plated on Geltrex-coated dishes for up to 12 days, both in standard ES medium without FGF2. mESCs were differentiated as embryoid bodies in standard ES medium without LIF.
NSC derivation, culture and differentiation were performed as described previously (Swistowska et al., 2010; Swistowski et al., 2009). H9 hESC-derived NSCs were from Millipore. BJ fibroblasts were from the ATCC and grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing sodium pyruvate and L-glutamine (Invitrogen) plus 10% foetal bovine serum (FBS) and penicillin and streptomycin. All other cell types used for RNA expression analysis were from Invitrogen and cultured briefly using the medium recommended by the manufacturer before RNA isolation (for details, see supplementary material Table S1).
To calculate the cell generation times, NSCs were imaged at 24 hours after seeding, using phase-contrast microscopy in a humidified chamber with 5% CO2, at 4-minute intervals for 8 hours. At 24 hours after seeding, hESCs were labelled with 2 μg/ml Hoechst 33342 (Molecular Probes H3570) and fluorescent images were recorded at 8-minute intervals with minimal excitation intensity for 24 hours. Cells were randomly selected from several fields, tracked by eye and division or death events were recorded. The mean generation time and frequency of death were calculated.
Analysis of conditioned media
For the analysis of conditioned media, I6 hESCs or NSCs were grown to high density on Geltrex-coated 6-cm-diameter dishes in normal growth medium as above, rinsed twice and then incubated in DMEM with Ham's F12 plus Glutamax for 10 hours. After collection, the medium was centrifuged to remove debris, and the protein component was precipitated using a 1:8:1 ratio of medium, acetone and trichloroacetic acid, respectively, at −80°C for 1 hour. Precipitates were centrifuged at 18,000 g for 15 minutes at 4°C and resuspended in a volume of SDS-PAGE loading buffer proportional to the relative amounts of cellular protein harvested from the dish. Total cell lysates were made from the monolayers by using mammalian protein extraction reagent (Pierce) containing protease inhibitor cocktail (Roche). The cell lysate protein levels were quantified according to the Biuret method and confirmed on test gels with GelCode Coomassie Brilliant Blue staining (Thermo Scientific). Precipitated samples were subjected to SDS-PAGE alongside the protein lysates and stained as above. Gels were scanned and lane intensities were quantified using densitometry in ImageJ software (NIH).
Measurement of respiration and acidification rates using a Seahorse XF-24 analyser
Respiration and acidification rates were measured on adherent cells using a Seahorse XF-24 analyser (Seahorse Bioscience, North Billerica, MA). For hESCs and NSCs, the XF24 V7 assay plates were coated with Geltrex (1:50). Cells were seeded at approximately 24 hours before measurement in their normal growth medium; NSCs were seeded at 7×104 cells per well and hESCs as colonies totalling ~3×104 cells per well. The assay was performed in bicarbonate-free DMEM (Sigma D-5030) supplemented with 15 mM glucose, 2 mM L-glutamine, 0.5 mM sodium pyruvate and 0.4% BSA. Cells were washed twice and preincubated in this medium for 1 hour before measurement. Oligomycin was used at 0.2 μg/ml; carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) was titrated in four injections to 1.5 μM for hESCs, NSCs and D-35s, and 2 μM for HDFs; and rotenone and antimycin A were added at 1 μM and 2 μM, respectively. Oxygen consumption values were calculated from 3-minute measurement cycles by the XFReader software Version 1.4 updated with a recent correction (Gerencser et al., 2009). Acidification rates were taken as the mean rate from the second and third baseline readings. After the assay, a standard protein assay was performed. Alternatively, with the D-35 cells, some wells were fixed with 4% paraformaldehyde for immunocytochemical staining, as described below.
ATP demand assay
ATP demand was measured using the Seahorse XF-24 analyser as above with the addition of 10 mM HEPES to the assay medium. The basal respiration rate was recorded and then the selective inhibitors were injected and respiration recorded for the following 2–5 minutes. Small changes from vehicle-only controls, which were included on each plate, were subtracted to yield the specific effects of the inhibitors. The inhibitors used for each cellular process, and the concentrations tested, include: protein synthesis (cycloheximide, 0.01–50 μg/ml); DNA and/or RNA synthesis (actinomycin D, 0.1–10 μM); proteasome (MG132, 0.1–50 μM); tubulin dynamics (nocodazole, 0.1–50 μM); actin dynamics (latrunculin A, 0.1–3 μM); Na+/K+-ATPase (ouabain, 1 mM); sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) (thapsigargin, 1–1000 nM); plasma-membrane Ca2+-ATPase (PMCA) (LaCl3, 1–4 mM); and xanthine oxidase (allopurinol, 10–1000 μM). Mitochondrial respiration was subsequently fully inhibited with rotenone and antimycin A, as described above. All compounds were from Sigma, except MG132 (Calbiochem).
Wide-field imaging for determination of plasma and mitochondrial membrane potentials
Imaging was conducted in eight-well LabTek I (Nunc, Rochester, NY) coverglass chambers. For hESCs, the glass was coated with 0.1% gelatin and the colonies were grown on a feeder layer of MEFs. For NSCs the glass was coated with poly-ornithine (20 μg/ml) and then Geltrex (1:100). Tetramethylrhodamine methyl ester (TMRM) (7.5 nM; Invitrogen), plasma membrane potential indicator (PMPI) (1:200; no. R8042 FLIPR membrane potential assay kit from Molecular Devices; Sunnyvale, CA) and Na-tetraphenylborate (1 μM) were continuously present in the experimental buffer (120 mM NaCl, 1.75 mM KCl, 0.2 mM KH2PO4, 10 mM TES, 2.5 mM NaHCO3, 0.6 mM Na2SO4, 1.3 mM CaCl2, 2 mM MgCl2, 15 mM glucose, 2 mM L-glutamine and 0.5 mM Na-pyruvate, pH 7.4) at 37°C. Imaging was performed on a Nikon Eclipse Ti-PFS inverted fluorescence microscope controlled by NIS Elements 3.1 (Nikon, Melville, NY) and equipped with a S-Fluor 20×/0.75 NA lens, a Lambda LB-LS17 light source (Sutter Instruments, Novato, CA) a Cascade 512B camera (Photometrics, Tucson, AZ) and a MS-2000 motorised stage (ASI; Eugene, OR). The filter sets given as excitation–dichroic mirror–emission in nm were: for PMPI 500/24–520LP (long pass)–542/27 and for TMRM: 582–593LP (all from Semrock, Rochester, NY)–610LP (Omega Optical, Brattleboro, VT). Images were taken at 10-second intervals, typically cycling in the 2–12 stage positions. To establish the K+ equilibrium potential, a cocktail of drugs (KEC) was added including K+-channel activators: flupirtine (10 μM), NS309 (10 μM) and diazoxide (100 μM); chloride channel co-transport inhibitors: R-(+)-IAA94 (100 μM), R-(+)-DIOA (10 μM), bumetanide (80 μM); and the sodium channel inhibitor tetrodotoxin (1 μM). For calculation of Δψm mitochondria were completely depolarised with a cocktail (MDC) of valinomycin (1 μM), oligomycin (2 μM), FCCP (1 μM) and myxothiazol (1 μM). Complete cell depolarisation was induced with a cocktail (CDC) of ionophores: gramicidin (10 μM), nigericin (10 μM), monensin (10 μM) and the glycolysis inhibitor iodoacetate (500 μM). This cocktail also included the components of the MDC. Calculation of Δψp and Δψm from the measured fluorescence intensities were performed as described in the supplementary methods.
Confocal imaging of mitochondrial volume ratios
Cells were grown in LabTek I chambers as described above and were loaded with calcein-AM (1 μM; Invitrogen) and MitoTracker Red (hESC, 50 nM; NSC, D-35 and HDF, 25 nM; Invitrogen) in the above experimental buffer for 20 minutes, then imaged in experimental buffer with a Zeiss LSM 510 laser scanning confocal microscope. Single planes of 1024×1024 pixel images at 44 nm/pixel resolution were recorded using a Plan-Apochromat 100×/1.4 NA oil lens. Calcein and MitoTracker Red were excited simultaneously using the argon 488 nm and helium–neon 543 nm laser lines at high intensity, and fluorescence emissions were collected in the range of 500–530 nm and above 560 nm, respectively. To record serial optical sections without recording photo-damaged areas, random images were acquired at different x- and y-locations, and the z-plane was advanced from the bottom of the culture to the highest focal plane in steps of 1–1.5 μm. Images were binarised by highpass filtering and locally adaptive thresholding using Image Analyst MKII software (Image Analyst Software, Novato, CA). To calculate mitochondria-to-cell-volume ratios, the total number of mitochondrial pixels was divided by the total number of cytosolic pixels in the complete image set and multiplied by 0.67 – a stereologic correction factor (Weibel and Paumgartner, 1978) that accounts for the projection and clipping of mitochondria by the optical section. See the supplementary methods.
Transmission electron microscopy
Sample preparation and processing are described in the supplementary methods. For determination of matrix volume ratios, mitochondria were selected in a uniform random approach and imaged at ×105,000 magnification. Matrix-to-whole-mitochondrial-volume ratios were calculated by the Cavaileri estimator using Stereo-Investigator (MBF Bioscience; Williston, VT).
mtDNA copy number assessment
Absolute mtDNA copy number was assessed by comparing PCR amplification of a mitochondrial amplicon [human, NADH-ubiquinone oxidoreductase chain 1 (ND1); mouse, cytochrome b] with a nuclear amplicon (human, Δ- and/or γ-globin; mouse, glucagon) from DNA isolated using a Qiagen DNA mini kit. Primer sequences are in supplementary material Tables S2 and S3. DNA templates were made of regions spanning each amplicon by PCR amplification. Dilutions from 1×108 down to 1×104 copies of the isolated DNA were used to generate a standard curve for quantification.
Western blots and immunocytochemistry
Total cell lysates were made using the mammalian protein extraction reagent (Pierce) containing protease inhibitor cocktail (Roche). Gels were run and proteins transferred using standard techniques. Antibodies were: anti-cytochrome c (no. 556433, BD Pharmingen); anti-VDAC (no. MSA03, MitoSciences); anti-MnSOD (no. sc-30080, Santa Cruz Biotechnology); anti-CuZnSOD (no. sc-11407, Santa Cruz Biotechnology). Immunocytochemistry and staining procedures were performed as described previously (Zeng et al., 2003).
RNA isolation and cDNA synthesis for real-time PCR
RNA was isolated using TRIzol (Invitrogen) and was treated with DNase-I (Sigma). cDNA was synthesised using the Superscript III reverse transcriptase system (Invitrogen). Real-time PCR was performed using the PerfeCTa SYBR Green 2× mix (Quanta Bioscience) on an Applied Biosystems 7900HT fast real-time PCR machine. Primer details and sequences are presented in supplementary material Tables S4 and S5.
Lentiviral transduction
cDNA encoding PGC-1β was obtained from Open Biosystems (clone ID: 40146993) and cloned into a pLenti CMV/TO Neo vector (Addgene: 17292). The shRNA targeting RIP140 was constructed using sense: 5′-gcagcagtattctcgagaaGTGTGCTGTCCttctcgagaatactgctgc-3′ and antisense 5′-gcagcagtattctcgagaaGGACAGCACACttctcgagaatactgctgc-3′ sequences and expressed in a pLenti X2 Puro vector (Addgene number 17296). The uppercase nucleotides indicate the target sequence. The empty pLenti CMV/TO Neo vector or an shRNA against luciferase (Addgene: 21472) were used as controls. This lentiviral system has been described previously (Campeau et al., 2009). All vector inserts were sequenced to confirm identity. For packaging lentiviruses, human embryonic kidney HEK-293FT cells were used (Invitrogen). Lentiviral supernatants were concentrated using ultracentrifugation and titers were used to infect ~80–90% of the cells, as judged by a control virus with a cytomegalovirus promoter driving the expression of green fluorescent protein (GFP). Cells were selected with neomycin or puromycin for 1 week before analysis. Two separate derivations were made for I6 NSCs and HDFs for bioenergetic analyses.
Supplementary Material
Acknowledgments
This work was supported by the California Institute for Regenerative Medicine Grant CL1-00501-1 (X.Z.) and the Larry L. Hillblom Foundation (X.Z.). RS1-00163-1 (D.T.M., laboratory of Dale E. Bredesen), National Institutes of Health grants PL1 AG032118 (A.L.O., A.A.G., C.V. and M.D.B.), P01 AG025901, P30 AG025708 and R01 AG033542 (M.D.B.), the W. M. Keck Foundation (A.A.G. and M.D.B.), and The Ellison Medical Foundation (AG-SS-2288-09) (A.L.O. and M.D.B.). We thank Linzhao Cheng at the Johns Hopkins University for providing the iPSC line. We thank Eric Campeau at the University of Massachusetts for providing the lentiviral vectors. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/3/348/DC1
Supplementary methods available online at http://www.buckinstitute.org/sites/default/files/Birket_et_al_Supplementary_material.pdf
References
- Arany Z., Lebrasseur N., Morris C., Smith E., Yang W., Ma Y., Chin S., Spiegelman B. M. (2007). The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 5, 35-46 [DOI] [PubMed] [Google Scholar]
- Armstrong L., Hughes O., Yung S., Hyslop L., Stewart R., Wappler I., Peters H., Walter T., Stojkovic P., Evans J., et al. (2006). The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum. Mol. Genet. 15, 1894-1913 [DOI] [PubMed] [Google Scholar]
- Armstrong L., Tilgner K., Saretzki G., Atkinson S. P., Stojkovic M., Moreno R., Przyborski S., Lako M. (2010). Human induced pluripotent stem cell lines show similar stress defence mechanisms and mitochondrial regulation to human embryonic stem cells. Stem Cells 28, 661-673 [DOI] [PubMed] [Google Scholar]
- Becker K. A., Ghule P. N., Therrien J. A., Lian J. B., Stein J. L., van Wijnen A. J., Stein G. S. (2006). Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell. Physiol. 209, 883-893 [DOI] [PubMed] [Google Scholar]
- Becker K. A., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2010). Human embryonic stem cells are pre-mitotically committed to self-renewal and acquire a lengthened G1 phase upon lineage programming. J. Cell. Physiol. 222, 103-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall S. C., Hughes C., Campbell J. L., Stewart M. H., Pittock P., Liu S., Bonneil E., Thibault P., Bhatia M., Lajoie G. A. (2009). An enhanced mass spectrometry approach reveals human embryonic stem cell growth factors in culture. Mol. Cell. Proteomics 8, 421-432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B., Benvenisty N. (2009). The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle 8, 3822-3830 [DOI] [PubMed] [Google Scholar]
- Braam S. R., Passier R., Mummery C. L. (2009). Cardiomyocytes from human pluripotent stem cells in regenerative medicine and drug discovery. Trends Pharmacol. Sci. 30, 536-545 [DOI] [PubMed] [Google Scholar]
- Brand M. D. (2005). The efficiency and plasticity of mitochondrial energy transduction. Biochem. Soc. Trans. 33, 897-904 [DOI] [PubMed] [Google Scholar]
- Brand M. D. (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466-472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. T., Shih Y. R., Kuo T. K., Lee O. K., Wei Y. H. (2008). Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 26, 960-968 [DOI] [PubMed] [Google Scholar]
- Dejosez M., Levine S. S., Frampton G. M., Whyte W. A., Stratton S. A., Barton M. C., Gunaratne P. H., Young R. A., Zwaka T. P. (2010). Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev. 24, 1479-1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbordes S. C., Placantonakis D. G., Ciro A., Socci N. D., Lee G., Djaballah H., Studer L. (2008). High-throughput screening assay for the identification of compounds regulating self-renewal and differentiation in human embryonic stem cells. Cell Stem Cell 2, 602-612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D. J., Dash C., Bazett-Jones D. P., Le Grice S., McKay R. D., Buetow K. H., et al. (2008). Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2, 437-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facucho-Oliveira J. M., St John J. C. (2009). The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. Rep. 5, 140-158 [DOI] [PubMed] [Google Scholar]
- Fan Y., Dickman K. G., Zong W. X. (2010). Akt and c-Myc differentially activate cellular metabolic programs and prime cells to bioenergetic inhibition. J. Biol. Chem. 285, 7324-7333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige J. N., Auwerx J. (2007). Transcriptional coregulators in the control of energy homeostasis. Trends Cell. Biol. 17, 292-301 [DOI] [PubMed] [Google Scholar]
- Fritah A., Steel J. H., Nichol D., Parker N., Williams S., Price A., Strauss L., Ryder T. A., Mobberley M. A., Poutanen M., et al. (2010). Elevated expression of the metabolic regulator receptor-interacting protein 140 results in cardiac hypertrophy and impaired cardiac function. Cardiovasc. Res. 86, 443-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser A. A., Neilson A., Choi S. W., Edman U., Yadava N., Oh R. J., Ferrick D. A., Nicholls D. G., Brand M. D. (2009). Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal. Chem. 81, 6868-6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M., Leedman P., Zu X., Russell V. (2002). Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 364, 309-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock C. R. (1966). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 30, 269-297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Miller A., Mangada J., Liu Y., Swistowski A., Zhan M., Rao M. S., Zeng X. (2009). Identification by automated screening of a small molecule that selectively eliminates neural stem cells derived from hESCs but not dopamine neurons. PLoS ONE 4, e7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C., Spiegelman B. M. (2006). Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27, 728-735 [DOI] [PubMed] [Google Scholar]
- Hanna J., Cheng A. W., Saha K., Kim J., Lengner C. J., Soldner F., Cassady J. P., Muffat J., Carey B. W., Jaenisch R. (2010). Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 107, 9222-9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton F. D., Thompson J. G., Kennedy C. J., Leese H. J. (1996). Oxygen consumption and energy metabolism of the early mouse embryo. Mol. Reprod. Dev. 44, 476-485 [DOI] [PubMed] [Google Scholar]
- Kim J., Lee J. H., Iyer V. R. (2008). Global identification of myc target genes reveals its direct role in mitochondrial biogenesis and its e-box usage in vivo. PLoS ONE 3, e1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E., Eggan K. (2010). Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Invest. 120, 51-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFramboise W. A., Petrosko P., Krill-Burger J. M., Morris D. R., McCoy A. R., Scalise D., Malehorn D. E., Guthrie R. D., Becich M. J., Dhir R. (2010). Proteins secreted by embryonic stem cells activate cardiomyocytes through ligand binding pathways. J. Proteomics 73, 992-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelliott C. J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H. M., Bjursell M., Parker N., Curtis K., Campbell M., Hu P., et al. (2006). Ablation of PGC-1beta results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 4, e369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Wang Y., Zeller K. I., Potter J. J., Wonsey D. R., O'Donnell K. A., Kim J. W., Yustein J. T., Lee L. A., Dang C. V. (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 25, 6225-6234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Tarr P. T., Yang R., Rhee J., Puigserver P., Newgard C. B., Spiegelman B. M. (2003). PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 278, 30843-30848 [DOI] [PubMed] [Google Scholar]
- Mali P., Chou B. K., Yen J., Ye Z., Zou J., Dowey S., Brodsky R. A., Ohm J. E., Yu W., Baylin S. B., et al. (2010). Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 28, 713-720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirhaeghe A., Crowley V., Lenaghan C., Lelliott C., Green K., Stewart A., Hart K., Schinner S., Sethi J. K., Yeo G., et al. (2003). Characterization of the human, mouse and rat PGC1 beta (peroxisome-proliferator-activated receptor-gamma co-activator 1 beta) gene in vitro and in vivo. Biochem. J. 373, 155-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E., Misteli T. (2006). Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 7, 540-546 [DOI] [PubMed] [Google Scholar]
- Murphy M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. G. (2006). Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem. 281, 14864-14874 [DOI] [PubMed] [Google Scholar]
- Passier R., van Laake L. W., Mummery C. L. (2008). Stem-cell-based therapy and lessons from the heart. Nature 453, 322-329 [DOI] [PubMed] [Google Scholar]
- Powelka A. M., Seth A., Virbasius J. V., Kiskinis E., Nicoloro S. M., Guilherme A., Tang X., Straubhaar J., Cherniack A. D., Parker M. G., et al. (2006). Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J. Clin. Invest. 116, 125-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigione A., Fauler B., Lurz R., Lehrach H., Adjaye J. (2010). The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28, 721-733 [DOI] [PubMed] [Google Scholar]
- Reubinoff B. E., Itsykson P., Turetsky T., Pera M. F., Reinhartz E., Itzik A., Ben-Hur T. (2001). Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 19, 1134-1140 [DOI] [PubMed] [Google Scholar]
- Rohas L. M., St-Pierre J., Uldry M., Jager S., Handschin C., Spiegelman B. M. (2007). A fundamental system of cellular energy homeostasis regulated by PGC-1alpha. Proc. Natl. Acad. Sci. USA 104, 7933-7938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G., Walter T., Atkinson S., Passos J. F., Bareth B., Keith W. N., Stewart R., Hoare S., Stojkovic M., Armstrong L., et al. (2008). Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 26, 455-464 [DOI] [PubMed] [Google Scholar]
- Sathananthan H., Pera M., Trounson A. (2002). The fine structure of human embryonic stem cells. Reprod. Biomed. Online 4, 56-61 [DOI] [PubMed] [Google Scholar]
- Seth A., Steel J. H., Nichol D., Pocock V., Kumaran M. K., Fritah A., Mobberley M., Ryder T. A., Rowlerson A., Scott J., et al. (2007). The transcriptional corepressor RIP140 regulates oxidative metabolism in skeletal muscle. Cell Metab. 6, 236-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturmey R. G., Leese H. J. (2003). Energy metabolism in pig oocytes and early embryos. Reproduction 126, 197-204 [DOI] [PubMed] [Google Scholar]
- Swistowska A. M., da Cruz A. B., Han Y., Swistowski A., Liu Y., Shin S., Zhan M., Rao M. S., Zeng X. (2010). Stage-specific role for shh in dopaminergic differentiation of human embryonic stem cells induced by stromal cells. Stem Cells Dev. 19, 71-82 [DOI] [PubMed] [Google Scholar]
- Swistowski A., Peng J., Han Y., Swistowska A. M., Rao M. S., Zeng X. (2009). Xeno-free defined conditions for culture of human embryonic stem cells, neural stem cells and dopaminergic neurons derived from them. PLoS ONE 4, e6233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swistowski A., Peng J., Liu Q., Mali P., Rao M. S., Cheng L., Zeng X. (2010). Efficient generation of functional dopaminergic neurons from human induced pluripotent stem cells under defined conditions. Stem Cells 28, 1893-1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synnergren J., Giesler T. L., Adak S., Tandon R., Noaksson K., Lindahl A., Nilsson P., Nelson D., Olsson B., Englund M. C., et al. (2007). Differentiating human embryonic stem cells express a unique housekeeping gene signature. Stem Cells 25, 473-480 [DOI] [PubMed] [Google Scholar]
- Thompson J. G., Partridge R. J., Houghton F. D., Cox C. I., Leese H. J. (1996). Oxygen uptake and carbohydrate metabolism by in vitro derived bovine embryos. J. Reprod. Fertil. 106, 299-306 [DOI] [PubMed] [Google Scholar]
- Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., Jones J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145-1147 [DOI] [PubMed] [Google Scholar]
- Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B. M. (2006). Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 3, 333-341 [DOI] [PubMed] [Google Scholar]
- van Riggelen J., Yetil A., Felsher D. W. (2010). MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301-309 [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Alexander P., Wu L., Hammer R., Cleaver O., McKnight S. L. (2009). Dependence of mouse embryonic stem cells on threonine catabolism. Science 325, 435-439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel E. R., Paumgartner D. (1978). Integrated stereological and biochemical studies on hepatocytic membranes. II. Correction of section thickness effect on volume and surface density estimates. J. Cell Biol. 77, 584-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Neilson A., Swift A. L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., et al. (2007). Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292, C125-C136 [DOI] [PubMed] [Google Scholar]
- Zeng X. (2007). Human embryonic stem cells: mechanisms to escape replicative senescence? Stem Cell Rev. 3, 270-279 [DOI] [PubMed] [Google Scholar]
- Zeng X., Rao M. S. (2008). Controlled genetic modification of stem cells for developing drug discovery tools and novel therapeutic applications. Curr. Opin. Mol. Ther. 10, 207-213 [PubMed] [Google Scholar]
- Zeng X., Chen J., Sanchez J. F., Coggiano M., Dillon-Carter O., Petersen J., Freed W. J. (2003). Stable expression of hrGFP by mouse embryonic stem cells: promoter activity in the undifferentiated state and during dopaminergic neural differentiation. Stem Cells 21, 647-653 [DOI] [PubMed] [Google Scholar]
- Zeng X., Cai J., Chen J., Luo Y., You Z. B., Fotter E., Wang Y., Harvey B., Miura T., Backman C., et al. (2004). Dopaminergic differentiation of human embryonic stem cells. Stem Cells 22, 925-940 [DOI] [PubMed] [Google Scholar]
- Zhang S. C., Wernig M., Duncan I. D., Brustle O., Thomson J. A. (2001). In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129-1133 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}