

Abstract
Elevated glucocorticoid levels result in the transdifferentiation of pancreatic acinar cells into hepatocytes through a process that requires a transient repression of WNT signalling upstream of the induction of C/EBP-β. However, the mechanism by which glucocorticoid interacts with WNT signalling is unknown. A screen of microarray data showed that the serine/threonine protein kinase SGK1 (serum- and glucocorticoid-regulated kinase 1) was markedly induced in the model B-13 pancreatic rat acinar cell line after glucocorticoid treatment (which converts them into hepatocyte-like ‘B-13/H’ cells) and this was confirmed at the level of mRNA (notably an alternatively transcribed SGK1C form) and protein. Knockdown of SGK1 using an siRNA designed to target all variant transcripts inhibited glucocorticoid-dependent transdifferentiation, whereas overexpression of the human C isoform (and also the human SGK1F isoform, for which no orthologue in the rat has been identified) alone – but not the wild-type A form – inhibited distal WNT signalling Tcf/Lef transcription factor activity, and converted B-13 cells into B-13/H cells. These effects were lost when the kinase functions of SGK1C and SGK1F were mutated. Inhibition of SGK1 kinase activity also inhibited glucocorticoid-dependent transdifferentiation. Expression of SGK1C and SGK1F resulted in the appearance of phosphorylated β-catenin, and recombinant SGK1 was shown to directly phosphorylate purified β-catenin in vitro in an ATP-dependent reaction. These data therefore demonstrate a crucial role for SGK1 induction in B-13 cell transdifferentiation to B-13/H hepatocytes and suggest that direct phosphorylation of β-catenin by SGK1C represents the mechanism of crosstalk between glucocorticoid and WNT signalling pathways.
Keywords: Dexamethasone, Stem cell, Reprogramming, LY294002, Liver, Pancreas
Introduction
Transdifferentiation is a form of metaplasia and describes the irreversible switching of cellular differentiation in fully differentiated cells (Burke and Tosh, 2005; Wallace et al., 2010a). Administration of glucocorticoid to rats results in the appearance of hepatocytes in the pancreas (Wallace et al., 2009). In mice with elevated levels of circulating glucocorticoid, extensive areas of the exocrine (acinar) pancreas express hepatocyte markers, suggestive of widespread acinar transdifferentiation to hepatocytes (Wallace et al., 2010b). This pathological response of acinar cells to elevated glucocorticoid in vivo also occurs to a limited degree in primary cultures of rat acinar cells (Lardon et al., 2004) and efficiently in the AR42J-B-13 (B-13) rat pancreatic exocrine cell line (Shen et al., 2000; Marek et al., 2003), the latter providing a model to study the mechanism(s) involved.
Previous work has shown that transdifferentiation of B-13 cells into hepatocyte-like cells (defined as B-13/H cells) is dependent on induction of the CCAAT enhancer binding protein-β (C/EBP-β) (Shen et al., 2000). More recently, we have shown that a transient repression of WNT3a expression, WNT signalling and Tcf/Lef transcriptional activity in B-13 cells is an upstream event to induction of C/EBP-β (Wallace et al., 2010c). Thus, siRNA knockdown of β-catenin – the intracellular messenger for WNT signalling (Hoppler and Kavanagh, 2007) – substituted for glucocorticoid and led to an induction of C/EBP-β expression and transdifferentiation (Wallace et al., 2010c). Overexpression of a dominant-negative β-catenin protein blocked glucocorticoid-dependent transdifferentiation (Wallace et al., 2010c).
However, precisely how glucocorticoids interact with the WNT signalling pathway in B-13 cells remains obscure. It is known that some nuclear receptors – the super-family of receptor proteins that includes the glucocorticoid receptor (GR) – interact with components of the WNT signalling pathway (Mulholland et al., 2005). However, there are only a small number of investigations examining interactions of glucocorticoids with WNT signalling, and interactions appear to depend on the cell type examined. Thus, glucocorticoid exposure reduces the expression of β-catenin in pituitary cells (Spangler and Delidow, 1998), whereas it has no effect in osteoblasts (Smith et al., 2002).
The serum- and glucocorticoid-regulated kinases (SGKs) belong to a family of related serine/threonine kinases including protein kinase A, protein kinase G and protein kinase C (AGC kinases) (Firestone et al., 2003; Pearce et al., 2010). The SGK subfamily consists of three genes, SGK1, SGK2 and SGK3 (Pearce et al., 2010). SGK1 has been shown to be involved in the regulation of a number of ion channels, with sodium re-absorption in the kidney the best studied (for a review, see Lang et al., 2006). In the kidney, activation of the mineralocorticoid receptor results in the transcriptional induction of SGK1 expression. Phosphorylation of SGK1 through active phosphoinositide 3-kinase (PI3K) and mTorC signalling then renders SGK1 functionally active as a kinase (Pearce et al., 2010). This results in phosphorylation of the ubiquitin ligase Nedd4-2, a block in epithelial Na+ channel (ENaC) ubiquitylation, a reduction in ENaC endocytosis and increased renal tubular epithelial Na+ transport into the cell (Lang et al., 2006). SGK1 might also increase expression of ENaCs (Boyd and Naray-Fejes-Toth, 2005). The predominant steroidal regulator of SGK1 expression in the kidney is the mineralocorticoid. Although glucocorticoids activate the mineralocorticoid receptor, rapid oxidation by HSD11B2 in the kidney ensures that the mineralocorticoid receptor primarily responds to mineralocorticoid and not glucocorticoid levels (Seckl and Walker, 2001).
Despite a clear functional role for SGK1 in the kidney, SGK1 is also constitutively expressed in a wide range of other tissues. Expression is also upregulated by a diverse range of factors in addition to glucocorticoids and mineralocorticoids, including 1,25 dihidroxyvitamin D3 (Akutsu et al., 2001), TGFβ (Waldegger et al., 1999), PDGF (Mizuno and Nishida, 2001), PPARγ activators (Hong et al., 2003) and osmotic stress (Waldegger et al., 1997). Placement of SGK1 in a functional context in other tissues is therefore problematical. Indeed, Sgk1−/− mice show no overt phenotype. Only when these mice are placed on a low NaCl diet does a lack of SGK1 have an effect, which is restricted to salt wasting (Wulff et al., 2002; Fejes-Tóth et al., 2008). The most notable feature of SGK1 that sets it apart from other AGC kinases is the rapid and marked change in expression levels seen with SGK1 after exposure of cells to transcriptional regulators (Firestone et al., 2003). Because many of the regulators of SGK1 expression are associated with stressful cellular conditions, it is likely that SGK1 has an ancillary role in fine-tuning kinase-dependent control under stress conditions. SGK2 and SGK3 are closely related to SGK1 in terms of amino acid sequence, but are not inducible by glucocorticoid or serum (Kobayashi et al., 1999).
The data in this paper demonstrate that transdifferentiation of B-13 cells is dependent on an interaction of glucocorticoid with the GR – and not the closely related mineralocorticoid receptor (MR) – and induction of SGK1. SGK1 was actively phosphorylated in B-13 cells by a PI3K-dependent mechanism and we show that SGK1 phosphorylates β-catenin in vitro. These data therefore suggest that B-13 cell transdifferentiation to B-13/H cells in response to glucocorticoid is dependent on SGK1 induction, and its crosstalk with the WNT signalling pathway is through direct phosphorylation of β-catenin by SGK1.
Results
B-13 transdifferentiation is dependent on interaction with the glucocorticoid receptor and not the mineralocorticoid receptor
Glucocorticoids activate both the GR and the MR (Arriza et al., 1987). The presence and activity of 11β-hydroxysteroid dehydrogenase (HSD11B) enzymes 1 and 2 determine the extent to which glucocorticoids remain active GR and MR ligands (Seckl and Walker, 2001). Expression and activity of HSD11B2 ensures that glucocorticoids do not activate the MR in selected tissues (such as the kidney) (Fig. 1A). Fig. 1B confirms that B-13 cells expressed the GR and expressed barely detectable levels of MR. Treatment with the glucocorticoid dexamethasone (DEX) resulted in transdifferentiation of B-13 cells to B-13/H cells, as judged by the expression of liver markers CYP2E1 and albumin (Fig. 1C) and to a marked change in morphology (see later). The GR antagonist mifepristone (RU486) prevented transdifferentiation (and morphological changes – data not shown), whereas the MR antagonist spironolactone had no effect (Fig. 1C), suggesting the effects of DEX on transdifferentiation are mediated entirely through the GR. This is supported by a failure of the natural MR agonist aldosterone (ALD) to promote transdifferentiation (Fig. 1C). Fludrocortisone (FLUD) is a mixed GR and MR agonist and Fig. 1C demonstrates that only RU486 – and not spironolactone – blocked transdifferentiation (confirming that MR does not have a redundant role in transdifferentiation when GR activity is inhibited).
Fig. 1.

Transdifferentiation of B-13 cells into B-13/H cells is dependent on glucocorticoid interaction with the glucocorticoid receptor, not the mineralocorticoid receptor. (A) schematic diagram of glucocorticoid metabolism and interaction with the GR and MR. In aldosterone-selective tissues such as the kidney and placenta, 11β-hydroxysteroid dehydrogenase 2 (HSD11B2) oxidises glucocorticoids to an inactive keto form, reducing glucocorticoid activation of the MR. In tissues such as the liver, 11β-hydroxysteroid dehydrogenase 1 (HSD11B1) promotes high concentrations of active glucocorticoid, ensuring these tissue are responsive to circulating glucocorticoid levels. (B) RT-PCR analysis of the expression of the indicated transcript using primers listed in Table 1. (C) Western blot showing the expression of the indicated liver markers plus β-actin as loading control. B-13 cells were cultured with the indicated additions made from 1000-fold ethanol stocks. Control cells received 0.1% (v/v) ethanol. Medium and additions were renewed every 2–3 days and cells were harvested for western blotting analysis (30 μg total protein/lane) after 14 days of treatment. FLUD, fludrocortisone; ALD, aldosterone. All data are typical of at least three separate experiments.
SGK1 is induced by DEX treatment in B-13 cells
Microarray data has shown that the transdifferentiation of B-13 cells into B-13/H cells is a co-ordinated change in gene expression that results in a qualitative and quantitative expression that is similar to that of primary hepatocytes (Wallace et al., 2009). Further interrogation of this microarray data indicated that there was a marked (>100-fold) increase in Sgk1 mRNA transcripts in B-13/H cells, contrasting with few changes in the expression levels of mRNAs encoding many components of the WNT and PI3K signalling pathways (supplementary material Fig. S1), which converge at glycogen synthase kinase 3 (GSK3) phosphorylation and potentially regulate β-catenin levels in B-13 cells (see later).
The induction of all known rat Sgk1 isoforms (for sequence comparison, see supplementary material Fig. S2) in B-13/H cells, with the Sgk1c mRNA isoform induced from undetectable levels in B-13 cells is shown in Fig. 2A. In the Tg(Crh) mouse model of Cushing's disease, which results in widespread expression of hepatocyte-specific gene expression in the acinar pancreas by 21 weeks of age (Wallace et al., 2010b), Sgk1c was also induced from undetectable levels in the pancreas (Fig. 2B). Induction of Sgk1 transcripts in B-13/H and Tg(Crh) also resulted in an induction of SGK1 proteins (Fig. 2C). Induction of Sgk1c mRNA and SGK1 protein occurs in B-13 cells before induction of C/EBP-β, hepatocyte marker gene expression and conversion to B-13/H cells (Fig. 2D,E). The possibility arises therefore, that SGK1 induction could be a crucial upstream event that controls B-13 transdifferentiation to B-13/H cells.
Fig. 2.

Transdifferentiation of B-13 cells into B-13/H cells is associated with an induction of SGK1 gene expression. (A) RT-PCR analysis of the indicated transcript using primers as outlined in Table 1 (see also supplementary material Fig. S2 illustrating 5′ region alignment of alternative transcribed Sgk1 cDNA sequences and positions of selective upstream primers). (B) RT-PCR analysis of the indicated mouse mRNA transcripts in pairs of age-matched wild-type (W/T) and Tg(Crh) female mouse liver, pancreas and kidney tissues. (C) Western blot of SGK1 expression (30 μg total protein/lane). (D) Time course for the induction of the indicated transcripts after treatment with DEX for the indicated number of days. (E) Time course for the induction of the indicated protein after treatment with DEX for the indicated number of days. All data are typical of at least three separate experiments.
SGK1 expression promotes B-13 transdifferentiation to B-13/H cells
A specific chemical inhibitor for SGK1 is not currently available. An inhibition of SGK1 expression was therefore used to determine whether SGK1 has a role in transdifferentiation. An siRNA designed to promote the degradation of mRNA encoding C/EBP-β inhibited DEX-dependent C/EBP-β induction and transdifferentiation (Fig. 3A), as predicted from previous work which has shown that overexpression of C/EBPβ in B-13 cells substitutes for the effects of DEX and promotes transdifferentiation (Shen et al., 2000). Transfection with Sgk1 siRNA similarly inhibited DEX-dependent SGK1 induction and transdifferentiation (Fig. 3B) whereas an siRNA designed to knock down expression of the low-affinity glucocorticoid binding protein (LAGS), used as a control, had no effect (Fig. 3C).
Fig. 3.
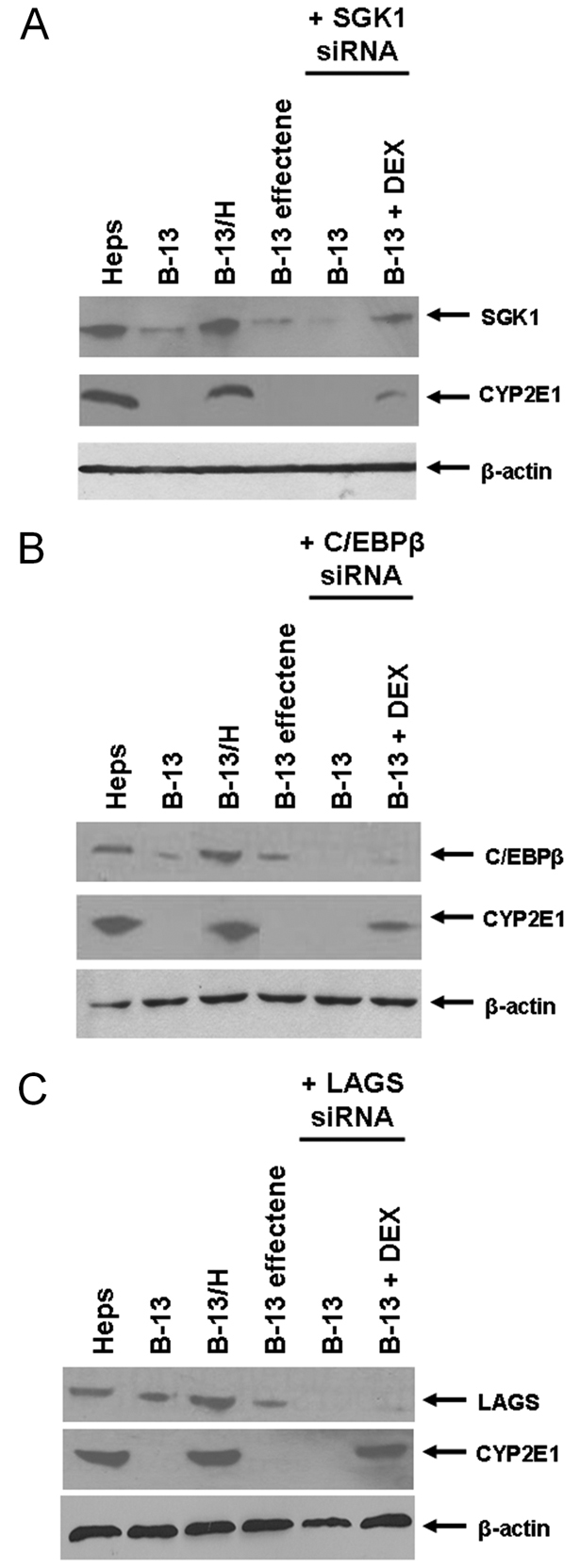
siRNA knockdown of SGK1 expression inhibits DEX-dependent transdifferentiation of B-13 cells to B-13/H cells. B-13 cells cultured in six-well plates were transfected with 4 μg of siRNA designed to inhibit the expression of (A) C/EBP-β, (B) SGK1 or (C) LAGS. After 7 days, cells were harvested and analysed by western blotting for the indicated protein. All data are typical of at least three separate experiments.
To further test the hypothesis that SGK1 induction is involved in transdifferentiation, B-13 cells were transfected with a series of human SGK1 expression constructs (SGK1 genes are highly homologous between species; rat and human SGK1A isoforms show 97% identical amino acid homology with only three functional amino acid differences over the entire 431 amino acids; see supplementary material Fig. S3). Fig. 4A demonstrates that expression of either the human SGK1C (orthologous to the C form in rat) or SGK1F (which has no orthologue in the rat) isoforms alone resulted in a transdifferentiation of B-13 cells. Fig. 4B demonstrates that SGK1C and SGK1F also significantly inhibited the transcriptional activity of Tcf/Lef to levels similar to those obtained with DEX treatment, in contrast to the SGK1A and SGK1D isoforms.
Fig. 4.

Expression of human SGK1C or SGK1F isoforms in B-13 cells promotes the transdifferentiation of B-13 cells to B-13/H cells. B-13 cells cultured in six-well plates were transfected with 2 μg of the indicated plasmid construct [plus 2 μg of either Topflash (T) or Fopflash (F) combined with 0.02 μg of transfection control Renilla reporter vector] or treated with 10 nM DEX for 14 days to generate B-13/H cells. 14 days after transfection, cells were harvested and analysed by western blotting (A,C) or analysed for expression of luciferase activities (B,D). KD, kinase-dead. *P<0.05, compared with Topflash/RL-TK-transfected (i.e. no expression construct) B-13 cells using ANOVA (two-tailed).
To establish whether the kinase function of SGK1C and SGK1F is required for promoting B-13 transdifferentiation, mutant variants of each protein were generated. Work by others has demonstrated that phosphorylation of SGK1A at Ser422 by mTorC2 (Garcia-Martinez and Alessi, 2008) enables 3-phosphoinositide-dependent protein kinase-1 (PDK1, activated via the PI3K pathway) to phosphorylate the T-loop (Biondi et al., 2001), which results in a functional kinase (Pearce et al., 2010). However, when these residues were mutated in SGK1 isoforms to alanines, the proteins were unstable when expressed in B-13 cells and expression could not be reliably and repeatedly confirmed (data not shown). Mutating the lysine residue in SGK1A at residue 127 impedes binding of ATP and also results in a kinase-dead (KD) protein (Park et al., 1999). SGK1 isoforms were therefore generated with this residue mutated to a methionine.
The kinase function of SGK1C and F isoforms is essential for promoting the transdifferentiation of B-13 cells to B-13/H cells because KD mutant SGK1C and SGK1F proteins failed to inhibit Tcf/Lef transcriptional function or induce expression of CYP2E1 or albumin (Fig. 4C,D) in contrast to the wild-type equivalents (Fig. 4A,B).
The PI3K pathway has an ancillary role in B-13 transdifferentiation to B-13/H cells
The PI3K pathway is activated by a range of growth factors and has been shown to lead to the phosphorylation of SGK1 in the T loop (at Thr256 of the wild-type protein) as part of its modification to an active kinase (Biondi et al., 2001). To further determine whether endogenous SGK1 phosphorylation and kinase activity is required for B-13 transdifferentiation, cells were treated with the PI3K inhibitor LY294002.
LY294002 treatment alone did not promote transdifferentiation to B-13/H cells because there was no change in morphology of the cells (Fig. 5A) or expression of hepatic markers, in contrast to DEX treatment (Fig. 5B). LY294002 might have had a weak glucocorticoid effect because there was a small induction of SGK1 and a less potent reduction in total β-catenin levels compared with DEX, but this did not result in any overt change in B-13 cell differentiation. However, LY294002 treatment inhibited the phosphorylation of SGK1 induced by DEX at both activating positions, reduced the levels of phosphorylated β-catenin and inhibited the degree to which B-13 cells expressed liver markers by ~50% (Fig. 5B), indicating that SGK1 phospho-activation correlates with B-13 transdifferentiation to B-13/H cells.
Fig. 5.

Inhibition of PI3K signalling inhibits SGK1 phosphorylation and B-13 transdifferentiation. (A) B-13 cells were treated with or without DEX (10 nM) or LY294002 (5 μM). After 14 days, cells appeared typically as shown. (B) Cells harvested and analysed for the indicated protein by western blotting. CPS, carbamoyl phosphate synthase I. Data are typical of at least three independent experiments. (C) Schematic diagram illustrating overlap between the WNT and PI3K pathways.
SGK1C and SGK1F phosphorylate β-catenin
Previous work has demonstrated that β-catenin phosphorylation, which targets the protein for degradation (Hoppler and Kavanagh, 2007), and reductions in the levels of β-catenin are early upstream events to C/EBP-β induction and B-13 transdifferentiation to B-13/H cells (Wallace et al., 2010c). It was therefore hypothesised that expression of SGK1C and SGK1F or induction of endogenous SGK1C resulted in phosphorylation of β-catenin. Thus, B-13 cells were transfected with SGK1C, SGK1F or SGK1A (as a control) and the effects on β-catenin examined. Expression of both SGK1C and SGK1F resulted in phosphorylation of β-catenin and reduced levels of total β-catenin, whereas the SGK1A isoform had no effect (Fig. 6A).
Fig. 6.

SGK1 can directly phosphorylate β-catenin. (A) B-13 cells cultured in six-well plates were transfected with 2 μg of the indicated plasmid construct or treated with 10 nM DEX for 14 days to generate B-13/H cells. After 14 days, cells were harvested and analysed for the indicated protein. (B) In vitro phosphorylation of β-catenin. Western blot for the indicated protein after incubation. Results presented are typical of three independent experiments.
To determine whether SGK1 could directly phosphorylate β-catenin (rather than activate other kinases that mediate the phosphorylation of β-catenin), purified recombinant SGK1 was incubated with purified recombinant β-catenin and phosphorylation was examined by western blotting. β-catenin phosphorylation only occurred when SGK1 and ATP were present (Fig. 6B).
Discussion
The data in this paper demonstrate that the transdifferentiation of B-13 cells into B-13/H cells is glucocorticoid dependent, mediated via the glucocorticoid receptor and dependent on an induction of SGK1 expression. siRNA-mediated knockdown of SGK1 and overexpression of SGK1 inhibited and promoted (without the requirement for glucocorticoid) transdifferentiation, respectively, unequivocally demonstrating a regulatory role for SGK1 in the response. Our data also suggest that the mechanism is likely to involve crosstalk with the WNT signalling pathway, which has previously been shown to regulate B-13 cell transdifferentiation (Wallace et al., 2010c), because overexpression of SGK1 inhibited transcriptional activity of distal WNT signalling, an effect that was absent when the SGK1 kinase function was blocked. The transdifferentiation of pancreatic acinar cells to hepatocyte-like cells in primary culture (Shen et al., 2000; Lardon et al., 2004; Sumitran-Holgersson et al., 2009) and in vivo (Wallace et al., 2009; Wallace et al., 2010c) is therefore likely to be a pathological response to stressful conditions that could be mediated, in part, by an induction of SGK1.
Interestingly, only selective variants of SGK1 were effective regulators of B-13 transdifferentiation. Expression of wild-type SGK1A had no effect. This suggests that the data are unlikely to be associated with a non-specific effect, such as the overexpression of a kinase. The various SGK1 forms differ only in their N-terminal sequences, suggesting that the ability to promote transdifferentiation is dependent on a unique interaction of the N-terminus (in SGK1C and SGK1F) with an unknown factor(s). Because SGK1 is capable of phosphorylating the intracellular WNT signalling mediator β-catenin, it is likely that the N-terminal sequence in these isoforms are crucial (in a cellular context) for effective β-catenin phosphorylation, perhaps by providing a direct interaction with β-catenin. There is however, no significant similarity between the N terminal sequences of SGK1C and SGK1F (data not shown). Despite this lack of N terminal similarity, both proteins required an intact functional kinase function for effective promotion of B-13 transdifferentiation to B-13/H cells.
The crucial role of SGK1 in transdifferentiation is also supported by the requirement for PI3K signalling. This would be expected to inhibit transdifferentiation of B-13 cells in the absence of glucocorticoid exposure through the PKB/Akt-dependent phosphorylation and inactivation of GSK3 kinase activity and therefore the accumulation of β-catenin (Liu et al., 2009) (see also Fig. 5C). However, the PI3K pathway also phosphorylates and activates SGK1 by PDK1 (Biondi et al., 2001; Alessi, 2001; Mora et al., 2004). Addition of the PI3K inhibitor LY294002 inhibited DEX-dependent transdifferentiation. This also suggests that induction of SGK1 is crucial for transdifferentiation, either to overcome the effects of the PI3K pathway on GSK3, or to generate an isoform of SGK1 specific for this function (such as the rat SGK1C orthologue, which is induced from undetectable levels by DEX treatment).
There might be targets of SGK1 in B-13 cells other than β-catenin that are involved in the transdifferentiation response because many of the targets for SGK1 phosphorylation are shared with other AGC kinase members (Lang et al., 2006). One potential target is forkhead box O1 which is the target for SGK1 in pre-adipocytes and is reported to regulate differentiation to fat cells (Di Petro et al., 2010). Another target might be N-myc downstream regulated genes (NDRGs), which are unique targets of SGK1 (Murray et al., 2004). Indeed, NDRG isoform 4A2 has recently been implicated in rat pancreatic duct cell differentiation (Wang and Hill, 2009).
This paper therefore shows that SGK1 is implicated in the transdifferentiation of B-13 cells into B-13/H hepatocyte-like cells and adds knowledge to the changes that occur in cells under pathophysiological conditions. These data also give an insight into potential ways to manipulate cell differentiation that might be of particular use in the drive to generate hepatocytes in vitro for a wide range of uses, from toxicological screening to bioartificial livers.
Materials and Methods
Cell isolation and culture
B-13 cells were routinely cultured in Dulbecco's modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal calf serum, 80 μg/ml penicillin and 80 μg/ml streptomycin as previously outlined and under which conditions the cells remained proliferative and phenotypically stable (Marek et al., 2003; Wallace et al., 2010c). Treatment with 10 nM DEX results in the transdifferentiation to hepatocyte-like cells which is maximal (75–85% of all cells) after 14 days, whereupon these cells are referred to as B-13/H cells/cultures (Wallace et al., 2010c). Rat hepatocytes were isolated from adult male 250–300 g body weight Sprague–Dawley rats by collagenase perfusion (Wright et al., 2001) with additional tissues harvested and snap frozen when required.
Animals
Tg(Crh) transgenic mice overexpressing a rat corticotrophin releasing factor transgene that results in chronic elevated circulating glucocorticoid and Cushing's disease (Stenzel-Poore et al., 1992; Wallace et al., 2010b) were obtained from the Jackson Laboratory (Bar Harbor, ME). Female adult mice were used in all studies (since only males could be used for successful mating with wild type females). Mice showed overt clinical signs of Cushing's syndrome within 15 weeks of age (e.g. hair loss, obesity, thinning skin). Animals were killed at 21 weeks of age by cervical dislocation and tissues removed for analyses. Littermate wild-type females were used as controls.
RT-PCR
Total RNA was purified from cells using Trizol (Invitrogen, Paisley, UK) and RT-PCR performed as previously outlined (Haughton et al., 2006). Primer sequences are given in Table 1.
Table 1.
DNA oligonucleotide sequences employed in RT-PCR or PCR genotyping

Western blotting
Western blotting was performed essentially as previously outlined (Marek et al., 2005) with the following antibodies: rabbit anti-human SGK1 (#S5188, raised to a synthetic peptide corresponding to the C-terminus of human SGK1A, amino acids 412–431) purchased from Sigma (Poole, UK); anti-SGK1-P422 (sc16745-R) obtained from Santa Cruz Biotechnology (Santa Cruz, CA). All other antibodies have previously been detailed (Wallace et al., 2010c). Detection was achieved using appropriate horseradish-peroxidase-conjugated anti-IgG secondary antibodies and chemiluminescence (ECL, GE Healthcare, Amersham, UK).
siRNA transfection
Cells were transfected with siRNAs purchased from Qiagen (Southampton, UK) using Effectene as previously described (Wallace et al., 2010c) using siRNA designed to knock down rat C/EBP-β (Rn-Cebpb-4/SI01498028); rat SGK1 (Rn-sgk1-6/SI04717524) or rat low-affinity glucocorticoid binding site (LAGS) also referred to as progesterone receptor membrane component 1 (Rn-Pgrmc1-2/SI02900023) a gene that encodes a glucocorticoid and progesterone binding protein of unknown function (Marek et al., 2009) and used as a control.
Plasmid DNA transfection
Cells were transfected with a range of plasmid DNA constructs (see Table 2) using Effectene (Qiagen, Southampton, UK) as outlined by the manufacturers. In all cases, cells were co-transfected with a Renilla expression vector (RL-TK) obtained from Promega (Southampton, UK) at a ratio of 6:1 (TK plasmid: RL-TK plasmid). Luciferase and Renilla activities were determined using the Dual-Luc kit (Promega, Southampton, UK) and a luminometer.
Table 2.
Plasmid DNA constructs employed

In vitro phosphorylation of β-catenin
5 ng purified (ΔNT60) N-terminal GST-tagged human SGK1 (Stressgen, Victoria, Canada) was added to 5 μg recombinant N-terminal GST-tagged full-length human β-catenin (SignalChem, Richmond, Canada) in a final volume of 30 μl containing 6.7 mM 3-(N-morpholino) propane sulfonic acid (MOPS) buffer, 1.7 mM EGTA, 0.67 mM EDTA, 4.2 mM sodium β-glycerophosphate, 6.7 mM MgCl2, 83 μM dithiothreitol and 1.7 μM β-methyl aspartic acid, pH 7.2. When required, ATP was added at a final concentration of 50 μM. Reactions were initiated by the addition of β-catenin and incubated for 60 minutes at 30°C before heating at 95°C in SDS-PAGE loading buffer to denature all components before being subjected to SDS-PAGE and western blotting analysis.
Statistics
One-way ANOVA was used to test for statistical significance.
Supplementary Material
Acknowledgments
Supported by a grant from Newcastle University. K.W. was supported by a BBSRC PhD Studentship award. E.A.F. is currently a recipient of an MRC ITTP Studentship. The technical assistance of Trevor Booth with confocal microscopy is gratefully acknowledged. The expertise and assistance of Trevor R. Jackson is acknowledged. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/3/405/DC1
References
- Akutsu N., Lin R., Bastien Y., Bestawros A., Enepekides D. J., Black M. J., White J. H. (2001). Regulation of gene expression by 1alpha,25-dihydroxyvitamin D3 and its analog EB1089 under growth-inhibitory conditions in squamous carcinoma cells. Mol. Endocrinol. 15, 1127-1139 [DOI] [PubMed] [Google Scholar]
- Alessi D. R. (2001). Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem. Soc. Trans. 29, 1-14 [DOI] [PubMed] [Google Scholar]
- Arriza J. L., Weinberger C., Cerelli G. (1987). Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science 237, 268-275 [DOI] [PubMed] [Google Scholar]
- Biondi R. M., Kieloch A., Currie R. A., Deak M., Alessi D. R. (2001). The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 20, 4380-4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd C., Naray-Fejes-Toth A. (2005). Gene regulation of ENaC subunits by serum- and glucocorticoid-inducible kinase-1. Am. J. Physiol. Renal. Physiol. 288, F505-F512 [DOI] [PubMed] [Google Scholar]
- Burke Z. D., Tosh D. (2005). Therapeutic potential of transdifferentiated cells. Clin. Sci. 108, 309-321 [DOI] [PubMed] [Google Scholar]
- Di Pietro N., Panel V., Hayes S., Bagattin A., Meruvu S., Pandolfi A., Hugendubler L., Fejes-Tóth G., Naray-Fejes-Tóth A., Mueller E. (2010). Serum- and glucocorticoid-inducible kinase 1 (SGK1) regulates adipocyte differentiation via forkhead box O1. Mol. Endocrinol. 24, 370-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes-Tóth G., Frindt G., Náray-Fejes-Tóth A., Palmer L. G. (2008). Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal. Physiol. 294, F1298-F1305 [DOI] [PubMed] [Google Scholar]
- Firestone G. L., Giampaolo J. R., O'Keeffe B. A. (2003). Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell. Physiol. Biochem. 13, 1-12 [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez J. M., Alessi D. R. (2008). mTOR complex-2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum and glucocorticoid induced protein kinase-1 (SGK1). Biochem. J. 416, 375-385 [DOI] [PubMed] [Google Scholar]
- Haughton E. L., Tucker S. J., Marek C. J., Durward E., Leel V., Bascal Z., Monaghan T., Koruth M., Collie-Duguid E., Mann D. A., et al. (2006). Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology 131, 194-209 [DOI] [PubMed] [Google Scholar]
- Hong G., Lockhart A., Davis B., Rahmoune H., Baker S., Ye L., Thompson P., Shou Y., O'Shaughnessy K., Ronco P., et al. (2003). PPARgamma activation enhances cell surface ENaCalpha via up-regulation of SGK1 in human collecting duct cells. FASEB J. 17, 1966-1968 [DOI] [PubMed] [Google Scholar]
- Hoppler S., Kavanagh C. L. (2007). Wnt signalling: variety at the core. J. Cell Sci. 120, 385-393 [DOI] [PubMed] [Google Scholar]
- Kobayashi T., Deak M., Morrice N., Cohen P. (1999). Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem. J. 344, 189-197 [PMC free article] [PubMed] [Google Scholar]
- Lang F., Böhmer C., Palmada M., Seebohm G., Strutz-Seebohm N., Vallon V. (2006). (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol. Rev. 86, 1151-1178 [DOI] [PubMed] [Google Scholar]
- Lardon J., De Breuck S., Rooman I., Van Lommel L., Kruhøffer M., Orntoft T., Schuit F., Bouwens L. (2004). Plasticity in the adult rat pancreas: transdifferentiation of exocrine to hepatocyte-like cells in primary culture. Hepatology 39, 1499-1507 [DOI] [PubMed] [Google Scholar]
- Liu P., Cheng H., Roberts T. M., Zhao J. J. (2009) Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627-644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek C. J., Cameron G. A., Elrick L. J., Hawksworth G. M., Wright M. C. (2003). Generation of hepatocytes expressing functional cytochromes P450 from a pancreatic progenitor cell line in vitro. Biochem. J. 370, 763-769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek C. J., Tucker S. J., Konstantinou D. K., Elrick L. J., Haefner D., Sigalas C., Murray G. I., Goodwin B., Wright M. C. (2005). Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem. J. 387, 601-608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek C. J., Wallace K., Durward E., Koruth M., Leel V., Leiper L. J., Wright M. C. (2009). Low affinity glucocorticoid binding site ligands as potential anti-fibrogenics. Comp. Hepatol. 8, 1-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H., Nishida E. (2001). The ERK MAP kinase pathway mediates induction of SGK (serum- and glucocorticoid-inducible kinase) by growth factors. Genes Cells 6, 261-268 [DOI] [PubMed] [Google Scholar]
- Mora A., Komander D., van Aalten D. M., Alessi D. R. (2004). PDK1, the master regulator of AGC kinase signal transduction. Semin. Cell Dev. Biol. 15, 161-170 [DOI] [PubMed] [Google Scholar]
- Mulholland D. J., Dedhar S., Coetzee G. A., Nelson C. C. (2005). Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr. Rev. 26, 898-915 [DOI] [PubMed] [Google Scholar]
- Murray J. T., Campbell D. G., Morrice N., Auld G. C., Shpiro N., Marquez R., Peggie M., Bain J., Bloomberg G. B., Grahammer F., et al. (2004). Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477-488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Leong M. L., Buse P., Maiyar A. C., Firestone G. L., Hemmings B. A. (1999). Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 18, 3024-3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce L. R., Komander D., Alessi D. R. (2010). The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9-22 [DOI] [PubMed] [Google Scholar]
- Seckl J. R., Walker B. R. (2001). Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 142, 1371-1376 [DOI] [PubMed] [Google Scholar]
- Shen C. N., Slack J. M., Tosh D. (2000). Molecular basis of transdifferentiation of pancreas to liver. Nat. Cell Biol. 2, 879-887 [DOI] [PubMed] [Google Scholar]
- Simon P., Schneck M., Hochstetter T., Koutsouki E., Mittelbronn M., Merseburger A., Weigert C., Niess A., Lang F. (2007). Differential regulation of serum- and glucocorticoid-inducible kinase 1 (SGK1) splice variants based on alternative initiation of transcription. Cell Physiol. Biochem. 20, 715-728 [DOI] [PubMed] [Google Scholar]
- Smith E., Coetzee G. A., Frenkel B. (2002). Glucocorticoids inhibit cell cycle progression in differentiating osteoblasts via glycogen synthase kinase-3beta. J. Biol. Chem. 277, 18191-18197 [DOI] [PubMed] [Google Scholar]
- Spangler P. R., Delidow B. C. (1998). Co-regulation of pituitary tumor cell adhesion and prolactin gene expression by glucocorticoid. J. Cell Physiol. 174, 115-124 [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore M. P., Cameron V. A., Vaughan J., Sawchenko P. E., Vale W. (1992). Development of Cushing's syndrome in corticotropin-releasing factor transgenic mice. Endocrinology 130, 3378-3386 [DOI] [PubMed] [Google Scholar]
- Sumitran-Holgersson S., Nowak G., Thowfeequ S., Begum S., Joshi M., Jaksch M., Kjaeldgaard A., Jorns C., Ericzon B. G., Tosh D. (2009). Generation of hepatocyte-like cells from in vitro transdifferentiated human fetal pancreas. Cell Transplant. 18, 183-193 [DOI] [PubMed] [Google Scholar]
- Waldegger S., Barth P., Raber G., Lang F. (1997). Cloning and characterization of a putative human serine/threonine protein kinase transcriptionally modified during anisotonic and isotonic alterations of cell volume. Proc. Natl. Acad. Sci. USA 94, 4440-4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldegger S., Klingel K., Barth P., Sauter M., Rfer M. L., Kandolf R., Lang F. (1999). h-Sgk serine-threonine protein kinase gene as transcriptional target of transforming growth factor beta in human intestine. Gastroenterol. 116, 1081-1088 [DOI] [PubMed] [Google Scholar]
- Wallace K., Marek C. J., Currie R. A., Wright M. C. (2009). Exocrine pancreas trans-differentiation to hepatocytes-a physiological response to elevated glucocorticoid in vivo. J. Steroid Biochem. Mol. Biol. 116, 76-85 [DOI] [PubMed] [Google Scholar]
- Wallace K., Fairhall E. A., Charlton K. A., Wright M. C. (2010a). AR42J-B-13 cell: an expandable progenitor to generate an unlimited supply of functional hepatocytes. Toxicology 278, 277-287 [DOI] [PubMed] [Google Scholar]
- Wallace K., Flecknell P. A., Burt A. D., Wright M. C. (2010b). Disrupted pancreatic exocrine differentiation and malabsorption in response to chronic elevated systemic glucocorticoid. Am. J. Pathol. 177, 1225-1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace K., Marek C. J., Hoppler S., Wright M. C. (2010c). Glucocorticoid-dependent trans-differentiation of pancreatic progenitor cells into hepatocytes is dependent on transient suppression of WNT signalling. J. Cell Sci. 123, 2103-2110 [DOI] [PubMed] [Google Scholar]
- Wang J. F., Hill D. J. (2009). Identification and action of N-myc downstream regulated gene 4 A2 in rat pancreas. J. Endocrinol. 201, 15-25 [DOI] [PubMed] [Google Scholar]
- Wright M. C., Issa R., Smart D. E., Trim N., Murray G. I., Primrose J. N., Arthur M. J., Iredale J. P., Mann D. A. (2001). Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology 121, 685-698 [DOI] [PubMed] [Google Scholar]
- Wulff P., Vallon V., Huang D. Y., Völkl H., Yu F., Richter K., Jansen M., Schlünz M., Klingel K., Loffing J., et al. (2002). Impaired renal Na(+) retention in the sgk1-knockout mouse. J. Clin. Invest. 110, 1263-1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}