

Abstract
Loss-of-function mutations in mucolipin 1 (MCOLN1) result in mucolipidosis type IV (MLIV), a lysosomal storage disorder characterized by severe mental and psychomotor retardation. MCOLN1 is a lysosomal ion channel that belongs to the transient receptor potential (TRP) superfamily. To better understand the cellular function of MCOLN1, a split-ubiquitin yeast two-hybrid screen was performed with the purpose of revealing new MCOLN1 interaction partners. The screen identified two members of the lysosome-associated protein transmembrane (LAPTM) family as novel interaction partners of MCOLN1. The binding between MCOLN1 and LAPTM members (LAPTMs) was confirmed by co-immunoprecipitation and yeast two-hybrid assays. In addition, MCOLN1 and LAPTMs extensively colocalize at late endosomes and lysosomes. Overexpression of LAPTM4b caused enlargement of lysosomes and defective lysosomal degradation, indicating that LAPTMs are important for proper lysosomal function. Interestingly, lysosomal swelling induced by LAPTM4b was rescued by expression of MCOLN1, suggesting a functional connection between the two proteins. Finally, depletion of endogenous LAPTMs by siRNA induced accumulation of concentric multi-lamellar structures and electron-dense inclusions that closely resemble the structures found in MLIV cells. Overall, our data provide new insight into the molecular mechanisms of MCOLN1 function and suggest a potential role for LAPTMs in MLIV pathogenesis.
Keywords: Mucolipin, LAPTM, TRPML, MLIV, Lysosomes
Introduction
Mucolipidosis type IV (MLIV) is an autosomal recessive disorder characterized by severe neurologic and ophthalmologic abnormalities (Altarescu et al., 2002; Amir et al., 1987; Bach, 2001). Symptoms appear during the first year of life and include intellectual disabilities, psychomotor delay, weak muscle tone (hypotonia), corneal clouding and progressive retinal degeneration (Berman et al., 1974; Riedel et al., 1985). In addition, MLIV patients show impaired secretion of gastric acid (achlorhydria) leading to defective iron absorption and anemia (Schiffmann et al., 1998). By their early teens, most affected individuals are unable to walk, present limited or no ability to speak and have severe vision loss or blindness.
MLIV is caused by loss-of-function mutations in MCOLN1 (also referred to as TRPML1), an ion channel that belongs to the TRP superfamily (Bargal et al., 2000; Bassi et al., 2000; Slaugenhaupt et al., 1999; Sun et al., 2000). MCOLN1 is a protein with six transmembrane-spanning domains with both amino- and C-terminal tails oriented toward the cytosol and the pore located between transmembrane segments 5 and 6. Electrophysiological studies indicate that MCOLN1 is an inwardly (from lumen to cytoplasm) rectifying channel permeable to Ca2+, Na+, K+ and Fe2+ or Mn2+, and whose activity is modulated by pH and Ca2+ (Cheng et al., 2010; Puertollano and Kiselyov, 2009). However, the selectivity and mechanisms of activation of MCOLN1 under physiological conditions remain unknown.
MCOLN1 is localized at late endosomes and lysosomes. Two acidic di-leucine consensus motifs located at the N- and C-terminal tails regulate trafficking of MCOLN1 to the late endosomal pathway through interactions with clathrin adaptors (Miedel et al., 2006; Pryor et al., 2006; Vergarajauregui and Puertollano, 2006). MCOLN1 undergoes several post-translational modifications that regulate its function. For example, palmitoylation and phosphorylation at the C-terminal tail modulate trafficking and channel activity, respectively, whereas cleavage at the first luminal loop inactivates the protein (Kiselyov et al., 2005; Vergarajauregui et al., 2008b; Vergarajauregui and Puertollano, 2006).
According to previous reports, analysis of fibroblasts from MLIV patients by electron microscopy revealed the accumulation of enlarged vacuolar structures that contain mucopolysaccharides and lipids forming characteristic multiconcentric lamellae (Bach et al., 1975; Bach et al., 1977; Crandall et al., 1982; Tellez-Nagel et al., 1976). These enlarged vacuoles are present not only in fibroblasts but in every tissue and organ of MLIV patients, suggesting a general impairment of lysosomal function. Accumulation of defective lysosomes appears to affect other cellular processes such as autophagy. Increased levels of insoluble p62, ubiquitinated aggregates and autophagosomes have been observed in MLIV fibroblasts, indicating that autophagy-mediated clearance of harmful cellular products is impaired in MLIV patients (Vergarajauregui et al., 2008a). Generation of two different MLIV animal models in Drosophila and mouse confirmed that absence of MCOLN1 results in defective autophagy (Micsenyi et al., 2009; Venkatachalam et al., 2008). However, these observations are based on studies characterizing cellular effects resulting from the loss of MCOLN1. Thus, it is unclear whether the observed phenomena directly result from the absence of MCOLN1 or whether they are a secondary consequence of lipid accumulation in lysosomes.
In order to gain insights into MCOLN1 function, a yeast two-hybrid screen was performed to identify proteins that interact with MCOLN1. Here, we report a novel interaction between MCOLN1 and the members of the LAPTM family. Although the cellular function of LAPTMs is not well understood, it has been suggested that LAPTMs might participate in the transport of small molecules across intracellular membranes (Hogue et al., 1996; Hogue et al., 1999). We found that MCOLN1 and LAPTMs colocalize to late endosomes and lysosomes and confirmed the interaction by co-immunoprecipitation in human cells. Overexpression of LAPTMs caused enlargement of lysosomes and defective lysosomal degradation, whereas depletion of endogenous LAPTMs induced accumulation of concentric multi-lamellar structures and electron-dense inclusions that closely resemble the structures found in MLIV cells. Overall, our data provide new insight for understanding MCOLN1 function and reveal a novel role for LAPTMs in the regulation of lysosomal function.
Results
Identification of LAPTMs as novel MCOLN1 binding partners
In order to further understand the cellular function of MCOLN1, we searched for novel binding partners of MCOLN1. Given that MCOLN1 is a highly hydrophobic transmembrane protein that oligomerizes and undergoes post-translational modifications, we used the split-ubiquitin membrane-based yeast two-hybrid system. This system uses the split-ubiquitin approach, in which reconstitution of two ubiquitin halves (Nub and Cub) is mediated by a protein–protein interaction, resulting in the release of a transcription factor and expression of reporter genes (Johnsson and Varshavsky, 1994). The advantage of this approach is that it allows us to use full-length MCOLN1 as bait and reveals interactions that take place at the organelle where the protein typically localizes (in this case the vacuole).
To generate the MCOLN1 bait, we cloned the full-length human MCOLN1 protein into the pBTE-STE vector, thus generating MCOLN1–Cub. The bait was screened against a human adult brain library of cDNAs fused to the mutated form of N-ubiquitin in the pPR3-N vector (NubG-x) and was carried out by Dualsystems Biotech AG (Schlieren, Switzerland). Among 277 positive clones isolated, two independent clones encoded members of a family of endosomal and lysosomal transmembrane proteins named LAPTMs. The clones included the first 217 amino acids (aa) of LAPTM4a and the N-terminal sequence (aa 27–47) of LAPTM4b, respectively. Both clones were in-frame with the N-terminal half of ubiquitin. The function of LAPTMs is not completely understood but it has been suggested that they are transporters involved in the subcellular compartmentalization of different compounds (Hogue et al., 1996; Hogue et al., 1999). MCOLN1 protein binding to LAPTMs was confirmed by performing additional yeast two-hybrid experiments. As seen in Fig. 1, MCOLN1 interacted with the three members of the LAPTM family (LAPTM4a, LAPTM4b and LAPTM5). By contrast, MCOLN3, another member of the mucolipin family responsible for the varitint-waddler phenotype in mice, did not show any significant binding to LAPTMs (Fig. 1).
Fig. 1.
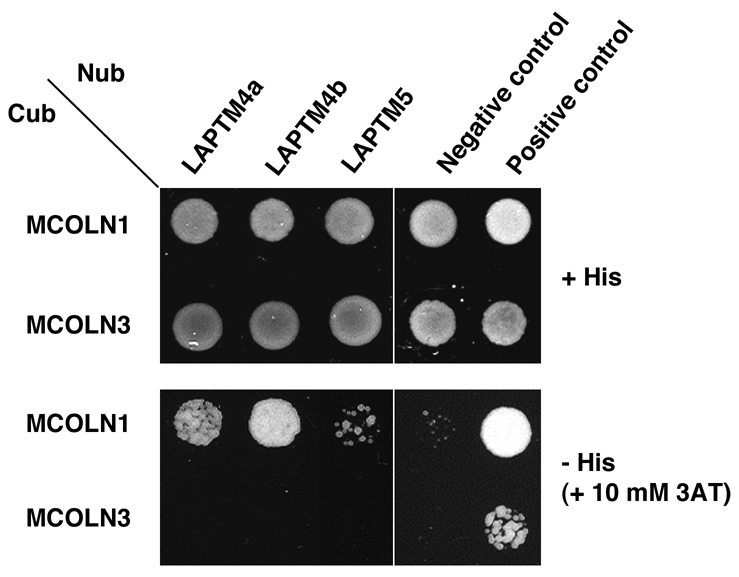
MCOLN1 interacts with the three members of the LAPTM family in yeast two-hybrid assays. A split-ubiquitin yeast two-hybrid assay was performed to corroborate the interaction of LAPTM proteins with MCOLN1. Interactions were tested by monitoring the growth of yeast cells expressing MCOLN1-Cub or MCOLN3-Cub bait with various Nub-LAPTM fusion proteins on agar plates lacking tryptophan and leucine (top), or tryptophan, leucine and histidine and containing 10 mM 3-aminotriazole (3AT; bottom). The vectors pAl-Alg5 and pDL2-Alg5 were used as positive and negative controls, respectively.
Confirmation of the LAPTM–MCOLN1 interaction by in vivo binding assays
To confirm the association between LAPTMs and MCOLN1, we coexpressed full-length MCOLN1-FLAG and GFP–LAPTMs in HeLa cells and performed immunoprecipitation using an anti-FLAG antibody directly coupled to protein-G–agarose beads. Co-immunoprecipitation of LAPTMs was detected by immunoblot using polyclonal anti-GFP antibodies. As shown in Fig. 2A, GFP–LAPTM4b was present in immunoprecipitates of cell lysates transfected with MCOLN1–FLAG cDNA but not in those collected after transfection with the FLAG vector alone, suggesting that both proteins interact in vivo. Co-immunoprecipitation experiments also confirmed the interaction of MCOLN1 with GFP–LAPTM4a and GFP–LAPTM5 (Fig. 2A,B). In support of our two-hybrid data, MCOLN3 did not interact with LAPTMs (Fig. 2B). Western blot profiles of LAPTM4a, LAPTM4b and LAPTM5 showed the presence of low molecular mass bands that probably correspond to proteolytic fragments (Fig. 2, arrows). In addition, MCOLN1 and LAPTMs did not interact with LAMP1, another lysosomal transmembrane protein used as a negative control (supplementary material Fig. S1). Overall, our data reveal that MCOLN1 interacts with the three members of the LAPTM family both in yeast and human cells.
Fig. 2.

Physical interaction of MCOLN1 with the three members of the LAPTM family. (A,B) The interaction between MCOLN1 and LAPTM family members was tested by co-immunoprecipitation. HeLa cells were transfected with various combinations of the indicated vectors. FLAG-tagged MCOLN1 and MCOLN3 were immunoprecipitated using the anti-FLAG mAb and samples were assessed by western blotting using anti-FLAG antibody or anti-GFP antibody. The results shown are representative of three independent experiments. The predicted molecular weight is approximately 53.7 kDa for GFP–LAPTM4a, 62 kDa for GFP–LAPTM4b, 56.8 kDa for LAPTM5 and 66 kDa for MCOLN1-FLAG. Arrowheads indicate full-length proteins, arrows correspond to proteolytic fragments and asterisks represent oligomers.
MCOLN1 and LAPTMs colocalize to late endosomes and lysosomes
Next, we asked whether MCOLN1 and LAPTMs colocalize, which would indicate that the proteins have the ability to interact at the physiological level. To analyze the colocalization between LAPTMs and MCOLN1, ARPE-19 cells were transiently co-transfected with GFP–LAPTMs and Cherry–MCOLN1 constructs and observed by confocal microscopy. As seen in Fig. 3, all the members of the LAPTM family (LAPTM4a, LAPTM4b and LAPTM5) showed a very high degree of colocalization with MCOLN1. In fact, nearly all LAPTM-containing vesicles were also labeled for MCOLN1. We have previously described that MCOLN1 localizes to late endosomes and lysosomes and identified two di-leucine-sorting consensus motifs that mediate the transport of MCOLN1 to late-endocytic compartments (Vergarajauregui and Puertollano, 2006). Therefore, these results suggest that the interaction between MCOLN1 and LAPTMs probably occurs at late endosomes and lysosomes. These data are also consistent with previous reports describing the distribution of LAPTM4a and LAPTM5 in late endosomes and/or lysosomes (Cabrita et al., 1999; Pak et al., 2006).
Fig. 3.

MCOLN1 and LAPTMs colocalize to late endosomes and lysosomes. ARPE-19 cells were transiently transfected with Cherry-MCOLN1 (red) and either GFP–LAPTM4a, GFP–LAPTM4b or GFP–LAPTM5 (green). Twenty hours after transfection, cells were fixed and analyzed by confocal fluorescence microscopy. Insets show a 3-fold magnification of the indicated region. Scale bar: 10 μm.
To examine the subcellular localization of LAPTM4b in more detail and to ensure that its distribution was not altered by coexpression of MCOLN1, we expressed GFP–LAPTM4b alone in ARPE-19 cells and performed co-staining with different endogenous markers. As shown in Fig. 4, most of the LAPTM4b-positive vesicles also contain the late-endosomal and -lysosomal marker CD63. By contrast, colocalization of LAPTM4b with early endosomal markers such as Hrs was very limited, thus confirming the distribution of LAPTM4b at the late-endocytic pathway (Fig. 4).
Fig. 4.

Distribution of LAPTM4b into late endosomes and lysosomes. ARPE-19 cells transiently transfected with GFP–LAPTM4b were fixed, permeabilized, immunostained with the indicated antibodies and analyzed by confocal fluorescence microscopy. Yellow indicates colocalization between LAPTM4b (green) and CD63 (red), whereas white indicates colocalization of LAPTM4b, Hrs (blue) and CD63. Insets show a 4-fold magnification of the indicated region. Scale bar: 10 μm.
LAPTM4b undergoes proteolytic cleavage in late endosomes and lysosomes
As mentioned previously, expression of LAPTMs in ARPE-19 cells resulted in two major protein products: the predicted full-length protein and a shorter form that we propose might correspond to a proteolytic fragment. To test this hypothesis, we followed the fate of newly synthesized LAPTM4b by performing ‘pulse-chase’ experiments upon treatment with brefeldin A (BFA). This approach has been previously used to monitor the transport of MCOLN1 to lysosomes (Vergarajauregui and Puertollano, 2006). ARPE-19 cells were transfected with GFP–LAPTM4b and, 6 hours after transfection, cells were incubated with BFA for 12 hours to accumulate the newly synthesized proteins in the endoplasmic reticulum (ER; pulse) (Lippincott-Schwartz et al., 1989). Proteins were then released from the ER by incubation in medium without BFA (chase) and cells were analyzed at different time-points by either immunofluorescence or western blot. As presented in Fig. 5A, LAPTM4b started showing colocalization with CD63 3 hours after BFA removal, whereas colocalization between LAPTM4b and CD63 was nearly 100% complete after 6 hours. As expected, delivery of LAPTM4b to late endosomes and lysosomes correlated with the appearance of the low molecular mass band, consistent with the idea that it is the result of proteolytic cleavage (Fig. 5B). The fragment was not observed when LAPTM4b was retained at the ER or shortly after release of the protein from the ER, but it was most abundant at 6 hours chase when most of the protein had reached the late endosomes and lysosomes. Appearance of the proteolytic fragment was inhibited by incubation with leupeptin, an inhibitor of lysosomal proteases, thus confirming that cleavage of LAPTM4b occurs at lysosomes. In addition, the amount of high molecular mass bands increased over time, suggesting that LAPTM4b might form homo-oligomers upon reaching the lysosomes.
Fig. 5.

LAPTM4b undergoes proteolytic cleavage following delivery to the lysosomes. ARPE-19 cells were grown on coverslips (A) or plates (B) and transfected with a plasmid encoding GFP–LAPTM4b. Six hours after transfection, cells were incubated for an additional 12 hours in the presence of 2 μg/ml BFA to accumulate the newly synthesized protein in the ER. BFA was then removed and the cells were incubated for the indicated periods of time at 37°C in the absence or presence of 20 μM leupeptin. Cells were then fixed, stained with the late-endosomal and -lysosomal marker CD63 and analyzed by confocal microscopy (A) or lysated and subjected to western blotting (B) to detect GFP–LAPTM4b and actin (loading control). Scale bar: 10 μm.
LAPTMs and MCOLN1 are transported to lysosomes independently of each other
Next, we addressed whether the binding of LAPTMs to MCOLN1 is required for the proper transport of LAPTMs to late endosomes and lysosomes. For that, we generated a stable cell line in which the levels of endogenous MCOLN1 were reduced by lentivirus-mediated shRNA interference. Quantitative RT-PCR showed that the lentivirus-mediated shRNA against MCOLN1 caused a greater than 80% reduction in MCOLN1 mRNA levels compared with a lentivirus-expressing control shRNA (data not shown). Delivery of LAPTMs to late endosomes and lysosomes was not affected by depletion of MCOLN1, as assessed by the appearance of LAPTM proteolytic fragments (Fig. 6A) and by confocal microscopy (data not shown). These results indicate that the binding of LAPTMs to MCOLN1 does not regulate sorting of LAPTMs along the endocytic pathway. Similarly, MCOLN1 efficiently reached late endosomes and lysosomes in cells depleted of LAPTM4a and LAPTM4b (LAPTM5 is not expressed in HeLa cells; Fig. 6B). The levels of LAPTM4a and LAPTM4b mRNAs were reduced by more than 90% in cells treated with specific siRNAs when compared with control cells (data not shown). Therefore, our results indicate that LAPTMs and MCOLN1 are sorted independently of each other.
Fig. 6.

Sorting of LAPTMs to late endosomes and lysosomes is independent of MCOLN1. (A) Stable HeLa cell lines expressing control non-target shRNA (shControl) or shRNA against MCOLN1 (shMCOLN1) were transfected with GFP–LAPTM4a, GFP–LAPTM4b or GFP–LAPTM5. Twenty-four hours after transfection, cells were analyzed by western blot using anti-GFP antibody. The presence of proteolytic fragments (arrows) indicates delivery of LAPTMs to the lysosomes. (B) HeLa cells treated with siRNAs against both LAPTM4a and LAPTM4b were transfected with a plasmid encoding GFP–MCOLN1. Sorting of MCOLN1 to late endosomes and lysosomes was assessed by colocalization with CD63. Scale bar: 10 μm.
Overexpression of LAPTM4b causes enlargement of late endosomes and lysosomes
Next, we sought to determine the effect of LAPTM4b overexpression on the late-endosomal pathway. In order to achieve high levels of protein expression, we generated recombinant adenoviruses expressing GFP–LAPTM4b (Ad-LAPTM4b) or an adenovirus expressing GFP alone (Ad-GFP) as a control. As shown in Fig. 7A, overexpression of GFP–LAPTM4b caused a significant change in the size and distribution of CD63-positive vesicles. Late endosomes and lysosomes appeared enlarged and, in some cases, clustered to the perinuclear region when compared with non-infected cells. Expression of Ad-GFP did not affect the extent of distribution of late endosomes or lysosomes. The average diameter of CD63-positive structures was 0.45 μm (±0.12 s.d., n=56) in mock-infected cells and 0.46 μm (±0.15 s.d., n=98) in Ad-GFP-infected cells. By contrast, the average diameter of late endosomes and lysosomes increased to 1.33 μm (±0.28 s.d., n=89) in LAPTM4b-expressing cells (supplementary material Fig. S2).
Fig. 7.

MCOLN1 rescues the enlargement of late endosomes and lysosomes induced by LAPTM4b overexpression. (A) ARPE-19 cells were infected with adenovirus encoding GFP–LAPTM4b (top panel) or GFP alone (bottom panel). Forty hours after infection, cells were fixed and immunostained with antibodies against CD63. Scale bar: 10 μm. (B) ARPE-19 cells were infected with adenovirus expressing GFP–LAPTM4b (top panel) or co-infected with adenovirus expressing GFP–LAPTM4b and MCOLN1–FLAG (bottom panel). Forty hours after infection, cells were fixed, immunostained with antibodies against FLAG and analyzed by confocal microscopy. Insets represent a 2-fold magnification. Scale bar: 10 μm.
Interestingly, the enlargement of late endosomes and lysosomes caused by the overexpression of LAPTM4b was rescued by coexpression of MCOLN1. As seen in Fig. 7B, co-infection of ARPE-19 cells with Ad-LAPTM4b and Ad-MCOLN1–FLAG prevented the accumulation of swelled late endosomes and lysosomes. The presence of MCOLN1 did not affect the localization of LAPTM4b with CD63 but induced a 60% reduction in the size of late endosomes and lysosomes, changing from 1.33 μm in LAPTM4b-expressing cells to 0.53 μm (±0.12 s.d., n=76) in cells co-expressing LAPTM4b and MCOLN1 (supplementary material Fig. S2). These results indicate that the interaction between MCOLN1 and LAPTM4b is functionally relevant in cells.
Autophagy is altered in LAPTM4b-expressing cells
To analyze whether the enlargement of late endosomes and lysosomes upon LAPTM4b overexpression correlates with defects in lysosomal function, we monitored autophagic flux. Autophagy is a crucial clearance mechanism that prevents accumulation of protein aggregates and abnormal organelles (Cuervo, 2004; Martinez-Vicente and Cuervo, 2007). It has been described that alterations in lysosomal function result in defective autophagosome degradation. Interestingly, we found a remarkable accumulation of LC3-positive structures in cells expressing LAPTM4b, whereas accumulation of autophagosomes was not observed in mock-infected cells or in cells expressing Ad-GFP (Fig. 8A). LC3 is considered to be one of the most specific markers for autophagosomes (Kabeya et al., 2000); therefore, our data indicate that autophagosome maturation is severely diminished by overexpression of LAPTM4b. Quantification of the number of autophagosomes per cell revealed that mock-infected and Ad-GFP-infected cells show an average of 20 LC3-positive vesicles per cell (18.82±4.93, n=40 in mock-infected cells and 19.46±5.14, n=46 in cells expressing Ad-GFP, where n represents the number of cells). By contrast, there was a greater than 6-fold increase in the average number of autophagomes in cells infected with Ad-LAPTM4b (123±28.39, n=36; Fig. 8B).
Fig. 8.

Overexpression of LAPTM4b causes accumulation of autophagosomes. (A) ARPE-19 cells were infected with adenovirus encoding GFP–LAPTM4b (top panel) or GFP alone (bottom panel). Forty hours after infection, cells were fixed and immunostained with antibodies against LC3. Scale bar: 10 μm. (B) The number of LC3-positive vesicles per cell was counted in control (mock-infected) cells or in cells expressing either Ad-GFP or Ad-LAPTM4b. Bars represent mean values + s.d. (C) ARPE-19 cells mock-infected or infected with Ad-GFP or Ad-LAPTM4b were harvested and subjected to western blotting to detect LC3. The blot shown is representative of three independent experiments. (D) Quantification of the LC3II:LC3I ratio. Results are the mean + s.d. from three independent experiments. (E) Lysates from mock-infected ARPE-19 cells or cells expressing either Ad-GFP or Ad-LAPTM4b were subjected to western blotting using a monoclonal antibody to ubiquitin.
Autophagy can also be monitored by immunoblot. LC3 is initially synthesized in an unprocessed form that is rapidly cleaved in its C-terminal region to generate soluble LC3I. During autophagy induction, the LC3I isoform is converted into LC3II by conjugation of a phosphatidylethanolamine group. This post-translational modification allows LC3 to translocate to autophagosomal membranes. It is well established that the LC3II:LC3I ratio correlates with the extent of autophagosome formation (Klionsky et al., 2007). We prepared lysates from mock-infected cells and from cells infected with adenovirus expressing GFP or GFP–LAPTM4b. As seen in Fig. 8C, a robust increase in the level of LC3II was observed in cells expressing GFP–LAPTM4b, confirming disruption of the autophagic process. Quantification of three independent experiments revealed an approximately threefold increase (2.85±0.2) in the LC3II:LC3I ratio in LAPTM4b-expressing cells (Fig. 8D). To further confirm that basal autophagic flux is interrupted upon overexpression of LAPTM4b, we examined the levels of ubiquitinated proteins. As expected, a remarkable accumulation of ubiquitinated proteins was observed by western blot (Fig. 8E) and immunofluorescence (supplementary material Fig. S3). Therefore, our results indicate that appropriate levels of LAPTM4b are crucial for optimal lysosomal function because overexpression of the protein results in enlargement of late endosomes and lysosomes and defective autophagosome maturation.
Depletion of LAPTMs induces accumulation of electron-dense and multi-laminar structures
In MLIV, absence of MCOLN1 leads to the buildup of membranous and electron-dense organelles containing undigested lipid products. This heterogeneous storage is readily identified by electron microscopy and suggests that proper MCOLN1 function is essential for the maintenance of lysosomal integrity. To further confirm the role of LAPTMs in lysosomal function, we depleted endogenous LAPTMs by treatment with specific siRNAs. Quantitative RT-PCR revealed that only LAPTM4a and LAPTM4b are expressed in HeLa cells. These results are in agreement with previous reports suggesting that LAPTM5 is preferentially expressed in hematopoietic cells (Adra et al., 1996; Scott et al., 1996). After two 72-hour consecutive transfections with a combination of LAPTM4a and LAPTM4b siRNAs, we were able to achieve a greater than 90% reduction in the levels of LAPTM4 (a+b) mRNA, whereas no variations were observed in cells transfected with control non-target siRNA. Ultrastructure analysis revealed a significant accumulation of lysosomal inclusions in LAPTM4 knockdown cells when compared with control cells (Fig. 9A,B). These storage bodies, composed of electron-dense materials and concentric multi-laminar (fingerprint) organelles (Fig. 9E; supplementary material Fig. S4), closely resemble those found in MLIV patients (Fig. 9D,G; supplementary material Fig. S4). Quantification experiments confirmed that accumulation of lysosomal inclusions in LAPTM4 knockdown cells is highly significant when compared with control cells (Fig. 10). For example, only 19% (n=54) of the cells treated with control siRNA showed more than 10 electron-dense structures, whereas 78% (n=50) of the LAPTM4-depleted cells have over 10 inclusions (Fig. 10A). Interestingly, simultaneous depletion of LAPTM4 (a+b) and MCOLN1 caused a greater than twofold increase in the size of the storage bodies when compared with cells lacking just LAPTM4 (Fig. 9C,F; Fig. 10B). Overall, our results confirm that endogenous LAPTMs are important for the maintenance of lysosomal integrity and indicate that defects in LAPTM function lead to accumulation of storage material in lysosomes that mimics the phenotype found in MLIV patients.
Fig. 9.

Depletion of LAPTMs results in accumulation of storage bodies. (A–D) Electron micrographs showing HeLa cells treated with non-target siRNA (siControl; A), HeLa cells depleted of endogenous LAPTM4a and LAPTM4b (B), HeLa cells depleted of both LAPTMs and MCOLN1 (C) and MLIV human fibroblasts (D). Note the accumulation of electron-dense and concentric multi-laminar membrane structures in B–D. Scale bar: 2 μm. (E–G) High-magnification images of the fingerprint structures shown in B–D. Scale bar: 200 nm.
Fig. 10.

Simultaneous depletion of LAPTMs and MCOLN1 increases lysosomal inclusion size. (A,B) Quantification of the number and size of the electron-dense and multiconcentric structures accumulated under the following conditions (n represents the number of cells analyzed per condition): HeLa cells treated with control non-target siRNA (siControl, n=54), HeLa cells treated with siRNAs against both LAPTM4a and LAPTM4b (siLAPTM, n=51), stable HeLa cell line infected with lentivirus-mediated shRNA against MCOLN1 (shMCOLN1, n=16), stable HeLa MCOLN1-deficient cell line transfected with siRNAs against LAPTM4a and LAPTM4b (shMCOLN1+ siLAPTM, n=14).
Discussion
Mutations in MCOLN1 are the cause of MLIV, a devastating lysosomal storage disorder that affects children in their first year of life. To better understand the cellular function of MCOLN1, we performed a split-ubiquitin yeast two-hybrid screen with the goal of revealing novel interaction partners of MCOLN1. As a result of the screen, we found that MCOLN1 physically interacts with the three members of the LAPTM family, and we observed extensive colocalization between MCOLN1 and LAPTMs in late-endosomes and lysosomes.
Initial studies identified LAPTM4a as a transporter that transfers nucleosides (and/or nucleoside metabolites) between the cytosol and intracellular organelles (Hogue et al., 1996). Further work suggested that LAPTMs might have a more general role in the transport of structurally unrelated amphiphilic molecules (Hogue et al., 1999). Thus, drug-sensitive yeast expressing mouse LAPTM4a were found to have increased resistance to several compounds such as daunorubicin, doxorubicin, erythromycin, progesterone and rhodamine 123, whereas they showed increased sensitivity to 5-fluorouracil, 5-fluorouridine and trifluoperazine. Moreover, the finding that LAPTM4a and LAPTM5 located to lysosomes (Cabrita et al., 1999; Pak et al., 2006) suggested that LAPTMs mediate transport of one or more substrates across lysosomal membranes.
In this study, we described for the first time that LAPTM4b also localizes to late endosomes and lysosomes. LAPTM4b is a candidate oncogene whose expression is increased in hepatocellular carcinoma, ovarian cancer and gastric cancer (Kasper et al., 2005; Liu et al., 2007; Yang et al., 2010). Furthermore, expression of LAPTM4b contributes to chemotherapy resistance in breast cancer by preventing transport of the anthracycline doxorubicin to the nucleus (Li et al., 2010). Therefore, this could suggest that sequestration of doxorubicin in the lysosomal lumen mediated by LAPTM4b is the cause of anthracycline chemoresistance.
We are also the first to describe that LAPTMs undergo proteolytic cleavage upon reaching lysosomes. The approximate molecular masses of the proteolytic products were 8.5 kDa for LAPTM4a, 17 kDa for LAPTM4b and 7 kDa for LAPTM5, indicating that the cleavage probably occurs at the first intraluminal loop. Interestingly, injection of a truncated version of mouse LAPTM4a (lacking the first 83 residues) into Xenopus oocytes stimulated uptake of thymidine, whereas full-length mouse LAPTM4a did not show transport activity (Hogue et al., 1996). These data suggest that LAPTMs might be activated by proteolytic cleavage only after being delivered to lysosomes. The first luminal loop of MCOLN1 is also proteolytically cleaved in lysosomes. However, in the case of MCOLN1, it has been suggested that proteolytic cleavage might induce inactivation of the protein (Kiselyov et al., 2005). In addition, we did not observe significant homology between the loops of MCOLN1 and LAPTMs, thus suggesting that the cleavage might be mediated by different lysosomal hydrolases.
Overexpression of LAPTM4b has a profound effect on the morphology and functionality of lysosomes. Infection of ARPE-19 cells with Ad-LAPTM4b caused enlargement of CD63-positive vesicles, accumulation of ubiquitinated proteins and defective autophagosome maturation. These data are consistent with the proposed role for LAPTMs in the transport of molecules across the lysosomal membrane, as the accumulation of certain substrates is likely to impair lysosomal function. The data are also in agreement with recent evidence showing that overexpression of LAPTM5 associates with spontaneous regression of neuroblastomas by inducing lysosomal cell death with impaired autophagy (Inoue et al., 2009). We and others have previously described that overexpression of MCOLN1 also causes accumulation of enlarged aberrant lysosomes and impaired autophagy, thus indicating that LAPTMs and MCOLN1 are required to maintain adequate lysosomal function (Vergarajauregi et al., 2008; Manzoni et al., 2004).
The importance of LAPTMs in lysosomal function was further corroborated by the accumulation of storage bodies in cells depleted of endogenous LAPTMs. Storage material included fine lamellated membrane structures (fingerprints), as well as electron-dense bodies. Interestingly, the same types of lysosomal inclusions are found in MLIV cells. It has been suggested that the ultrastructural appearance of storage material might be specific to the type of lipid accumulated in each lysosomal storage disorder (Alroy and Ucci, 2006). For example, fingerprint structures often result from the accumulation of gangliosides and other glycolipids, and have been described in MLIV, Fabry disease, fucosidosis, GM1 and GM2 gangliosidosis, galactosialidosis and sialidosis. Therefore, the appearance of concentric membranes in the absence of either MCOLN1 or LAPTMs could indicate accumulation of the same type of storage material.
The presented evidence suggests that MCOLN1 and LAPTMs are functionally connected. The fact that MCOLN1 and LAPTMs interact and colocalize to late endosomes and lysosomes, the ability of MCOLN1 to rescue the enlargement of lysosomes caused by LAPTM4b overexpression, the accumulation of the same type of storage material upon depletion of either LAPTMs or MCOLN1 and the increase in the size of lysosomal inclusions by simultaneous depletion of both MCOLN1 and LAPTMs suggest that MCOLN1 might regulate LAPTM function. We speculate that the release of ions by MCOLN1 might provide energy for the LAPTM-dependent transport of specific molecules across lysosomal membranes. Alternatively, binding of LAPTMs to MCOLN1 might induce changes in the conformation and/or activity of the transporters.
Our model has important implications for the understanding of MLIV. Alterations in the transport activity of LAPTMs caused by the absence of MCOLN1 might lead to the storage disorder observed in MLIV cells. Studies have shown that most lysosomal storage diseases are caused by deficiencies of soluble lysosomal hydrolases or lysosomal transporters that cause accumulation of specific molecules in the lumen of lysosomes. In support of this, expression of LAPTM4a and LAPTM5 in MLIV cells is increased 3-fold and 7-fold, respectively (Bozzato et al., 2008). This increase might represent an attempt by MLIV cells to compensate for the lack of LAPTM activity.
Further studies will try to identify the physiological ligands transported by LAPTMs as well as discover compounds that modulate LAPTM activity. This information will further improve our understanding of MLIV and might open new and exciting avenues for the development of a treatment for the disease.
Materials and Methods
Antibodies and reagents
The primary antibodies used were: mouse monoclonal anti-CD63 (H5C6; BD Pharmingen, San Jose, CA), mouse monoclonal anti-actin (clone Ab-5; BD Biosciences, San Jose, CA), mouse monoclonal anti-GFP (Covance, Princeton, NJ), mouse monoclonal anti-Lamp1 (clone H4A3, Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit polyclonal anti-LC3 (Sigma, St Louis, MO), rabbit anti-GFP and mouse monoclonal anti-ubiquitin (clone FK2) (MBL International, Woburn, MA), rabbit polyclonal anti-Hrs (Novus Biologicals, Littleton, CO) and mouse monoclonal anti-FLAG M2 (Sigma). The secondary antibodies used were: goat anti-mouse or goat anti-rabbit conjugated to Alexa Fluor 488 or 555, goat anti-mouse conjugated to Alexa Fluor 633 (Molecular Probes, Eugene, OR) and HRP-conjugated anti-mouse or anti-rabbit IgG (Amersham Bioscience, Piscataway, NJ). FuGENE-6 reagent and protease inhibitor cocktail tablets were obtained from Roche Applied Science (Indianapolis, IN). BFA and leupeptin were from Sigma and were used according to the manufacturer's instructions.
Split-ubiquitin yeast two-hybrid assays
Screening of an adult human brain cDNA library with full-length MCOLN1 was performed by Dualsystems Biotech AG (Schlieren, Switzerland). Full-length MCOLN1 was inserted into the bait expression vector pBT3-STE (Dualsystems Biotech, Switzerland) with Cub-LexA-VP16 downstream of MCOLN1. The bait vector carries the LEU2 gene for auxotrophic selection. A human adult brain cDNA library of 50.3×106 clones fused to a mutated version of the amino-terminal portion of ubiquitin (NubG-x) in their amino-terminal sequence in the pPR3-N (prey vector) was used to screen for potential positive interactions. The prey vector carries the TRP1 (TRNAP1) gene for auxotrophic selection. The screening was carried out by the mating of NMY32 yeast transformed with the bait and prey constructs. A total of 277 positive clones representing 23 distinct human proteins were isolated. One clone was found to encode the first 217 amino acids of LAPTM4a and a second one encodes amino acids 27–47 of LAPTM4b. Selection of positive interactions was performed by the ability of yeast to grow on selective synthetic dropout (SD) agar plates [SD –tryptophan –leucine –histidine (Clontech, Palo Alto, CA)] containing 10 mM 3-amino-1,2,4-triazole (3AT). The selected positive clones were sequenced and the resulting nucleotide sequences were analyzed using the NCBI BLAST algorithm.
Plasmids and adenovirus preparation
The complete open reading frames (ORFs) of human LAPTM4a and LAPTM5 were PCR-amplified from a placenta cDNA library using specific primers and cloned into the EcoRI and SalI sites of the pEGFP-C2 vector (Clontech). The ORF of human LAPTM4b was PCR-amplified from a cDNA clone from OriGene (NM_018407) using specific primers to introduce an EcoRI site at the 5′ end and an XhoI site at the 3′ end and cloned into the EcoRI and SalI sites of pEGFP-C2. To generate Cherry-MCOLN1, full-length MCOLN1 was cloned into the EcoRI and SalI sites of the pCherry vector (Clontech). MCOLN3–FLAG was cloned into the EcoRI–SalI sites of the pCMV-TAG-2B vector. Cloning of MCOLN1–FLAG has been described previously (Vergarajauregui et al., 2008b). MCOLN1, MCOLN3, LAPTM4a, LAPTM4b and LAPTM5 were cloned into the SfiI sites of the pBT3-STE, pPR3-N and pPR3-STE vectors (Dualsystems Biotech AG, Switzerland) according to the manufacturer's directions. We used pDL2-Alg5 as a negative control for the split-ubiquitin two-hybrid system. To check if the bait was properly expressed and functional, we used the control prey pAl-Alg5. All constructs were confirmed by DNA sequencing. Adenoviruses expressing GFP–LAPTM4b and MCOLN1–FLAG were prepared, amplified and purified by Welgen (Worcester, MA).
shMCOLN1 cell line
Control and MCOLN1 lentiviral shRNAs were obtained from Sigma (MISSION shRNA Lentiviral Transduction Particles). HeLa cells were infected with either the MCOLN1-lentivirus (MISSION shRNA TRCN0000083297) or the control lentivirus (MISSION Non-Target shRNA Control SHC002) together with 8 μg/ml of Polybrene. Clones were selected using 1 μg/ml puromycin. Q-RT-PCR was used to assay for knockdown of MCOLN1.
Cell culture, transfection, siRNA knockdown and adenoviral infection
HeLa and ARPE19 cells (American Type Culture Collection, Manassas, VA) were grown at 37°C in DMEM medium or in a 1:1 mixture of DMEM and Ham's F12 media (Invitrogen, Carlsbad, CA), respectively, supplemented with 10% fetal bovine serum, 2 mM Glutamax, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified 5% CO2 atmosphere. Cells were transiently transfected using FuGENE-6 reagent according to the manufacturer's instructions. Transfected cells were analyzed 24–36 hours post-transfection.
For siRNA knockdown, we performed two 72-hour consecutive transfections with either ON-TARGETplus smart pool siRNA duplexes against LAPTM4a and LAPTM4b (L-013439-02-0005 and L-017710-02-0005, respectively) or ON-TARGETplus non-targeting pool siRNA duplexes using DharmaFect transfection reagent (Dharmacon-ThermoScientific, Lafayette, CO). For infection experiments, cells were infected with adenoviruses according to the manufacturer's instructions. Analyses were performed 36–40 hours post-infection.
BFA pulse and chase
To analyze trafficking of newly synthesized LAPTM4b, ARPE-19 cells grown on coverslips were transfected with the corresponding cDNA for 6 hours and then incubated with 2 μg/ml BFA (Sigma-Aldrich) for 12 hours to accumulate the newly synthesized proteins in the ER. The cells were then washed and incubated for different times at 37°C in regular culture medium in the absence or presence of 20 μM leupeptin. Cells were then fixed, permeabilized and analyzed by confocal microscopy and western blot.
Real-time quantitative reverse-transcription polymerase chain reaction
RNA (2–4 μg) was reverse-transcribed in a 20 μl reaction using oligo(dT)20 and SuperScript III First-Strand Synthesis System (Invitrogen) according to the manufacturer's instructions. PCR was performed using 5 μl SYBR GreenER qPCR SuperMix (Invitrogen), 2 μl cDNA, 1 μl gene-specific primer mix (QuantiTect Primer Assays, QIAGEN, Valencia, CA) and 2 μl water for a total reaction volume of 10 μl. Quantification of gene expression was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). The thermal profile of the reaction was: 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds followed by 60°C for 1 minute. All samples were run in triplicates. Amplification of the sequence of interest was compared with a reference probe (β-2-microglobulin) and normalized against a standard curve of cell-line mRNA. The 7900HT Fast Real-Time PCR System Software was used for data analyses (Applied Biosystems).
Immunofluorescence microscopy
For immunofluorescence, ARPE-19 cells were grown on coverslips and fixed in methanol and acetone (1:1 v/v) for 10 minutes at −20°C. Incubation with primary antibodies diluted in PBS, 0.1% (w/v) saponin and 0.1% BSA was carried out for 1 hour at room temperature. Unbound antibodies were removed by washing with PBS for 5 minutes and cells were subsequently incubated with a secondary antibody (Alexa-555-, Alexa-633- or Alexa-488-conjugated goat anti-rabbit or anti-mouse IgG) diluted in PBS, 0.1% (w/v) saponin and 0.1% BSA for 30–60 minutes at room temperature. After a final rinse with PBS, coverslips were mounted onto glass slides with Fluoromount G (Southern Biotechnology Associates, Birmingham, AL). Fluorescence images were acquired on an LSM 510 confocal microscope (Carl Zeiss, Thornwood, NY).
Electron microscopy
Cells were cultured in 60 mm Permanox dishes (Nalgene Nunc, Rochester, NY) and processed for transmission electron microscopy. In brief, cultures were fixed with glutaraldehyde and paraformaldehyde, post-fixed with osmium tetroxide, stained en bloc with uranyl acetate, ethanol-dehydrated and Epon-embedded. Chemicals were from Electron Microscopy Sciences (Hatfield, PA). Sections 60 nm thick were cut parallel to the adherent surface on a Sorvall MT2 ultramicrotome, stained with uranyl acetate and lead citrate and viewed with a JEM 1200 EXII electron microscope (JEOL USA, Peabody, MA) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques, Danvers, MA). Lysosomal inclusions in each cell condition were measured with the ImageJ program and quantified by the number of structures per cell and by the average size of each structure (number of pixels per structure).
Immunoprecipitation and western blot
Cells were washed with ice-cold PBS, extracted in ice-cold lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA and 1% NP40 supplemented with protease inhibitor cocktails), centrifuged for 10 minutes at 16,000 g and the supernatants were collected. For immunoprecipitation, cell supernatants were incubated with 3 μl of anti-FLAG antibody and protein G-Sepharose beads (Amersham Bioscience). Immunoprecipitates bound to the column were collected, washed 3 times with lysis buffer and the proteins were eluted with Laemmli sample buffer. Samples were analyzed by SDS-PAGE (4–20% gradient gels) under reducing conditions and transferred to nitrocellulose. The membrane was then blocked with 1× PBS, 0.05% Tween 20 and 10% non-fat milk and incubated with the appropriate antibodies. Enhanced chemiluminescence reagent (PerkinElmer, Waltham, MA) was used for protein detection.
Supplementary Material
Acknowledgments
We thank Mathew P. Daniels and Patricia S. Connelly from the NHLBI Electron Microscopy Core Facility for assistance. We also appreciate the editorial advice of the NIH Fellows Editorial Board. This project was supported by the Intramural Research Program of the NIH, National Heart, Lung and Blood Institute (NHLBI). The authors have no conflicts of interest to declare. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/3/459/DC1
References
- Adra C. N., Zhu S., Ko J. L., Guillemot J. C., Cuervo A. M., Kobayashi H., Horiuchi T., Lelias J. M., Rowley J. D., Lim B. (1996). LAPTM5: a novel lysosomal-associated multispanning membrane protein preferentially expressed in hematopoietic cells. Genomics 35, 328-337 [DOI] [PubMed] [Google Scholar]
- Alroy J., Ucci A. A. (2006). Skin biopsy: a useful tool in the diagnosis of lysosomal storage diseases. Ultrastruct. Pathol. 30, 489-503 [DOI] [PubMed] [Google Scholar]
- Altarescu G., Sun M., Moore D. F., Smith J. A., Wiggs E. A., Solomon B. I., Patronas N. J., Frei K. P., Gupta S., Kaneski C. R., et al. (2002). The neurogenetics of mucolipidosis type IV. Neurology 59, 306-313 [DOI] [PubMed] [Google Scholar]
- Amir N., Zlotogora J., Bach G. (1987). Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics 79, 953-959 [PubMed] [Google Scholar]
- Bach G. (2001). Mucolipidosis type IV. Mol. Genet. Metab. 73, 197-203 [DOI] [PubMed] [Google Scholar]
- Bach G., Cohen M. M., Kohn G. (1975). Abnormal ganglioside accumulation in cultured fibroblasts from patients with mucolipidosis IV. Biochem. Biophys. Res. Commun. 66, 1483-1490 [DOI] [PubMed] [Google Scholar]
- Bach G., Ziegler M., Kohn G., Cohen M. M. (1977). Mucopolysaccharide accumulation in cultured skin fibroblasts derived from patients with mucolipidosis IV. Am. J. Hum. Genet. 29, 610-618 [PMC free article] [PubMed] [Google Scholar]
- Bargal R., Avidan N., Ben-Asher E., Olender Z., Zeigler M., Frumkin A., Raas-Rothschild A., Glusman G., Lancet D., Bach G. (2000). Identification of the gene causing mucolipidosis type IV. Nat. Genet. 26, 118-123 [DOI] [PubMed] [Google Scholar]
- Bassi M. T., Manzoni M., Monti E., Pizzo M. T., Ballabio A., Borsani G. (2000). Cloning of the gene encoding a novel integral membrane protein, mucolipidin- and identification of the two major founder mutations causing mucolipidosis type IV. Am. J. Hum. Genet. 67, 1110-1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman E. R., Livni N., Shapira E., Merin S., Levij I. S. (1974). Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J. Pediatr. 84, 519-526 [DOI] [PubMed] [Google Scholar]
- Bozzato A., Barlati S., Borsani G. (2008). Gene expression profiling of mucolipidosis type IV fibroblasts reveals deregulation of genes with relevant functions in lysosome physiology. Biochim. Biophys. Acta 1782, 250-258 [DOI] [PubMed] [Google Scholar]
- Cabrita M. A., Hobman T. C., Hogue D. L., King K. M., Cass C. E. (1999). Mouse transporter protein, a membrane protein that regulates cellular multidrug resistance, is localized to lysosomes. Cancer Res. 59, 4890-4897 [PubMed] [Google Scholar]
- Cheng X., Shen D., Samie M., Xu H. (2010). Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 584, 2013-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall B. F., Philippart M., Brown W. J., Bluestone D. A. (1982). Review article: mucolipidosis IV. Am. J. Med. Genet. 12, 301-308 [DOI] [PubMed] [Google Scholar]
- Cuervo A. M. (2004). Autophagy: many paths to the same end. Mol. Cell. Biochem. 263, 55-72 [DOI] [PubMed] [Google Scholar]
- Hogue D. L., Ellison M. J., Young J. D., Cass C. E. (1996). Identification of a novel membrane transporter associated with intracellular membranes by phenotypic complementation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 271, 9801-9808 [DOI] [PubMed] [Google Scholar]
- Hogue D. L., Kerby L., Ling V. (1999). A mammalian lysosomal membrane protein confers multidrug resistance upon expression in Saccharomyces cerevisiae. J. Biol. Chem. 274, 12877-12882 [DOI] [PubMed] [Google Scholar]
- Inoue J., Misawa A., Tanaka Y., Ichinose S., Sugino Y., Hosoi H., Sugimoto T., Imoto I., Inazawa J. (2009). Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PLoS ONE 4, e7099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson N., Varshavsky A. (1994). Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 91, 10340-10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720-5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper G., Vogel A., Klaman I., Grone J., Petersen I., Weber B., Castanos-Velez E., Staub E., Mennerich D. (2005). The human LAPTM4b transcript is upregulated in various types of solid tumours and seems to play a dual functional role during tumour progression. Cancer Lett. 224, 93-103 [DOI] [PubMed] [Google Scholar]
- Kiselyov K., Chen J., Rbaibi Y., Oberdick D., Tjon-Kon-Sang S., Shcheynikov N., Muallem S., Soyombo A. (2005). TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J. Biol. Chem. 280, 43218-43223 [DOI] [PubMed] [Google Scholar]
- Klionsky D. J., Cuervo A. M., Seglen P. O. (2007). Methods for monitoring autophagy from yeast to human. Autophagy 3, 181-206 [DOI] [PubMed] [Google Scholar]
- Li Y., Zou L., Li Q., Haibe-Kains B., Tian R., Desmedt C., Sotiriou C., Szallasi Z., Iglehart J. D., Richardson A. L., et al. (2010). Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat. Med. 16, 214-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Yuan L. C., Bonifacino J. S., Klausner R. D. (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56, 801-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhang Q. Y., Qian N., Zhou R. L. (2007). Relationship between LAPTM4B gene polymorphism and susceptibility of gastric cancer. Ann. Oncol. 18, 311-316 [DOI] [PubMed] [Google Scholar]
- Manzoni M., Monti E., Bresciani R., Bozzato A., Barlati S., Bassi M. T., Borsani G. (2004). Overexpression of wild-type and mutant mucolipin proteins in mammalian cells: effects on the late endocytic compartment organization. FEBS Lett. 567, 219-224 [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M., Cuervo A. M. (2007). Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352-361 [DOI] [PubMed] [Google Scholar]
- Micsenyi M. C., Dobrenis K., Stephney G., Pickel J., Vanier M. T., Slaugenhaupt S. A., Walkley S. U. (2009). Neuropathology of the Mcoln1(−/−) knockout mouse model of mucolipidosis type IV. J. Neuropathol. Exp. Neurol. 68, 125-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedel M. T., Weixel K. M., Bruns J. R., Traub L. M., Weisz O. A. (2006). Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J Biol. Chem. 281, 12751-12759 [DOI] [PubMed] [Google Scholar]
- Pak Y., Glowacka W. K., Bruce M. C., Pham N., Rotin D. (2006). Transport of LAPTM5 to lysosomes requires association with the ubiquitin ligase Nedd4, but not LAPTM5 ubiquitination. J. Cell Biol. 175, 631-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor P. R., Reimann F., Gribble F. M., Luzio J. P. (2006). Mucolipin-1 is a lysosomal membrane protein required for intracellular lactosylceramide traffic. Traffic 7, 1388-1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R., Kiselyov K. (2009). TRPMLs: in sickness and in health. Am. J. Physiol. Renal Physiol. 296, F1245-F1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel K. G., Zwaan J., Kenyon K. R., Kolodny E. H., Hanninen L., Albert D. M. (1985). Ocular abnormalities in mucolipidosis IV. Am. J. Ophthalmol. 99, 125-136 [DOI] [PubMed] [Google Scholar]
- Schiffmann R., Dwyer N. K., Lubensky I. A., Tsokos M., Sutliff V. E., Latimer J. S., Frei K. P., Brady R. O., Barton N. W., Blanchette-Mackie E. J., et al. (1998). Constitutive achlorhydria in mucolipidosis type IV. Proc. Natl. Acad. Sci. USA 95, 1207-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L. M., Mueller L., Collins S. J. (1996). E3, a hematopoietic-specific transcript directly regulated by the retinoic acid receptor alpha. Blood 88, 2517-2530 [PubMed] [Google Scholar]
- Slaugenhaupt S. A., Acierno J. S., Jr, Helbling L. A., Bove C., Goldin E., Bach G., Schiffmann R., Gusella J. F. (1999). Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am. J. Hum. Genet. 65, 773-778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Goldin E., Stahl S., Falardeau J. L., Kennedy J. C., Acierno J. S., Jr, Bove C., Kaneski C. R., Nagle J., Bromley M. C., et al. (2000). Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 9, 2471-2478 [DOI] [PubMed] [Google Scholar]
- Tellez-Nagel I., Rapin I., Iwamoto T., Johnson A. B., Norton W. T., Nitowsky H. (1976). Mucolipidosis IV. Clinical, ultrastructural, histochemical, and chemical studies of a case, including a brain biopsy. Arch. Neurol. 33, 828-835 [DOI] [PubMed] [Google Scholar]
- Venkatachalam K., Long A. A., Elsaesser R., Nikolaeva D., Broadie K., Montell C. (2008). Motor deficit in a Drosophila model of mucolipidosis type IV due to defective clearance of apoptotic cells. Cell 135, 838-851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S., Puertollano R. (2006). Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 7, 337-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S., Connelly P. S., Daniels M. P., Puertollano R. (2008a). Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 17, 2723-2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergarajauregui S., Oberdick R., Kiselyov K., Puertollano R. (2008b). Mucolipin 1 channel activity is regulated by protein kinase A-mediated phosphorylation. Biochem. J. 410, 417-425 [DOI] [PubMed] [Google Scholar]
- Yang H., Xiong F. X., Lin M., Yang Y., Nie X., Zhou R. L. (2010). LAPTM4B-35 overexpression is a risk factor for tumor recurrence and poor prognosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 136, 275-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.