

Abstract
Purpose
Stimulation of toll-like receptor-9 (TLR9) by CpG oligodeoxynucleotides (CpG-ODN) has been shown to counteract the immunosuppressive microenvironment and to inhibit tumor growth in glioma models. These studies, however, have used high doses of CpG-ODN which can induce toxicity in a clinical setting. The goal of this study was to evaluate the anti-tumor efficacy of multiple low-dose intratumoral CpG- ODN in a glioma model.
Experimental Design
Mice bearing four-day old intracranial GL261 gliomas received a single or multiple (two or four) intratumoral injections of CpG-ODN (3 μg) every 4 days. Tumor growth was measured by bioluminescent imaging, brain histology, and animal survival. Flow cytometry and cytotoxicity assays were used to assess anti-glioma immune response.
Results
Two and four intracranial injections of low-dose CpG-ODN, but not a single injection, eradicated gliomas in 70% of mice. Moreover, surviving animals exhibited durable tumor free remission (> 3 months), and were protected from intracranial rechallenge with GL21 gliomas, demonstrating the capacity for long-term anti-tumor immunity. Although most inflammatory cells appeared to increase, activated NK cells (i.e. NK+CD107a+) were more frequent than CD8+CD107a+ in the brains of rechallenged CpG-ODN-treated animals and demonstrated a stronger in vitro cytotoxicity against GL261 target cells. Leukocyte depletion studies confirmed that NK cells played an important role in the initial CpG-ODN anti-tumor response, but both CD8 and NK cells were equally important in long-term immunity against gliomas.
Conclusions
These findings suggest that multiple low-dose intratumoral injections of CpG-ODN can eradicate intracranial gliomas possibly through mechanisms involving NK mediated effector function.
Keywords: Brain neoplasm, immunotherapy, myeloid-derived suppressor cells, toll-like receptor-9
Introduction
Despite aggressive treatment, the prognosis of patients with malignant glioma, the most common type of primary brain neoplasm, remains dismal (1). In addition to the development of targeted therapies, immunotherapy is being studied as an adjunct treatment option for these tumors. The selectivity of immunotherapy and the long-lasting memory of the immune system suggest that immunotherapy can potentially prevent tumor recurrence that most often occurs within the site of therapy. Several early-stage clinical trials using vaccination approach have been conducted with modest clinical efficacy in selected patients (2–5). Although further research in this area is ongoing, the ability of gliomas to escape immune response will continue to be a significant obstacle to this strategy as gliomas have developed several mechanisms to avoid the multiple effector arms of the immune system (6).
One strategy to overcome the local immunosuppressive tumor microenvironment is through the activation of the innate immune system (7–9). TLR-family members are pattern-recognition receptors that collectively recognize lipid, carbohydrate, peptide and nucleic-acid structures that are broadly expressed by micro-organisms. Among their key functions, TLRs enhance the uptake of micro-organisms by phagocytic cells and mediate leukocyte recruitment to infected tissues. This broad immune activation has made TLR agonists attractive candidates for vaccine adjuvants for cancer therapy. TLR9 is also found in brain microglia and macrophages (MP) (10), and direct injections of high doses of CpG oligonucleotides (CpG-ODN, 10-100 μg), a TLR9 ligand, into intracranial tumors triggers long-term immunity and tumor rejection in glioma and neuroblastoma-bearing mice (11). CpG-ODN as a single intratumoral injection has also been studied in patients with recurrent gliomas with tolerable toxicity and a partial tumor response in a few patients (7, 12). Although higher CpG-ODN doses may improve clinical efficacy, significant inflammatory response in an already edematous brain may hinder this approach. Because repeated administration of CpG-ODN has resulted in sustained immune activation (13–15), we hypothesized that multiple low-dose CpG-ODN injections can also be used as an alternative to high-dose treatment strategy for gliomas.
Here we report that multiple injections of low-dose CpG-ODN induced long-term tumor remission in mice bearing established gliomas. The observed immune-mediated tumor eradication correlated with increased activated NK cell activity and protected animals from tumor rechallenge. Our data indicate that multiple low-dose intratumoral injections of CpG-ODN eradicated intracranial gliomas through activation of NK cells, and induced anti-tumor immunity through induction of both NK and CD8 cells.
Materials and Methods
Cell lines and cell cultures
GL261, murine glioma cell line of C57BL/6 origin, was stably transfected with firefly luciferase expression vector and positive clones (GL261-ffluc) were selected using zeocin (1mg/ml) and G418, respectively. B16-F10, a melanoma cell line of C57BL/6 origin was a gift from Dr. K. Aboody at City of Hope. All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 370C in a humidified 5% CO2 atmosphere.
Oligodeoxynucleotides (ODN)
Single-stranded oligodeoxynucleotides CpG-ODN (5’-TGACTGTAACGTTCGAGATGA-3’) and control ODN (5 ’-TGACTGTAAGGTTAGAGATGA-3’) were purchased from Integrated DNA Technologies (IDT, Coralville, IA). CpG-ODN and control ODN were reconstituted in sterile water at concentration of 1 μg/μl and stored in −80°C for future use.
Tumor Implantation, Tumor Treatments, and Tumor Rechallenge
All animals were housed and handled in accordance to the guidelines of City of Hope Institutional Animal Care and Use Committee (IACUC). Intracranial tumor implantation was performed as described previously (16). GL261-ffluc cells were harvested by trypsinization, counted, and resuspended in PBS. C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) weighing 15–25 g were anesthetized by intraperitoneal (i.p.) administration of ketamine (132mg/kg) and xylazine (8.8mg/kg), and immobilized in a stereotactic head frame. Through a small burr hole, 3 μl of PBS containing 1 × 105 tumor cells was injected unilaterally at the coronal suture, 1 mm lateral to the midline and 3 mm deep into the frontal lobes, using a Hamilton syringe (Fisher Scientific, Pittsburgh, PA).
Four days after intracranial implantation of GL261-ffluc cells, mice received one or multiple (two or four) intracranial injections of CpG-ODN (3μg), control ODN (3μg), and PBS every four days. The intracranial treatment was administered through the initial burr hole aiming to target the tumor site (5–7mice/group).
Three months after initial treatment with CpG-ODN, all surviving animals were imaged to confirm the tumor eradication and then rechallenged with either GL261 or B16-F10 (1x105 cells) (6mice/group). In some experiments, absence of tumor was also confirmed by tissue histology (2–3 mice/experiment).
Flow Cytometry Analysis
Tumors, brain tissue at the injection sites, spleen, and blood samples were harvested for flow cytometry. Samples were prepared for flow cytometry as described previously (17). Cell suspensions from brain tissue, and spleen were forced through a 40 μm filter. Spleen and blood samples were incubated in Gey’s buffer (pH 7.2) for 10 min. For extracellular staining of immune markers, freshly prepared samples were resuspended in 0.1 M PBS containing 1% FBS and 2mM EDTA and incubated with FcγIII/IIR-specific antibody to block nonspecific binding. Samples were then stained with different combinations of antibodies (CD11b, CD45, Gr-1, CD8, NK1.1, CD107a, CD4, CD25) or isotype controls for 1 h at 4°C. For intracellular staining cells were fixed in 2% paraformaldehyde (Cell Sciences, Canton, MA) and permealized in methanol before incubation with FoxP3 or IFNγ. All antibodies (Ab) and isotype controls were purchased from BD Biosciences (San Jose, CA) or eBiosciences (San Diego, CA). Fluorescence data were collected on CyAn fluorescence cell sorter (BDIS, San Jose, CA). Inflammatory cells were gated and separated from the rest of sorted cells base on forward vs. side-scatter analysis and their staining characteristics. FlowJo 8.4.7 software (Tree Star, Inc., Ashland, OR) was used for data analysis and the proportion of each cell type was measured as percent of total inflammatory cells (4mice/group).
Luciferase-Based Cytotoxicity Assay
Blood samples were collected from cardiac puncture. Peripheral blood was diluted 1:3 in culture medium and layered on an equal volume Ficoll PaqueTM PLUS (GE Healthcare, Piscataway, NJ). The tubes were spun down for 20 min at 400 × g, at RT. The PBMCs in the interphase were collected and washed twice in 12 ml culture medium at 200×g, 5 min. Brains were harvested, minced, and forced through 40 μm cell strainer. Both brain and blood samples were stained with NK1.1, CD8, and PI for live/dead selections (eBiosciences, San Diego, CA). Samples were then washed and sorted with MoFlo MLS cell sorter (Beckman Coulter, Fullerton, CA). Effector cells (CD8+CD3+PI- and NK1.1+CD3-PI-) from both brain and blood samples were isolated. Target GL261-ffluci cells were cultured in DMEM supplemented with 10% FBS. Effector (E) and target (T) cells were added with following ratios 1:1 and 10:1. The plates were spun down at 100 × g for 3 min and incubated in 37°C for 4 h. Luminescence was measured by a Wallac 1420 VICTOR Luminometer (PerkinElmer Life and Analytical Sciences, Inc, Wellesley, MA) and percent cytotoxicity was measured as described previously (18).
Depletion Assays
Anti-NK1.1 (clone PK136) was purchased from eBioscience (eBioscience Inc., San Diego, CA). Anti-CD8 Ab (clone H35) was a kind gift from Dr. Don Diamond (City of Hope, Duarte, CA) and was purified as previously described (19). Control normal mouse IgG was purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology Inc., Santa Cruz, CA).
For initial leukocyte depletion studies, mice were treated with anti-CD8, anti-NK1.1, or control IgG (200 μg/mouse, i.p.) one day prior to tumor implantation and each CpG-ODN treatments. Leukocyte depletion was confirmed with FACS analysis of peripheral blood from treated animals (data not shown).
To check for memory immune response, tumor-bearing mice that had survived multiple CpG-ODN treatments were depleted of CD8 or NK cells (200 μg/mouse, i.p.) one day prior to tumor rechallenge.
Biophotonic Tumor Imaging
Tumor-bearing animals were injected i.p. with D-luciferin substrate (4.29 mg/mouse) (Xenogen, Palo Alto, CA). Mice were anesthetized by isoflurane (1.5 L/min oxygen + 4% isoflurane) and kept in an induction chamber. Images were captured with the Xenogen IVIS In Vivo Imaging System (Xenogen, Palo Alto, CA). Light emission was measured over an integration time of 1 min, 12 min after injection of luciferin. Luciferase activity was analyzed using Living Image Software (Xenogen, Palo Alto, CA) to quantify tumor region flux (photons per second) and to confirm tumor growth.
Statistical Analysis
Statistical comparison in all different experimental conditions was performed with the prism software using two-way analysis of variance (ANOVA) or Student’s T test. Survival was plotted using a Kaplan-Meir survival curve and statistical significance was determined by the Log-rank (Mantel-Cox) test. All plots and statistical analysis was performed with Prism software. A P value of less than 0.05 was considered significant.
Results
Multiple low-dose CpG-ODN injections improves survival of glioma-bearing mice
Mice bearing four-day old intracranial tumors were treated with one or multiple (2 or 4) injections of CpG-ODN, control ODN, and PBS every four days (Figure 1). Tumor eradication in CpG-ODN treated mice was confirmed by biophonic imaging of the GL261-ffLuc tumor (Figure 1A), and for a subset of mice, by tumor histology (data not shown). Four intratumoral injections of low-dose CpG-ODN eradicated intracranial gliomas in 71% of mice, while multiple injections of control ODN or PBS did not induce a survival benefit (Figure 1B). The efficacy of CpG-ODN appeared to depend on the number of injections as two CpG-ODN treatments also improved survival (cure rate of 71%), whereas no survival benefit was observed with a single low-dose CpG-ODN injection (Figure 1C).
Figure 1.

Multiple low-dose CpG oligonucleotide (CpG-ODN) injections improve survival of glioma-bearing mice. Mice bearing four-day old intracranial GL261-ffLuc tumors were treated with single (x1), two (x2) or four (x4) intratumoral injections of low-dose CpG-ODN (3μg/injection), control ODN (3μg/injection), or phosphate buffered saline (PBS) every four days (arrows). (A) Intracranial tumor burden was assessed by biophotonic imaging of mice at day 7, 14 and 21-post tumor injection. (B) Kaplan-Meier analysis demonstrates improved survival for mice that received four injections of CpG-ODN but not control ODN. (C) Survival was also improved in mice receiving two injections of CpG-ODN but not control ODN. n=7 mice/group, P values determined by Student’s T test. Data is representative of two separate experiments.
Effect of CpG ODN on cytotoxic CD8 and NK cells
In order to investigate inflammatory cellular responses to tumor growth and its differences between one vs. multiple CpG-ODN injections, mice bearing 4 day-old intracranial tumors were treated with CpG-ODN, control ODN, and PBS once or four times (every four days) similar to the survival experiments. Twenty-four hours after the last injection, brain, spleen, and blood were harvested and analyzed by flow cytometry (Figure 2). Several interesting observations were made in these experiments. First, irrespective of treatment type, the frequency of intratumoral NK cells (45–60%) was much higher than CD8 cells (0.5–1%), suggesting a stronger innate immune response to initial tumor implantation. Second, brain, blood, and spleen NK cells significantly decreased in frequency as intracranial tumors grew (Figure 2, black bars, lower panel in one vs. four PBS-treated groups, tumors harvested at day 5 and 17, respectively) highlighting the presence of local and systemic immunosuppressive tumor factors. Third, the relative frequency of brain, blood, and spleen NK cells (but not CD8 cells) in multiple CpG-ODN-treated group was lower than the single-injected mice (Figure 2, red bars, lower panel in one vs. four CpG-ODN-treated groups) and similar to CpG-ODN-treated normal mice (data not shown). Since most of these animals were tumor free, this observation suggests that CpG-ODN-induced NK cell response may have directly correlated to the extent of tumor burden. Finally, despite an increase in CD8+ cells, NK cells continue to be more frequent in CpG-ODN-cured animal brains (~2% vs. 20%).
Figure 2.
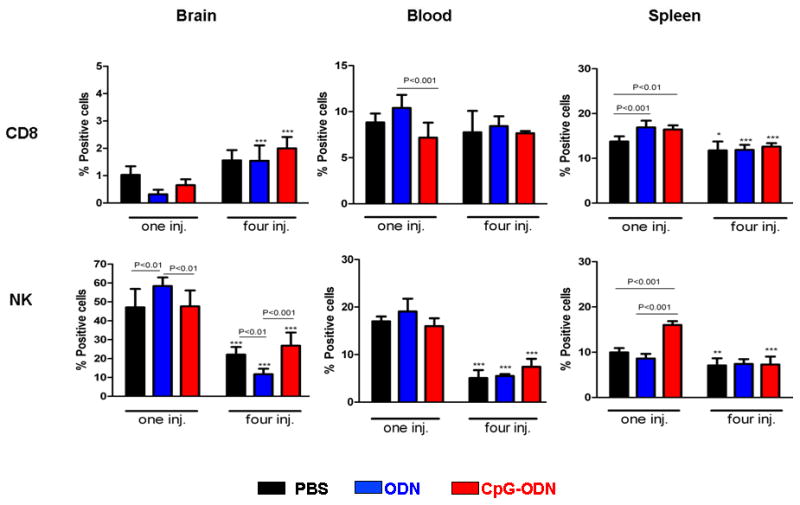
Effect of CpG-ODN on CD8 and NK cells. Mice bearing four day-old intracranial GL261 tumors were treated with intratumoral CpG-ODN (3μg/injection), control ODN (3μg/injection), and PBS once (one inj.) or four (four inj.) times as described in Fig. 1. Twenty-four hr after the last injection, brain, spleen, and blood were harvested and analyzed by flow cytometry for the percentage of CD8 (top panel) and NK (bottom panel) cells. P values for significant differences between treatment groups are provided above each bar graph. Significant differences between one and four injections for each treatment is represented as *: p<0.05, **: p<0.001, ***: p<0.0001. n=4 mice for each treatment group. Data is representative of two separate experiments.
Effect of CpG-ODN on immunosuppressive cells
Since previous reports have linked CpG-ODN to an increase in immunosuppressive cells (20), we next evaluated the effect of the number of CpG-ODN injections on regulatory T cells (Tregs; CD4+CD25+FOXP3+) and myeloid-derived suppressor cells (CD11b+Gr-1+, MDSCs). Both Tregs (brain) and MDSCs (brain and blood) increased with tumor growth (Figure 3, black bars in one vs. four PBS-treated groups), underscoring the role of these cells in tumor immunosuppression. In contrast to Tregs, MDSCs were more prevalent in the blood and significantly increased in the brain, blood and spleens in response to a single CpG-ODN injection (Figure 3, one injection CpG-ODN-treated group). These observations suggested that perhaps an influx of MDSCs may have abrogated the stimulatory effects of a single injection of CpG-ODN.
Figure 3.
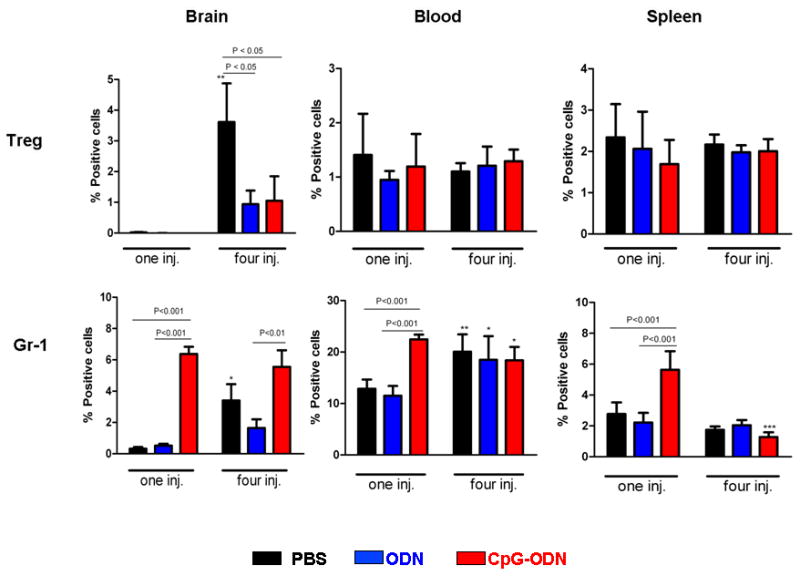
Effect of CpG-ODN on regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSC, Gr-1). Mice bearing four day-old intracranial GL261 tumors were treated with CpG-ODN (3μg/injection), control ODN (3μg/injection), and PBS once (one inj.) or four (four inj.) times as described in Fig. 1. Twenty-four hr after the last injection, brain, spleen, and blood were harvested and analyzed by flow cytometry for Treg and MDSCs by flow cytometry. P values for significant differences between treatment groups are provided above each bar graph. Significant differences between one and four injections for each treatment is represented as *: p<0.05, **: p<0.001, ***: p<0.0001. n=4 mice for each treatment group. Data is representative of two separate experiments.
Intracranial rechallenge of CpG-ODN-treated mice with tumor cells increases NK activity and cytotoxicity
To determine which cells were mostly responsible for tumor eradication, non-tumor bearing normal and CpG-ODN-treated mice that had survived for at least three months were rechallenged with an intracranial injection of GL261 cells. Three days after rechallenge, brain and blood samples from normal (naïve) (N), GL261-challenged naïve (T), CpG-ODN-treated survived (CS), and GL261-rechallenged CpG-ODN-treated survived mice (CR) were harvested and compared for cytotoxicity activity. Significant increase (P<0.001) in IFNγ production in both brain and blood of CR group, and to a lesser degree in the T group (P< 0.05), confirmed antitumor cytotoxicity activity in response to tumor challenge in both naïve and CpG-ODN-treated mice (Figure 4A). Although in naïve mice CD8 activation (CD8+CD107a+) appeared to be more pronounced than NK activation (NK+CD107a+) in the brain, the reverse was seen in CR group, suggesting that NK cells may be the primary cells responsible for anti-tumor immunity (Figure 4B). Furthermore, to compare NK and CD8 anti-glioma cytolytic activity, these cells were isolated from blood and brains of CR mice and incubated with GL261 target cells. Brain NK cell cytotoxicity was significantly higher than CD8 cells, suggesting that local activation of NK cells may be responsible for GL261 rejection in the rechallenged CpG-ODN-treated mice (Figure 4C).
Figure 4.
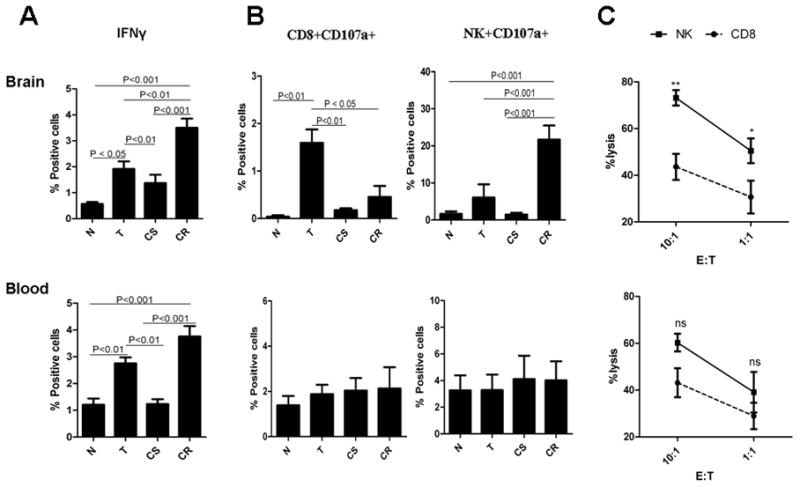
Effect of tumor rechallenge on CD8 and NK cell activation. Normal mice and CpG-ODN-treated mice (4 injections) that had survived for at least three months after initial tumor inoculation were rechallenged with an intracranial injection of GL261 cells. Brain and blood samples from normal (naïve) (N), GL261-challenged naïve (T), CpG-ODN-treated survived (CS), and GL261-rechallenged CpG-ODN-treated survived mice (CR) were harvested and compared by flow cytometry for (A) IFNγ production, (B) activation of CD8 and NK cells (CD107a expression), and (C) CD8 and NK cytotoxicity activity against GL261 target cells. ns: not significant, *: p<0.05; **: p<0.001. n=4 mice for each treatment group. Data is representative of two separate experiments.
Anti-glioma effect of low-dose CpG-ODN is primarily mediated by NK cells
To verify if the anti-glioma effect of CpG-ODN was solely due to activated NK cells, depleting doses of CD8 or NK-specific Abs were given one day prior to tumor implantation and prior to each CpG-ODN treatment. The CpG-ODN anti-tumor effect was completely abrogated by NK depletion (Figure 5A and B), while depletion of CD8 cells partially reduced tumor growth rate (Figure 5), confirming that the initial antitumor response by multiple intracranial low-dose CpG-ODN was mostly mediated through NK cell activation.
Figure 5.
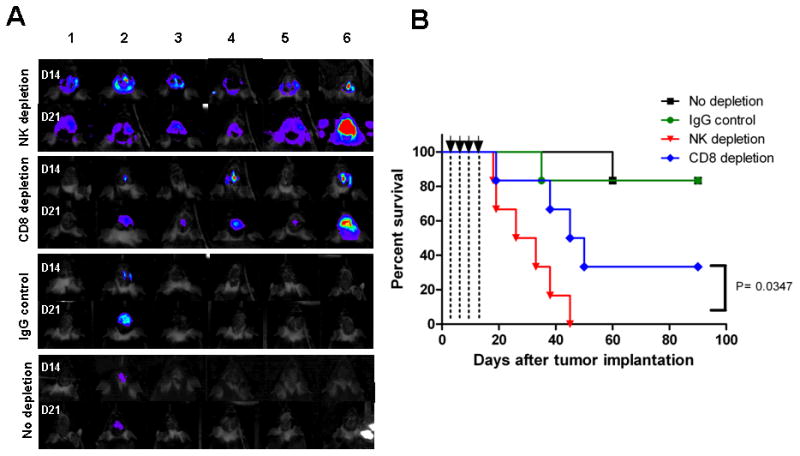
Anti-glioma effect of low-dose CpG-ODN is primarily mediated by NK cells. naïve mice were depleted of CD8 or NK cells by i.p. injection of relevant mAb or control IgG (200 μg/injection) one day prior to tumor implantation and each CpG-ODN treatment (3μg/injection, arrows). (A) Intracranial tumor burden was assessed by biophotonic imaging of mice at day 14 and 21 post tumor implantation. NK-depleted mice exhibited more rapid tumor growth. (B) Kaplan-Meier analysis demonstrates lower survival rate for mice that were depleted of NK cells (p=0.348). n=6 mice for each group. Data is representative of two separate experiments.
CpG ODN-treated mice develop immunity against gliomas
To determine if CpG-ODN treatment induced immunity against gliomas, CpG-ODN-treated GL261-bearing mice that had survived for at least three months, along with naïve mice, were rechallenged with an intracranial injection of GL261 glioma or B16 F10 melanoma cells. These “cured” mice not only developed full immunity against GL261 rechallenge (Figure 6A), but also partial immunity against B16 F10 melanomas (Figure 6B), suggesting that the antitumor response seen with multiple intracranial CpG-ODN injections was not tumor specific, and most likely mediated through NK cell activation. To assess NK anti-glioma cytolytic activity in these mice, NK cells from CpG-ODN-treated rechallenged and GL261-injected naïve mice were incubated with GL261 target cells. Cytotoxicity of NK cells from CpG-ODN-treated rechallenged mice was significantly higher than NKs from GL261-injected naïve mice. These findings strongly suggested that NK cells were reactivated with glioma rechallenge and possibly responsible for anti-tumor immunity seen with multiple low-dose CpG-ODN treatments (Figure 6C).
Figure 6.
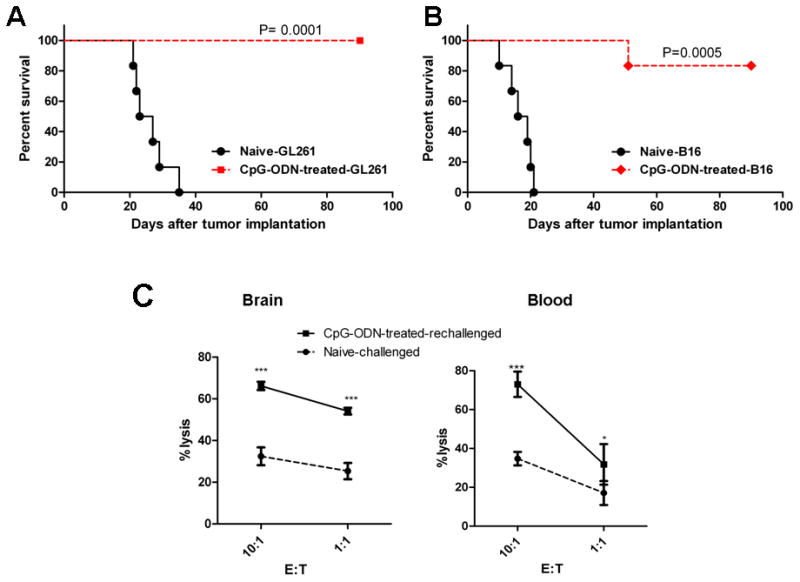
CpG-ODN-treated mice develop immunity against tumor rechallenge. Normal naïve mice and CpG-ODN-treated GL261-bearing mice that had survived for at least three months after the initial tumor inoculation were rechallenged with an intracranial injection of (A) GL261 glioma (1x105) or (B) B16 F10 melanoma (1x105) cells. Tumor growth was assessed by measuring animal survival (A and B) while NK cytotoxicity assay was measured by incubating isolated brain and blood NK cells with GL261 target cells. *: p<0.05, **: p<0.001, ***: p<0.0001. n=7 mice/group for survival experiments and 4 for cytotoxicity assays. Data is representative of two separate experiments.
Both NK and CD8 cells contribute to the CpG-ODN-induced immunity against gliomas
Finally, to determine if the developed immunity to GL261 gliomas was solely due to reactivation of memory NK cells, CpG ODN-treated survived mice were depleted of either CD8 or NK cells one day prior to tumor rechallenge. Implanted intracranial GL261 tumors were rapidly rejected in both groups (Supplementary Figure 1) indicating that either both NK and CD8 cells played a role in anti-glioma memory, or that systemic administration of anti NK or CD8 Ab was inadequate in depleting anti-tumor memory cells that may have resided in the brain after tumor rejection.
Discussion
TLR9 agonists are potent stimulators of both innate and adaptive immune systems. They induce cytokines, activate NK cells, monocytes, and elicit T-cell responses leading to anti-tumor effects (21). Based on promising preclinical data, CpG-ODNs are currently being tested as single agents, vaccine adjuvants, or in combination with other therapies in patients with various cancers (22). The efficacy of CpG-ODN immunotherapy has also been studied in brain tumors. Initial reports by Carpentier et al. demonstrated an 88% cure rate in rats bearing intracranial CNS-1 gliomas after multiple direct intratumoral CpG-ODN injections. Interestingly, cured animals were protected from tumor rechallenge 12 weeks after the initial therapy, suggesting induction of antitumor immunity in an organ that is considered “immune-privileged” (23, 24). Since then, however, a number of investigators have reported variable CpG-ODN anti-glioma responses ranging from no efficacy (25), partial response (9), to complete tumor eradication (26). Although differences in CpG-ODN constructs and glioma models could account for these inconsistent anti-tumor responses, most glioma studies have evaluated high doses of CpG-ODN (10-100 μg/mice or 100-200 μg/rat). Because of its pro-inflammatory response, high intracranial CpG-ODN could limit the clinical efficacy of this agent as brain edema, which is frequently present in patients with malignant brain tumors, may directly influence its dose-limiting toxicity. In fact, in clinical trials utilizing convection-enhanced intratumoral CpG-28 delivery conducted in patients with recurrent malignant glioma, a number of patients treated at highest dose (20 mg) experienced neurological worsening or seizures most likely related to the pro-inflammatory effects of CpG-28 (7, 12). One way to overcome this limitation is to use CpG-ODN in combination with radiation or as a vaccine adjuvant (25, 27). Here, however, we report that multiple low-dose intratumoral injections of CpG-ODN can be as effective in eradicating established intracranial gliomas, without causing significant brain edema or inflammation. This approach was well-tolerated by animals, and suggests that clinically improved efficacy with decreased therapy-related side-effects can be achieved by repetitive injections of low-dose CpG-ODN.
The exact anti-tumor mechanism of CpG-ODN is not clear but most likely due to both tumor cell apoptosis and immune activation. TLR9 is not only expressed on inflammatory cells, but also is present on gliomas (9, 28). CpG-ODN is efficiently taken up by glioma cells and induces apoptosis in vitro (9, 29). In vivo, direct intracranial CpG-ODN injection induces an inflammatory response mediated through activated T cells, NK cells, and glioma-infiltrating macrophages and microglia (30, 31). Using depletion studies, Roda et al showed that both NK and T cells contributed to tumor elimination (32). Our study confirmed that low-dose CpG-ODN also stimulated both CD8 and NK cells, but NK cells appeared to play a more significant role in the initial anti-tumor response. NK cell depletion completely abrogated the therapeutic effect of low-dose CpG-ODN, while CD8 depletion only retarded tumor growth. Furthermore, when CpG-ODN-cured mice were rechallenged with intracranial tumors, a strong local NK cell response was noted in the brain. Since NK levels had returned to baseline in CpG-ODN-cured mice, this finding suggests that perhaps activation of memory NK cells may have been responsible for anti-tumor immunity.
NK cells are important components of the innate immune system as they are cytotoxic towards tumor cells or virally infected cells without prior exposure to antigens and rapidly secrete IFNγ and other cytokines that activate the innate as well as adaptive immune responses (33). Although traditionally NK cells have been classified as cells of the innate immune system, in recent years the presence of immunological “memory” NK cells has been reported (34, 35). Recent reports by Sun et al have shown that NK cells demonstrate similar memory type characteristics as cytotoxic T cells (35). In our study, the initial CpG-ODN injection caused an increase in NK population which subsided after the fourth treatment and correlated with tumor rejection. Also, CpG-ODN-treated “cured” mice had low systemic levels of NK cells similar to naïve mice. But with tumor rechallenge, a robust NK cell response was noted in the brain and these mice were protected from not only glioma, but also from an unrelated tumor type. Interestingly, even after CD8 depletion, CpG-ODN-treated “cured” mice remained protected against intracranial tumor rechallenge. This anti-tumor NK cell response mimics the expansion, contraction, memory maintenance, and recall which are characteristic of memory NK cell responses (35).
Intratumoral NK infiltration was seen within the first week of tumor implantation, but these cells significantly decreased in frequency as tumors continued to grow. NK cell population not only retracted in the tumors but also in blood and spleen of glioma-bearing mice, most likely as a result of immunosuppressive factors secreted by gliomas (6). In addition to inhibitory cytokines, some inflammatory cell types within gliomas can play a role in immune tolerance. Of note, Tregs can actively suppress T and NK cell immune responses (36–38). Consistent with other reports, we detected a significant expansion of tumor Tregs in tumor-bearing mice that decreased in response to CpG-ODN therapy. A significant increase in the CD4 and CD8 to Treg ratio was also reported to be important in induction of anti-glioma response to high-dose CpG-ODN by Grauer et al (26). In our studies, however, Tregs accounted for only a small fraction of inflammatory cells and were not as prevalent as MDSCs which can also play a role in tumor immune escape.
Myeloid-derived suppressors cells are a heterogeneous population of cells of myeloid origin that have been reported to suppress the immune system and promote tumor growth (39). Studies have linked MDSCs to suppression of T cells (40–44), NK T cells (45), and NK cells (46, 47). Makarenkova et al showed that MDSC expression of L-arginine (arginase-mediated) may be the cause of T cell dysfunction following surgery and trauma (48). Recently, other studies have linked MDSCs to the suppression of NK cytotoxicity (46, 47). In one report, isolated CD11b+Gr-1+ cells from tumor bearing mice abolished NK cells ability to kill target cells (47). Also, Li et al recently showed that in tumor-bearing mice MDSCs can suppress NK cytototoxicty by inhibiting NKG2D expression and IFNγ production (46). Consistent with other reports, our study showed that single CpG-ODN injection resulted in significant increase in CD11b+Gr-1+ cells (20, 49). Because multiple CpG-ODN injections were not associated with a significant Gr-1+ flux in spleen or blood, we postulate that the induction of MDSCs after the first CpG-ODN injection may have suppressed the initial anti-glioma NK response. This hypothesis will be tested in future studies.
In summary, we demonstrated that multiple low-dose intratumoral injections of CpG-ODN can eradicate intracranial gliomas and induce immunity against tumor rechallenge. Activation of NK cells following CpG-ODN treatment or rechallenge of CpG-ODN “cured” mice, and partial immunity against B16 melanomas strongly suggested the involvement of an NK response. Furthermore, suppression of MDSC response following multiple CpG-ODN injections may have played a role in NK cell activation and induction of memory response. Future experiments will assess the role of MDSC depletion on CpG-ODN immunotherapy of gliomas.
Statement of Translational Relevance.
The prognosis of patients with malignant gliomas, the most common type of primary brain neoplasm, remains dismal even after aggressive multimodality treatment. Although immunotherapy is being investigated as an adjunct treatment, the ability of gliomas to escape immune response will continue to be a significant obstacle to this strategy. One approach to overcome the local immunosuppressive tumor microenvironment is the activation of the innate immune system by toll-like receptor agonists such as CpG oligonucleotides (CpG-ODN). Direct intratumoral injections of CpG-ODN at high doses can eradicate intracranial gliomas in animals and is currently being investigated in clinical trials. Because high-dose CpG-ODN can induce a significant inflammatory response in the brain, we evaluated the efficacy of low-dose injections in a murine glioma model. Here we report that multiple intracranial injections of CpG-ODN were non-toxic, effectively eradicated established gliomas, and triggered long-term immunity through activation of NK and CD8 cells. These findings have direct application to the design of future anti-glioma clinical trials with CpG-ODN.
Supplementary Material
Both NK and CD8 cells contribute to the CpG-ODN-induced immunity against gliomas. GL261-bearing mice that were tumor free for at least two months after the four low-dose CpG-ODN treatments were depleted of NK or CD8 one day prior to tumor rechallenge with GL261-ffluci (1x105) cells. (A) Tumor progression was assessed by biophotonic imaging of mice at day 2, 6, 8, 10 and 12-post tumor injection. (B) Luciferase expression as photon flux (photons/s) shows tumor rejection in both NK and CD8 depleted groups. Values are expressed as photon flux (photons/s) per mg of protein. n=5 mice for each group. Data is representative of two separate experiments.
Acknowledgments
Grant Support: This work was supported by NIH (R21CA131765-01A2) and James S. McDonnell Foundation (to BB). The City of Hope Flow Cytometry Core was equipped in part through funding provided by ONR N00014-02-1 0958, DOD 1435-04-03GT-73134, and NSF DBI-9970143.
Literature Cited
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 3.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20:267–75. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamanaka R, Abe T, Yajima N, et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172–9. doi: 10.1038/sj.bjc.6601268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64:4973–9. doi: 10.1158/0008-5472.CAN-03-3505. [DOI] [PubMed] [Google Scholar]
- 6.Gomez GG, Kruse CA. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene therapy & molecular biology. 2006;10:133–46. [PMC free article] [PubMed] [Google Scholar]
- 7.Carpentier A, Laigle-Donadey F, Zohar S, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8:60–6. doi: 10.1215/S1522851705000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalpke AH, Schafer MK, Frey M, et al. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–63. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- 9.El Andaloussi A, Sonabend AM, Han Y, Lesniak MS. Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia. 2006;54:526–35. doi: 10.1002/glia.20401. [DOI] [PubMed] [Google Scholar]
- 10.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–24. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 11.Carpentier AF, Auf G, Delattre JY. CpG-oligonucleotides for cancer immunotherapy : review of the literature and potential applications in malignant glioma. Front Biosci. 2003;8:e115–27. doi: 10.2741/934. [DOI] [PubMed] [Google Scholar]
- 12.Carpentier A, Metellus P, Ursu R, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 12:401–8. doi: 10.1093/neuonc/nop047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heikenwalder M, Polymenidou M, Junt T, et al. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004;10:187–92. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- 14.Klinman DM, Conover J, Coban C. Repeated administration of synthetic oligodeoxynucleotides expressing CpG motifs provides long-term protection against bacterial infection. Infect Immun. 1999;67:5658–63. doi: 10.1128/iai.67.11.5658-5663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner I, Sethi S, Xiang W, Giese A, Ebner S, Kretzschmar H. Repeated peripheral administrations of CpG oligodeoxynucleotides lead to sustained CNS immune activation. Immunopharmacol Immunotoxicol. 2007;29:413–24. doi: 10.1080/08923970701675028. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia. 2009;57:1458–67. doi: 10.1002/glia.20863. [DOI] [PubMed] [Google Scholar]
- 17.VanHandel M, Alizadeh D, Zhang L, et al. Selective uptake of multi-walled carbon nanotubes by tumor macrophages in a murine glioma model. Journal of neuroimmunology. 2009;208:3–9. doi: 10.1016/j.jneuroim.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Brown CE, Wright CL, Naranjo A, et al. Biophotonic cytotoxicity assay for high-throughput screening of cytolytic killing. J Immunol Methods. 2005;297:39–52. doi: 10.1016/j.jim.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Daftarian P, Song GY, Ali S, et al. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res. 2004;64:5407–14. doi: 10.1158/0008-5472.CAN-04-0169. [DOI] [PubMed] [Google Scholar]
- 20.Kortylewski M, Kujawski M, Herrmann A, et al. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009;69:2497–505. doi: 10.1158/0008-5472.CAN-08-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uhlmann E, Vollmer J. Recent advances in the development of immunostimulatory oligonucleotides. Curr Opin Drug Discov Devel. 2003;6:204–17. [PubMed] [Google Scholar]
- 22.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 23.Carpentier AF, Chen L, Maltonti F, Delattre JY. Oligodeoxynucleotides containing CpG motifs can induce rejection of a neuroblastoma in mice. Cancer Res. 1999;59:5429–32. [PubMed] [Google Scholar]
- 24.Carpentier AF, Xie J, Mokhtari K, Delattre JY. Successful treatment of intracranial gliomas in rat by oligodeoxynucleotides containing CpG motifs. Clin Cancer Res. 2000;6:2469–73. [PubMed] [Google Scholar]
- 25.Wu A, Oh S, Gharagozlou S, et al. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J Immunother. 2007;30:789–97. doi: 10.1097/CJI.0b013e318155a0f6. [DOI] [PubMed] [Google Scholar]
- 26.Grauer OM, Molling JW, Bennink E, et al. TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol. 2008;181:6720–9. doi: 10.4049/jimmunol.181.10.6720. [DOI] [PubMed] [Google Scholar]
- 27.Meng Y, Carpentier AF, Chen L, et al. Successful combination of local CpG-ODN and radiotherapy in malignant glioma. Int J Cancer. 2005;116:992–7. doi: 10.1002/ijc.21131. [DOI] [PubMed] [Google Scholar]
- 28.Meng Y, Kujas M, Marie Y, et al. Expression of TLR9 within human glioblastoma. Journal of neuro-oncology. 2008;88:19–25. doi: 10.1007/s11060-008-9536-2. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Weinschenk T, Schluesener HJ. Uptake, cellular distribution and novel cellular binding proteins of immunostimulatory CpG oligodeoxynucleotides in glioblastoma cells. Mol Cell Biochem. 2005;272:35–46. doi: 10.1007/s11010-005-6605-0. [DOI] [PubMed] [Google Scholar]
- 30.Chu RS, Askew D, Noss EH, Tobian A, Krieg AM, Harding CV. CpG oligodeoxynucleotides down-regulate macrophage class II MHC antigen processing. J Immunol. 1999;163:1188–94. [PubMed] [Google Scholar]
- 31.Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med. 1997;186:1623–31. doi: 10.1084/jem.186.10.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roda JM, Parihar R, Carson WE., 3rd CpG-containing oligodeoxynucleotides act through TLR9 to enhance the NK cell cytokine response to antibody-coated tumor cells. J Immunol. 2005;175:1619–27. doi: 10.4049/jimmunol.175.3.1619. [DOI] [PubMed] [Google Scholar]
- 33.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 34.O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7:507–16. doi: 10.1038/ni1332. [DOI] [PubMed] [Google Scholar]
- 35.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–61. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fecci PE, Mitchell DA, Whitesides JF, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 37.Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–7. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- 38.Grauer OM, Nierkens S, Bennink E, et al. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121:95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Apolloni E, Bronte V, Mazzoni A, et al. Immortalized myeloid suppressor cells trigger apoptosis in antigen-activated T lymphocytes. J Immunol. 2000;165:6723–30. doi: 10.4049/jimmunol.165.12.6723. [DOI] [PubMed] [Google Scholar]
- 41.Bronte V. Myeloid-derived suppressor cells in inflammation: Uncovering cell subsets with enhanced immunosuppressive functions. Eur J Immunol. 2009 doi: 10.1002/eji.200939892. [DOI] [PubMed] [Google Scholar]
- 42.Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24:431–46. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Dolcetti L, Marigo I, Mantelli B, Peranzoni E, Zanovello P, Bronte V. Myeloid-derived suppressor cell role in tumor-related inflammation. Cancer Lett. 2008;267:216–25. doi: 10.1016/j.canlet.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689–95. doi: 10.4049/jimmunol.168.2.689. [DOI] [PubMed] [Google Scholar]
- 45.Yanagisawa K, Exley MA, Jiang X, Ohkochi N, Taniguchi M, Seino K. Hyporesponsiveness to natural killer T-cell ligand alpha-galactosylceramide in cancer-bearing state mediated by CD11b+ Gr-1+ cells producing nitric oxide. Cancer Res. 2006;66:11441–6. doi: 10.1158/0008-5472.CAN-06-0944. [DOI] [PubMed] [Google Scholar]
- 46.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182:240–9. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 47.Liu C, Yu S, Kappes J, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109:4336–42. doi: 10.1182/blood-2006-09-046201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176:2085–94. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 49.Morecki S, Gelfand Y, Yacovlev E, Eizik O, Shabat Y, Slavin S. CpG-induced myeloid CD11b+Gr-1+ cells efficiently suppress T cell-mediated immunoreactivity and graft-versus-host disease in a murine model of allogeneic cell therapy. Biol Blood Marrow Transplant. 2008;14:973–84. doi: 10.1016/j.bbmt.2008.06.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Both NK and CD8 cells contribute to the CpG-ODN-induced immunity against gliomas. GL261-bearing mice that were tumor free for at least two months after the four low-dose CpG-ODN treatments were depleted of NK or CD8 one day prior to tumor rechallenge with GL261-ffluci (1x105) cells. (A) Tumor progression was assessed by biophotonic imaging of mice at day 2, 6, 8, 10 and 12-post tumor injection. (B) Luciferase expression as photon flux (photons/s) shows tumor rejection in both NK and CD8 depleted groups. Values are expressed as photon flux (photons/s) per mg of protein. n=5 mice for each group. Data is representative of two separate experiments.