

Abstract
Vitamin C, or ascorbic acid, decreases paracellular endothelial permeability in a process that requires rearrangement of the actin cytoskeleton. To define the proximal mechanism of this effect, we tested whether it might involve enhanced generation and/or sparing of nitric oxide (NO) by the vitamin. EA.hy926 endothelial cells cultured on semi-porous filter supports showed decreased endothelial barrier permeability to radiolabeled inulin in response to exogenous NO provided by the NO donor spermine NONOATE, as well as to activation of the downstream NO pathway by 8-bromo-cyclic GMP, a cell-penetrant cyclic GMP analog. Inhibition of endothelial nitric oxide synthase (eNOS) with Nω-nitro-L-arginine methyl ester increased endothelial permeability, indicating a role constitutive NO generation by eNOS in maintaining the permeability barrier. Inhibition of guanylate cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one also increased endothelial permeability and blocked barrier tightening by spermine NONOATE. Loading cells with what are likely physiologic concentrations of ascorbate decreased endothelial permeability. This effect was blocked by inhibition of either eNOS or guanylate cyclase, suggesting that it involved generation of NO by eNOS and subsequent NO-dependent activation of guanylate cyclase. These results show that endothelial permeability barrier function depends on constitutive generation of NO and that ascorbate-dependent tightening of this barrier involves maintaining NO through the eNOS/guanylate cyclase pathway.
Keywords: paracellular transport, nitric oxide, endothelial permeability, endothelial nitric oxide synthase, guanylate cyclase
The vascular endothelium forms a barrier to transfer of serum proteins out of the blood vessel. The permeability of this barrier is increased during inflammation by the action of molecules such as thrombin, TNF-α, histamine, and various oxidants [1,2]. This breakdown in barrier function is due to formation of gaps between the cells that is caused by loss of integrity of the adherens and tight junctions between the cells and by a rearrangement and tightening of the actin cytoskeleton that pulls the cells apart [2,3]. Counteracting increased endothelial permeability are agents that tighten the endothelial barrier such as those that increase intracellular cyclic AMP [4], increase cyclic GMP [5,6], or release lipid-derived prostaglandin E2 [7] and sphingosine 1-phosphate [8].
Recent studies from this laboratory showed that intracellular vitamin C, or ascorbic acid, also decreases paracellular transfer of the polysaccharide inulin and of ascorbate itself across endothelial monolayers prepared from both large vessels and microcapillaries [9]. The decrease required less than an hour for completion and was not dependent on collagen synthesis. It was associated with changes in the actin-distribution in the cells and was blocked by the microtubule agent colchicine, suggesting that it required rearrangement of the cell cytoskeleton and thus cell shape.
Apart from involvement of the cytoskeleton, the proximal cellular mechanism of how ascorbate tightens the endothelial barrier has not been established. Ascorbate is a one-electron donor that reduces and thus detoxifies reactive oxygen and nitrogen species, serves as a co-factor for various dioxygenase enzymes by maintaining catalytic non-heme iron in the active or reduced ferrous state, and preserves endothelial nitric oxide (NO) by several mechanisms. First, it can reduce nitrite, the oxidized form of NO, back to NO, at least in vitro [10]. Second, it directly scavenges radical species that might either destroy NO or react with it to generate a more potent oxidant. An example of the latter is scavenging of superoxide by ascorbate to prevent reaction of superoxide with NO and subsequent generation of peroxynitrite, a strong oxidant and nitrosating agent [11]. Third and probably most important, ascorbate potentiates endothelial nitric oxide synthase (eNOS) activity and thus NO generation in endothelial cells [12]. It does this by recycling tetrahydrobiopterin [13,14], a necessary reducing factor for the active site iron of eNOS. These effects of ascorbate to preserve NO or promote its generation in endothelial cells are also evident at the clinical level, where ascorbate supplements have been shown to reverse NO-dependent endothelial dysfunction in atherosclerosis and other diseases (reviewed in [15]).
NO derived from eNOS decreases both basal [16] and stimulated [5] endothelial permeability, although the latter is not always observed [1,17]. This effect of NO is likely mediated through its stimulation of guanylyl cyclase to generate guanosine 3′,5′-cyclic monophosphate (cGMP) [5,6]. We reasoned, therefore, that ascorbate could do the same through its ability to preserve NO. Indeed, we found that EA.hy926 endothelial cells required NO to maintain basal endothelial permeability and that further ascorbate-dependent tightening of the endothelial barrier was prevented by inhibition of NO synthesis and generation of cyclic GMP, the downstream messenger of NO.
Materials and methods
Materials
Sigma/Aldrich Chemical Co. (St. Louis, MO) supplied the reagent chemicals, including ascorbate, dehydroascorbic acid (DHA), N-2-hydroxyethylpiperazine N′-2-ethanesulfonic acid (Hepes), Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME), 8-bromoguanosine 3′,5′-cyclic monophosphate (8-bromo-cGMP), and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ). ODQ was initially dissolved in a small amount of dimethylsulfoxide, and then diluted with culture medium such that the final dimethylsulfoxide concentration was 0.06%. Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA) supplied the [carboxyl-14C]inulin (molecular weight range 5000–5500, 2 mCi/g).
Cell Culture
EA.hy926 cells (originally provided by Dr. Dr. Cora Edgell, University of North Carolina, Chapel Hill, NC, USA) were cultured in Dulbecco’s minimal essential medium and 10% (v/v) heat-inactivated fetal bovine serum, which contained 20 mM D-glucose and HAT media supplement (Sigma/Aldrich Chemical Co., St. Louis, MO). These cells are a hybridoma line derived from human umbilical vein endothelial cells. They retain features of endothelial cells in culture, including a cobblestone appearance with formation of capillary-like tubes in culture [18], expression of factor VIII antigen [19], oxidative modification of human low density lipoprotein [20], and calcium-dependent endothelial nitric oxide synthase activity [20,21]. Cells were cultured at 37 °C in humidified air containing 5% CO2.
Assay of trans-endothelial ascorbate and inulin transfer
EA.hy926 cells were cultured to confluence in 6-well plates on polyethylene terephthalate cell culture inserts (0.4 micron pores at a density of 2 ± 0.2 × 106 pores per cm2, Falcon BD Biosciences, Franklin Lakes, NJ). After attaining confluence, cells were cultured an additional 5–6 days with 1.7 ml of medium in the upper well and 2.8 ml of medium in the lower well. All agents and [carboxyl-14C]inulin were added above the cells/filter and incubation was carried out at 37 °C for the times indicated. Medium above and below the cells/filter was sampled for assay for liquid scintillation counting of radiolabeled inulin.
The permeability of the endothelial cell layer to [carboxyl-14C]inulin and ascorbate was determined as previously described [22], with minor modification [9]. The permeability coefficient for [carboxyl-14C]inulin was calculated and corrected for the rate of [carboxyl-14C]inulin transfer across filters after removal of cells [23]. This adjusts for any changes in permeability due to deposition of the matrix laid down by the cells during culture.
Assay of cellular ascorbate and GSH
To measure intracellular ascorbate and GSH, cells adherent to filters were rinsed 3 times with Krebs-Ringer Hepes buffer (KRH) that consisted of 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2, pH 7.4. After the last rinse was removed, 0.1 ml of 25 % (w/v) metaphosphoric acid was added to the cell monolayer, the cells were scraped from the filter with a rubber spatula, and the acid was partially neutralized with 0.35 ml of 0.1M Na2HPO4 and 0.05 mM EDTA, pH 8.0. The cell lysate was removed, centrifuged at 3 °C for 1 min at 13,000 × g, and the supernatant was taken for assay of ascorbate and GSH. Assay of ascorbic acid was performed in duplicate by high performance liquid chromatography as previously described [24]. Cellular GSH content was measured by the method of Hissin and Hilf [25]. Intracellular ascorbate concentrations were calculated based on the intracellular distribution space of 3-O-methylglucose in EA.hy926 cells relative to the cell protein content, which was previously measured to be 3.6 ± 1.2 μl/mg protein [21].
Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using SigmaStat 2.0 software (Jandel Scientific, San Rafael, CA). Differences between treatments were assessed by two-way analysis of variance with post-hoc testing using Tukey’s or Dunnett’s test, as appropriate.
Results
Changes in endothelial permeability in response to NO were assessed using exogenous NO generated by treating EA.hy926 cells with spermine NONOATE, which has a half-life in physiologic buffers of 39 min [26,27]. Transfer of radiolabeled inulin across EA.hy926 cells that had been cultured for 6–7 days on semi-permeable membrane supports was progressively decreased to about 50% of baseline when the cells were treated with increasing concentrations of spermine NONOATE during the 1 h transfer assay (Fig. 1A, circles). To assess whether the effect of NO might be mediated by its activation guanylate cyclase and subsequent generation of cGMP, cells were treated with the cell-penetrant cGMP analog 8-bromo-cGMP. After a 30 min treatment with increasing concentrations of 8-bromo-cGMP, followed by the transfer assay, transfer of inulin was again progressively decreased to about 50% of basal (Fig. 1A, squares).
Figure 1. eNOS-derived NO decreases endothelial barrier permeability in EA.hy926 cells.
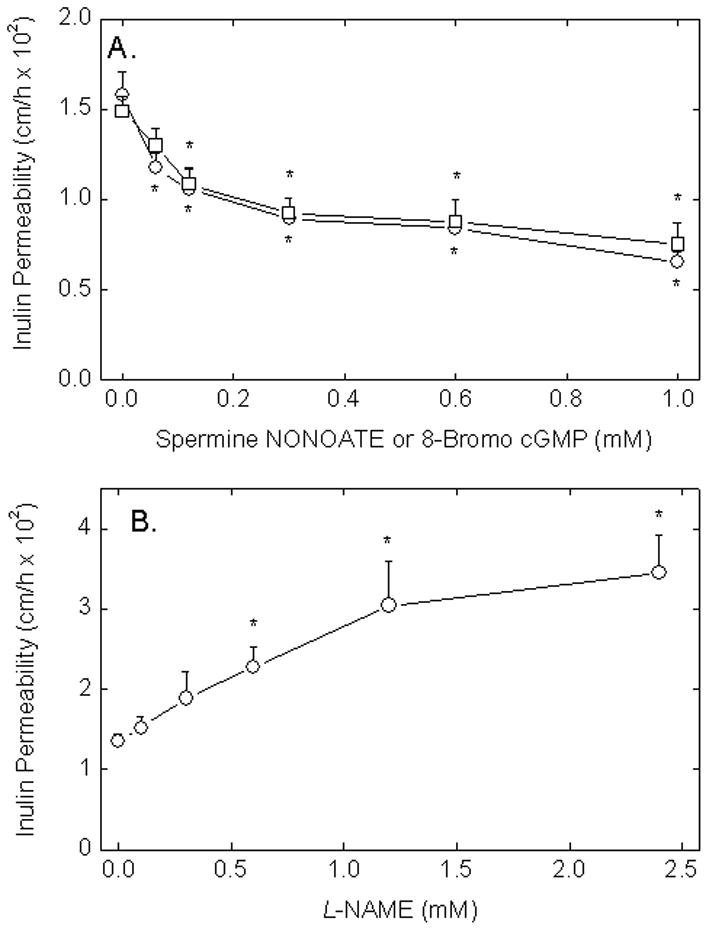
Panel A. Cells in culture on porous membrane filters were incubated at 37 °C with increasing concentrations of spermine NONOATE (circles) or 8-bromo-cGMP (squares). Spermine NONOATE was added just before the 60 min inulin transfer assay due to its instability, whereas cGMP was added 30 min before the assay to allow it to enter cells. The transfer assay was carried out as described in Materials and methods. Panel B. Cells were treated at 37 °C with increasing concentrations of L-NAME for 30 min followed by the 60 min inulin transfer assay. Results for each panel are shown from 3–5 experiments, with an “*” indicating p < 0.05 compared to the untreated sample.
To assess whether basal endogenous NO generation by eNOS affects endothelial permeability, EA.hy926 cells were treated with increasing concentrations of the eNOS inhibitor L-NAME for 30 min before carrying out the inulin transfer assay. As shown in Fig. 1B, increasing concentrations of L-NAME more than doubled endothelial permeability with a plateau above 1.2 mM. These results suggest that paracellular permeability in this cell system is responsive to NO derived from eNOS and that the decrease in permeability due to NO could be due to its ability to generate cGMP within the cells.
To further assess the role of cGMP as a mediator of basal NO-induced tightening of the endothelial barrier, cells were treated for 30 min with increasing concentrations of ODQ, a potent and specific inhibitor of guanylate cyclase [28]. As shown in Fig. 2A, inulin transfer was increased by ODQ in a concentration-dependent manner with a significant effect observed at 0.6 μM and above. This suggests that basal endothelial permeability is modulated by the ability of the cells to generate cGMP. To determine whether the effect of spermine NONOATE might be mediated by cGMP, cells were treated for 30 min with 0.3 μM ODQ before addition of spermine NONOATE and the transfer assay. This concentration of ODQ was selected as one that alone did not significantly increase endothelial permeability (Fig. 2A). As shown in Fig. 2B, the expected decrease in endothelial permeability by the NO donor was completely prevented by this low concentration of ODQ. This adds further support to the notion that NO acts through cGMP to tighten the endothelial barrier.
Figure 2. Inhibition of guanylate cyclase increases basal endothelial permeability and prevents NO-dependent decreases in permeability.
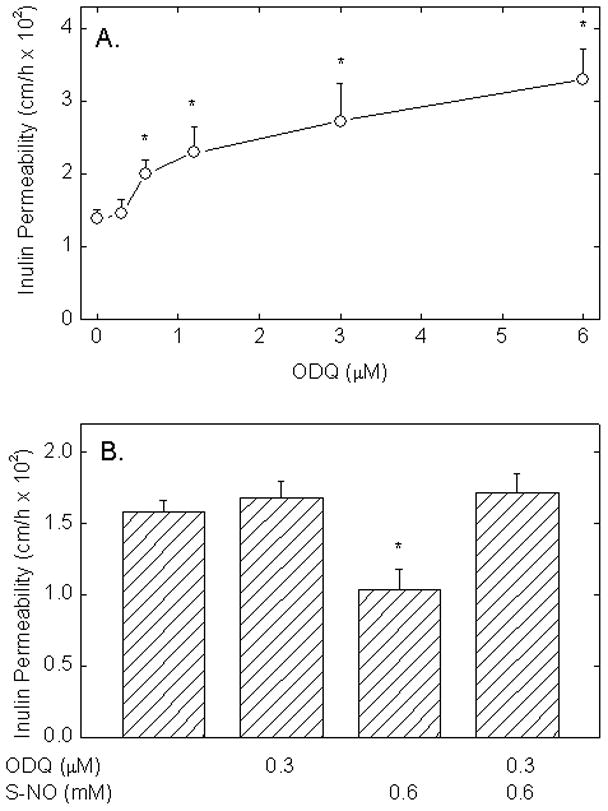
Panel A. EA.hy926 cells cultured on filters were treated for 30 min with the indicated concentrations of ODQ, followed by the radioactive inulin transfer assay. Panel B. Cells were incubated with or without ODQ as indicated for 30 min, spermine NONOATE was added as indicated, and the inulin transfer assay was carried out. Results are shown from 4 experiments for Panel A and from 5 experiments from Panel B, with an “*” indicating p< 0.05 compared to the control sample in Panel A and compared to all other samples in Panel B.
To assess the effect of intracellular ascorbate on endothelial permeability, cells were loaded with increasing concentrations of DHA. DHA is rapidly taken up on glucose transporters and reduced to ascorbate within the cells, leaving very low ascorbate concentrations outside the cells [29]. As shown in Fig. 3A, although these cells contained only traces of ascorbate in culture, treating them with increasing concentrations of DHA for 90 min at 37 °C resulted a linear increase in intracellular ascorbate concentrations. This time of loading was the same as that used in the inulin transfer assay (Fig. 3B), which showed a significant decrease in endothelial permeability at intracellular ascorbate concentrations as low as 0.3 mM. The decrease in endothelial permeability reached about 50% when the intracellular ascorbate concentration was just over 4 mM. There was no decrease in cellular GSH following ascorbate loading by DHA (results not shown), indicating that the reducing capacity of the cells was not overwhelmed. Together, these results show that endothelial barrier tightening is quite sensitive to intracellular ascorbate.
Figure 3. Intracellular ascorbate decreases endothelial permeability.
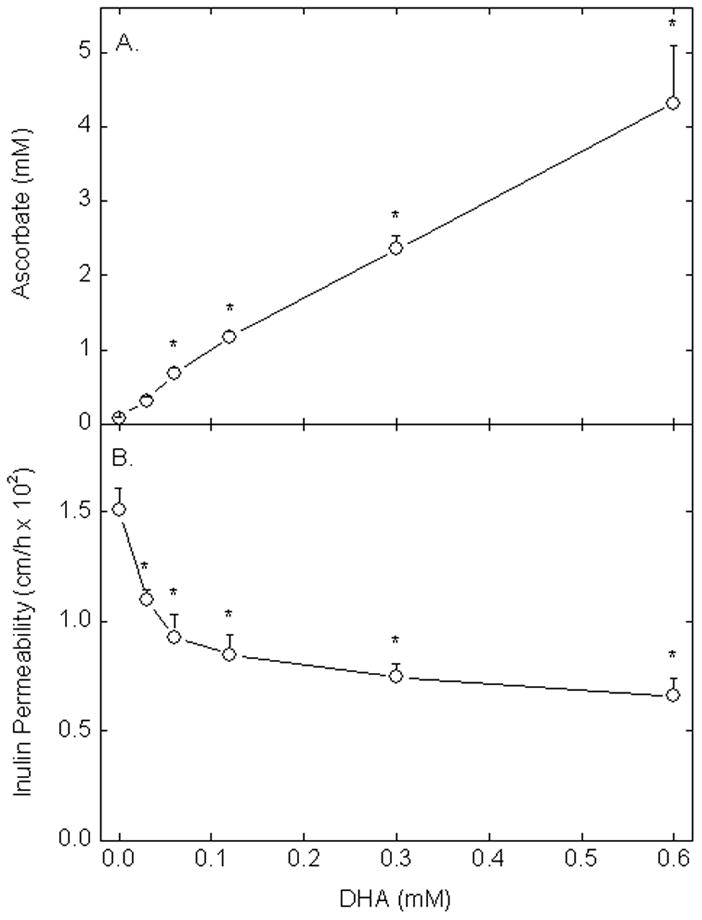
Panel A: EA.hy926 cells cultured on membrane filters were incubated for 90 min at 37 °C with increasing concentrations of DHA and taken for measurement of intracellular ascorbate. Panel B. Cells incubated with DHA as in Panel A for 30 min were then incubated with radiolabeled inulin for an additional 60 min in the transfer assay. Results are shown from 3 experiments in Panel A and from 4 experiments in Panel B, with an “*” indicating p < 0.05 compared to the sample not treated with DHA.
The expected decrease in endothelial permeability due to loading the cells with 0.3 mM DHA under the conditions of Fig. 3 was prevented when the cells were simultaneously treated with increasing concentrations of either L-NAME (Fig. 4A) or ODQ (Fig. 4B). Although L-NAME at 0.3 mM had only a small effect on basal endothelial permeability (Fig. 1B), the ability of this L-NAME concentration to block the ascorbate effect suggests that ascorbate acts by a mechanism dependent on NO. Similarly, although 0.3 μM ODQ had no effect alone on endothelial permeability (Fig. 2), this concentration of ODQ also blocked the expected decrease due to intracellular ascorbate. On the other hand, intracellular ascorbate also prevented the expected increases in endothelial permeability at higher concentrations of both inhibitors.
Figure 4. Prevention of the ascorbate-dependent decrease in endothelial permeability by inhibition of eNOS or guanylate cyclase.
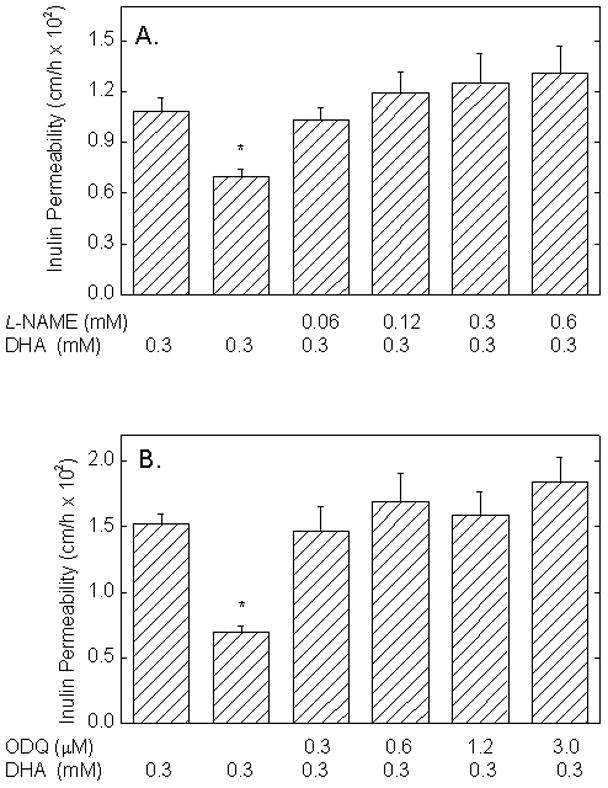
Cells cultured on filters were treated with the indicated concentrations of L-NAME and DHA (Panel A) or of ODQ (Panel B) with or without DHA as indicated for 30 min at 37 °C before addition of radiolabeled inulin and the 60 min transfer assay. Results are shown from 6 experiments for Panel A and from 4 experiments for Panel B, with an “*” indicating p < 0.05 compared to the other treatments in the same panel.
Discussion
The present results show that tightening of the endothelial permeability barrier by intracellular ascorbate requires NO derived from eNOS, as well as downstream function of guanylate cyclase. That maintenance of basal endothelial permeability depends on NO and subsequent generation of cGMP has been well documented in cultured endothelial cells [17]. The role of eNOS-derived NO has since been confirmed in vivo by a study in eNOS knockout mice and mice given L-NAME to inhibit eNOS, which showed clearly that constitutive levels of NO are needed to prevent formation of gaps between endothelial cells and increased albumin extravasation [16]. The mechanism by which cGMP mediates endothelial barrier tightening, at least in endothelial cells derived from human umbilical veins (as are EA.hy926 cells), appears to be cGMP inhibition of the cyclic AMP-degrading phosphodiesterase III [1,5]. The resulting increase in cyclic AMP is well known to tighten the endothelial permeability barrier by several mechanisms [2].
Immortalized endothelial cells such as EA.hy926 lack ascorbate in culture without added ascorbate, whereas primary culture endothelial cells will gradually lose it with passage number [30]. Absence of ascorbate in endothelial cells causes a modest increase in oxidative stress that is prevented by ascorbate loading [31]. This oxidative stress will increase NO destruction and will compound in decrease in NO synthesis by eNOS due to ascorbate deficiency. The ascorbate-dependent decrease in endothelial permeability in the present studies occurred at intracellular ascorbate concentrations that are likely to be in the physiologic range. Although ascorbate concentrations in freshly prepared endothelial cells have not been measured, EA.hy926 cells treated for 16–18 h with increasing amounts of ascorbate were able to maintain intracellular concentrations of just over 1 mM [32]. In the present work, tightening of the endothelial barrier in this cell line occurred at intracellular ascorbate concentrations as low as 0.3 mM, suggesting that relatively low ascorbate concentrations are needed to maintain a tight endothelial permeability in the basal state.
That intracellular ascorbate tightens the endothelial barrier by an NO and cGMP-dependent mechanism is supported by our findings that the effect was completely blocked by inhibition of either eNOS with L-NAME, or of guanylate cyclase by ODQ. The former suggests that eNOS-derived NO is required and the latter suggests that the effect is mediated by NO stimulation of guanylate cyclase to generate cGMP. Although the decrease in endothelial permeability due to intracellular ascorbate was blocked by inhibition of eNOS or guanylate cyclase, ascorbate prevented the expected increase in endothelial permeability due to L-NAME and ODQ. This could reflect preservation or recycling of residual NO by ascorbate.
In conclusion, tightening of the endothelial permeability barrier by intracellular ascorbate requires the NO/cGMP system, suggesting a role of the vitamin in preserving constitutive NO generation and persistence in endothelial cells.
Research Highlights.
Nitric oxide and a cell penetrant cyclic GMP analog increase basal endothelial barrier permeability
Generation by endothelial nitric oxide synthase supports basal endothelial barrier permeability
Ascorbic acid tightens the endothelial barrier at intracellular concentrations likely to be in the physiologic range
Ascorbate tightening of the endothelial permeability barrier requires activity of endothelial nitric oxide synthase and guanylate cyclase
Acknowledgments
This work was supported by NIH grant DK050435 and by the Cell Culture Core of the Vanderbilt Diabetes Research and Training Center (DK020593).
Abbreviations used
- cAMP
3′,5′ cyclic adenosine monophosphate
- cGMP
3′,5′ cyclic guanosine monophosphate
- DHA
dehydroascorbic acid
- eNOS
endothelial nitric oxide synthase
- Hepes
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- KRH
Krebs-Ringer Hepes
- PBS
phosphate-buffered saline
- L-NAME
Nω-nitro-L-arginine methyl ester
- NO
nitric oxide
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Hinsbergh WM. Endothelial permeability for macromolecules. Mechanistic aspects of pathophysiological modulation. Arterioscler Thromb Vasc Biol. 1997;17:1018–1023. doi: 10.1161/01.atv.17.6.1018. [DOI] [PubMed] [Google Scholar]
- 2.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 3.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 4.Moy AB, Bodmer JE, Blackwell K, Shasby S, Shasby DM. cAMP protects endothelial barrier function independent of inhibiting MLC20-dependent tension development. Am J Physiol. 1998;274:L1024–L1029. doi: 10.1152/ajplung.1998.274.6.L1024. [DOI] [PubMed] [Google Scholar]
- 5.Draijer R, Atsma DE, van der LA, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circ Res. 1995;76:199–208. doi: 10.1161/01.res.76.2.199. [DOI] [PubMed] [Google Scholar]
- 6.Westendorp RG, Draijer R, Meinders AE, van Hinsbergh VW. Cyclic-GMP-mediated decrease in permeability of human umbilical and pulmonary artery endothelial cell monolayers. J Vasc Res. 1994;31:42–51. doi: 10.1159/000159030. [DOI] [PubMed] [Google Scholar]
- 7.Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313:2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- 9.May JM, Qu ZC, Qiao H. Transfer of ascorbic acid across the vascular endothelium: mechanism and self-regulation. Am J Physiol Cell Physiol. 2009;297:C169–C178. doi: 10.1152/ajpcell.00674.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirvish SS, Wallcave L, Eagen M, Shubik P. Ascorbate-nitrite reaction: possible means of blocking the formation of carcinogenic N-nitroso compounds. Science. 1972;177:65–68. doi: 10.1126/science.177.4043.65. [DOI] [PubMed] [Google Scholar]
- 11.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am J Physiol Cell Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 12.Heller R, Müscher-Paulig F, Gräbner R, Till U. L-ascorbic acid potentiates nitric oxide synthesis in endothelial cells. J Biol Chem. 1999;274:8254–8260. doi: 10.1074/jbc.274.12.8254. [DOI] [PubMed] [Google Scholar]
- 13.Huang A, Vita JA, Venema RC, Keaney JF., Jr Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. J Biol Chem. 2000;275:17399–17406. doi: 10.1074/jbc.M002248200. [DOI] [PubMed] [Google Scholar]
- 14.Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J Biol Chem. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- 15.May JM. How does ascorbic acid prevent endothelial dysfunction? Free Radic Biol Med. 2000;28:1421–1429. doi: 10.1016/s0891-5849(00)00269-0. [DOI] [PubMed] [Google Scholar]
- 16.Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol. 2005;289:L371–L381. doi: 10.1152/ajplung.00175.2004. [DOI] [PubMed] [Google Scholar]
- 17.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 18.Bauer J, Margolis M, Schreiner C, Edgell CJ, Azizkhan J, Lazarowski E, Juliano RL. In vitro model of angiogenesis using a human endothelium-derived permanent cell line: contributions of induced gene expression, G-proteins, and integrins. J Cell Physiol. 1992;153:437–449. doi: 10.1002/jcp.1041530302. [DOI] [PubMed] [Google Scholar]
- 19.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pech-Amsellem MA, Myara I, Pico I, Maziere C, Maziere JC, Moatti N. Oxidative modifications of low-density lipoproteins (LDL) by the human endothelial cell line EA.hy 926. Experientia. 1996;52:234–238. doi: 10.1007/BF01920713. [DOI] [PubMed] [Google Scholar]
- 21.Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 22.Siflinger-Birnboim A, del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111–117. doi: 10.1002/jcp.1041320115. [DOI] [PubMed] [Google Scholar]
- 23.Utoguchi N, Ikeda K, Saeki K, Oka N, Mizuguchi H, Kubo K, Nakagawa S, Mayumi T. Ascorbic acid stimulates barrier function of cultured endothelial cell monolayer. J Cell Physiol. 1995;163:393–399. doi: 10.1002/jcp.1041630219. [DOI] [PubMed] [Google Scholar]
- 24.May JM, Qu ZC, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 25.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74:214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 26.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 27.Fitzhugh AL, Keefer LK. Diazeniumdiolates: pro- and antioxidant applications of the “NONOates”. Free Radic Biol Med. 2000;28:1463–1469. doi: 10.1016/s0891-5849(00)00251-3. [DOI] [PubMed] [Google Scholar]
- 28.Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 29.May JM, Qu ZC. Ascorbic acid prevents increased endothelial permeability caused by oxidized low density lipoprotein. Free Radic Res. 2010;44:1359–1368. doi: 10.3109/10715762.2010.508496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ek A, Ström K, Cotgreave IA. The uptake of ascorbic acid into human umbilical vein endothelial cells and its effect on oxidant insult. Biochem Pharmacol. 1995;50:1339–1346. doi: 10.1016/0006-2952(95)02024-1. [DOI] [PubMed] [Google Scholar]
- 31.Smith AR, Visioli F, Hagen TM. Vitamin C matters: increased oxidative stress in cultured human aortic endothelial cells without supplemental ascorbic acid. FASEB J. 2002;16:1102–1104. doi: 10.1096/fj.01-0825fje. [DOI] [PubMed] [Google Scholar]
- 32.May JM, Qu ZC, Li X. Ascorbic acid blunts oxidant stress due to menadione in endothelial cells. Arch Biochem Biophys. 2003;411:136–144. doi: 10.1016/s0003-9861(02)00715-4. [DOI] [PubMed] [Google Scholar]