

Abstract
Pancreatic cancer is the fourth leading cause of cancer death in the United States. Indeed, it has been estimated that 37,000 Americans will die from this disease in 2010. Late diagnosis, chemoresistance, and radioresistance of these tumors are major reasons for poor patient outcome, spurring the search for pancreatic cancer early diagnostic and therapeutic targets. ErbB4 (HER4) is a member of the ErbB family of receptor tyrosine kinases (RTKs), a family that also includes the Epidermal Growth Factor Receptor (EGFR/ErbB1/HER1), Neu/ErbB2/HER2, and ErbB3/HER3. These RTKs play central roles in many human malignancies by regulating cell proliferation, survival, differentiation, invasiveness, motility, and apoptosis. In this report we demonstrate that human pancreatic tumor cell lines exhibit minimal ErbB4 expression; in contrast, these cell lines exhibit varied and in some cases abundant expression and basal tyrosine phosphorylation of EGFR, ErbB2, and ErbB3. Expression of a constitutively-dimerized and -active ErbB4 mutant inhibits clonogenic proliferation of CaPan-1, HPAC, MIA PaCa-2, and PANC-1 pancreatic tumor cell lines. In contrast, expression of wild-type ErbB4 in pancreatic tumor cell lines potentiates stimulation of anchorage-independent colony formation by the ErbB4 ligand Neuregulin1β. These results illustrate the multiple roles that ErbB4 may be playing in pancreatic tumorigenesis and tumor progression.
Keywords: ErbB4/HER4, ErbB2/HER2/Neu, Pancreatic Cancer, Signal Transduction, Tumor Suppressor, Oncogene
Introduction
ErbB4 (HER4) is a member of the ErbB family of receptor tyrosine kinases (RTK), a family that also includes the Epidermal Growth Factor (EGF) Receptor (EGFR/ErbB1/HER1), Neu/ErbB2/HER2, and ErbB3/HER3. These family members share organizational homology; they all contain an extracellular ligand-binding domain, a hydrophobic single-pass transmembrane domain, and an intracellular tyrosine kinase domain. The agonists for these receptors are members of the EGF family of peptide hormones, which consists of more than 20 different members [1-3]. The signaling network composed of these RTKs and their complementary peptide hormones regulates many cellular functions, including proliferation, survival, differentiation, motility, growth arrest, and apoptosis [4-6]. Moreover, deregulation of this network, typically due to inappropriate receptor or ligand expression, often plays a significant role in tumorigenesis [7-11].
EGFR, ErbB2, and ErbB3 overexpression is observed in many tumor types and this overexpression is associated with the progression and malignancy of these tumors [2, 7, 12-15]. The roles that ErbB4 plays in tumorigenesis remain topics of some controversy. Some studies indicate that ErbB4 expression is an adverse prognostic factor in breast cancer [16-20]. Moreover, ErbB4 ligands stimulate the proliferation of some human tumor cell lines [17, 21, 22]. Thus, these studies support the hypothesis that ErbB4 is an oncogene. However, other studies indicate that ErbB4 expression correlates with a favorable outcome for breast cancer patients [23-28]. Furthermore, the ErbB4 ligand neuregulin (NRG) induces differentiation of the mammary epithelium into secretory lobuloalveoli in vivo [22] and the ErbB4 ligands NRG and heparin-binding EGF-like growth factor (HB-EGF) induce growth arrest and differentiation in some human breast cancer cell lines in vitro [29-32]. Moreover, betacellulin, an ErbB4 ligand endogenous to the pancreas, induces differentiation of intra islet precursor cells to β-cells in vivo [33] and together with activin-A causes differentiation of exocrine AR42J rat pancreatic tumor cells into insulin-secreting cells [34, 35]. These data indicate that ErbB4 signaling may couple to terminal differentiation and growth arrest and that ErbB4 may be a tumor suppressor. Consistent with this model, published and unpublished data from our laboratory indicate that the constitutively-active ErbB4 Q646C mutant inhibits clonogenic proliferation by human breast and prostate tumor cell lines [25, 26, 36]. Introduction of a glutamate residue into the transmembrane domain of ErbB4 results in constitutive ErbB4 dimerization, tyrosine phosphorylation, and coupling to apoptosis in a variety of cancer cell lines [37]. The s80 ICD, formed when the ErbB4 intracellular domain is released from the membrane by α and γ–secretase following ligand stimulation, forms tyrosine phosphorylated homodimers that inhibit cellular proliferation [38, 39].
Pancreatic cancer is one of the predominant cancers in developed countries. It is the fourth leading cause of cancer death in the United States and the sixth leading cause of cancer death in Europe [40]. Indeed, it has been estimated that approximately 43,000 people in the United States will be diagnosed with pancreatic cancer in 2010 and that approximately 37,000 Americans will die from this disease [41]. The median survival time of pancreatic cancer patients usually does not exceed 6 months [42]. Late diagnosis, chemoresistance, and radioresistance of these tumors are the main reasons for poor patient outcome [43, 44].
The deregulation of several signaling networks has been associated with the malignant growth transformation of pancreatic tumor cells. Examples include a gain-of-function mutation of the c-K-ras oncogene [45], a dominant negative mutation of the p53 tumor suppressor gene [46, 47], a loss-of-function mutation of the p16 tumor suppressor gene, deletion of the DPC4 tumor suppressor gene [47] and overexpression of growth factors [48-50] and their receptors, including EGFR [50], ErbB2 [51], and ErbB3 [52].
The roles that ErbB4 plays in pancreatic cancer have not been determined. However, ErbB4 transcription is decreased in the early stages of pancreatic cancer, indicating that loss of ErbB4 expression may be a prerequisite for tumorigenesis [53]. Indeed, ErbB4 expression in pancreatic tumor cells correlates with favorable staging [54]. However, an alternative explanation for the expression data is that ErbB4 expression may merely be a marker for the proliferative or differentiation state of these cells. To address this issue, we have determined the level of ErbB4 expression and basal ErbB4 signaling (basal ErbB4 tyrosine phosphorylation) in four human pancreatic tumor cell lines. We have also assessed the effect of a constitutively- dimerized and constitutively-active ErbB4 mutant on clonogenic proliferation of these cell lines. Finally, we have evaluated the effect of wild-type ErbB4 expression on the stimulation of anchorage-independent colony formation by the ErbB4 ligand Neuregulin 1β (NRG1β). The data presented indicate that ErbB4 has multiple functions and suggests that ErbB4 functions as a context-sensitive tumor suppressor and oncogene.
Materials and Methods
Cell lines and cell culture
Mouse C127 fibroblasts and the Ψ2 and PA317 recombinant retrovirus packaging cell lines are generous gifts of Dr. Daniel DiMaio (Yale University, New Haven, CT, USA). These cells were cultured essentially as described previously [55, 56]. CaPan-1, HPAC, MIA PaCa-2, and PANC-1 pancreatic tumor cell lines were obtained from American Type Culture Collection and were cultured as recommended. HEK293A and HEK293FT cells were obtained from Invitrogen (Carlsbad, CA) and were cultured as recommended. Cell culture media and supplements were obtained from Invitrogen. Fetal bovine serum and G418 were obtained from Gemini Bioproducts (Woodland, CA). Plasticware and Giemsa stain were obtained from Fisher Scientific (Pittsburgh, PA). Recombinant Neuregulin 1β (NRG1β) was obtained from Peprotech (Rocky Hill, NJ). Other biochemicals were obtained from Sigma Scientific (St. Louis, MO).
Recombinant retroviruses and lentiviruses
Briefly, the recombinant amphotropic retroviruses pLXSN (Vector) [57], pLXSN-ErbB4 (ErbB4 WT) [58], and pLXSN-ErbB4 Q646C [59] were packaged using the ψ2 ecotropic retrovirus packaging cell line [60] and the PA317 amphotropic retrovirus packaging cell line [61] essentially as described [55]. The recombinant retroviral expression vector pLXSN is a generous gift of Dr. Daniel DiMaio (Yale University, New Haven, CT, USA).
Standard molecular biology techniques and the shuttle vector pENTR1A (Invitrogen) were used to subclone wild-type ErbB4 from pCH4M2 [62] and the kinase-deficient ErbB4 K751M mutant from pLXSN-ErbB4 K751M [25] into the recombinant lentiviral expression vector pLenti6/V5/DEST (Invitrogen). The resulting pLenti-ErbB4 (ErbB4 WT) and pLenti-ErbB4 K751M (ErbB4 Kin Def) constructs and the pLenti6/V5/DEST vector control were packaged by transient cotransfection with the packaging vectors pLP1, pLP2, and pLP/VSVG into the FT lentiviral packaging cell line (Invitrogen). Recombinant lentiviruses were recovered from the medium conditioned by these transfected HEK293FT cells. Following infection with the recombinant lentiviruses, MIA PaCa-2 human pancreatic tumor cells were incubated with 6 μg/mL blasticidin to select for stably-infected, recombinant cell lines.
Recombinant adenoviruses and adenovirus infections
Standard molecular biology techniques and the shuttle vector pENTR1A (Invitrogen) were used to subclone the wild-type ErbB4 cDNA and the ErbB4 Q646C mutant from pLXSN-ErbB4 or pLXSN-ErbB4 Q646C [59] to the adenoviral expression vector pAD/CMV/V5-DEST (Invitrogen). The constructs were packaged by transient transfection into the HEK293A adenovirus packing cell line (Invitrogen). Low-titer adenovirus stocks recovered from the cytoplasm and conditioned medium of the transiently-transfected HEK293A cells were amplified by infecting naive HEK293 cells and recovering the adenovirus stocks from the cytoplasm and conditioned medium of the infected cells.
Immunoblot assays for ErbB receptor expression, multimer formation, and tyrosine phosphorylation
The four pancreatic tumor cell lines were incubated for 24 hrs in basal or complete medium. For 1 hour just prior to lysis for the ErbB expression analysis, the cells that had been incubated in complete medium received fresh medium supplemented with sodium orthovanadate at a final concentration of 30 mM. Receptor expression and tyrosine phosphorylation were assayed by immunoblotting essentially as described [58, 59, 63]. EGFR was precipitated using an anti-EGFR mouse monoclonal antibody (SC-120 - Santa Cruz Biotechnology); ErbB2 was precipitated using an anti-ErbB2 rabbit monoclonal antibody (SC-284 - Santa Cruz Biotechnology); ErbB3 was precipitated using an anti-ErbB3 rabbit polyclonal antibody (SC-285 - Santa Cruz Biotechnology); ErbB4 was precipitated using an anti-ErbB4 rabbit polyclonal antibody (SC-283 - Santa Cruz Biotechnology). For analysis of multimer formation, the precipitates were resolved on a 5% acrylamide non-reducing SDS-PAGE gel. For all other experiments, the precipitates were resolved on a 7.5% acrylamide reducing gel. Anti-phosphotyrosine immunoblotting was performed using the 4G10 anti-phosphotyrosine mouse monoclonal antibody (05-321 – Millipore). Other blotting antibodies included a sheep anti-EGFR polyclonal antibody (06-129 – Millipore), a rabbit anti-ErbB2 polyclonal antibody (SC-284 - Santa Cruz Biotechnology), a rabbit anti-ErbB3 polyclonal antibody (SC-285 - Santa Cruz Biotechnology), a rabbit anti-ErbB4 polyclonal antibody (SC-283 - Santa Cruz Biotechnology) and a mouse anti-ErbB4 monoclonal antibody (SC-8050 – Santa Cruz Biotechnology).
Ligand stimulation of ErbB receptor tyrosine phosphorylation was assayed using cells starved in basal medium for 24 hours. Cells were stimulated for 5 minutes on ice with 10 nM NRG1β or PBS (diluent control). Cells were lysed and ErbB expression and tyrosine phosphorylation were assayed as described earlier.
ErbB4 Tumor Suppression Assays
We have previously described a sensitive quantitative clonogenic proliferation (colony formation) assay for tumor suppression by ErbB4 [25, 26, 36]. Tester human tumor cell lines are infected with recombinant retroviruses that express a neomycin resistance gene alone (retrovirus vector control) or together with a gene of interest. Cells are subjected to selection with G418 and drug-resistant colonies are counted. C127 mouse fibroblasts are infected in parallel as controls for differences in viral titer. This strategy allows us to assess whether a gene of interest inhibits clonogenic proliferation (colony formation) by the tester cell lines. Such inhibition is indicative of tumor suppressor activity for the gene of interest and is presumably due to anti-proliferative or pro-apoptotic effects of the gene of interest.
The tumor suppressor activity of the constitutively-active ErbB4 Q646C mutant [59] in human pancreatic tumor cell lines was assayed essentially as described [25, 26, 36]. Briefly, cells infected with the recombinant retroviruses were selected using 200 to 1100 μg/mL G418. During selection some of the plates were treated with either 10 nM NRG1β or 5 nM TRAIL. Following section, drug-resistant colonies were stained using Giemsa and the culture plates were digitized using a flatbed document scanner.
Drug-resistant colonies were counted manually and the retrovirus titer for each combination of retrovirus and cell line was determined by dividing the number of colonies by the volume of retrovirus used in the infection. For each of the pancreatic tumor cell lines and for each trial, relative clonogenic colony formation efficiency was calculated for each retrovirus stock by dividing the retroviral titer in the pancreatic tumor cell line by the corresponding retroviral titer in the C127 cells. These values are reported as mean percentages calculated from at least three independent sets of infections (trials). The tumor suppressor activity of the tester retroviruses in each pancreatic tumor cell line was calculated by dividing the clonogenic colony formation efficiency of the tester retrovirus by the clonogenic colony formation efficiency of the vector control retrovirus. This value was then subtracted from 100%. These values are expressed as mean percentages calculated from at three least independent sets of infections (trials). The standard error was calculated for each mean and is reported.
Anchorage-independence Assays
The anchorage-independent proliferation of MIA PaCa-2 cell lines was assayed essentially as described [36]. Briefly, twenty thousand MIA PaCa-2 cells stably infected with the pLenti/V5/DEST (Vector), pLenti-ErbB4 (Wild Type ErbB4), and pLenti-ErbB4 K751M (Kinase Deficient ErbB4) recombinant lentiviruses were seeded in 60 mm dishes in semisolid medium consisting of 0.3% low melting point agarose and either 10 nM NRG1β or the PBS diluent control. Cells were maintained for 15 days to permit the growth of anchorage-independent colonies. Digital photomicrographs were taken of six randomly-selected fields for each experimental condition and the diameter of each anchorage-independent colony was measured by hand. The number of small (< 90 μm diameter), medium (90 – 180 μm diameter), and large (> 180 μm diameter) colonies was tabulated for each experimental condition and was pooled from five independent trials. For each experimental condition approximately 300 to 350 colonies were measured. The effects of lentiviral infection and NRG1β stimulation on anchorage-independent colony proliferation were determined by using a Chi-square test to compare the colony size distribution for each experimental condition.
Results
Pancreatic tumor cell lines display minimal ErbB4 expression and tyrosine phosphorylation
To determine the role that ErbB4 expression and signaling play in pancreatic tumors, we first measured ErbB receptor expression and tyrosine phosphorylation in the CaPan-1, HPAC, PANC-1 and MIA PaCa-2 human pancreatic tumor cell lines. PANC-1 cells exhibit robust EGFR expression and MIA PaCa-2 cells exhibit somewhat less EGFR expression (Figure 1). HPAC cells exhibit only modest EGFR expression and CaPan-1 cells exhibit minimal EGFR expression. All four cell lines exhibited much greater EGFR tyrosine phosphorylation in the absence of starvation (Figure 1). Surprisingly, HPAC cells exhibited the most EGFR tyrosine phosphorylation, whereas PANC-1 and MIA PaCa-2 cells exhibited lesser amounts of EGFR tyrosine phosphorylation and CaPan-1 cells exhibited minimal EGFR tyrosine phosphorylation (Figure 1).
Figure 1. EGFR expression and tyrosine phosphorylation vary in the four human pancreatic tumor cell lines.

EGFR was immunoprecipitated from each pancreatic tumor cell line. Controls included ψ2 mouse fibroblasts engineered to express EGFR (Ψ2 EGFR) and ψ2 vector control cells (2Ψ LXSN). Samples were resolved by SDS-PAGE and electroblotted onto nitrocellulose. The blot was probed with an anti-phosphotyrosine antibody and a parallel blot was probed with an anti-EGFR antibody. The mobility and the reported molecular weight of the protein markers are indicated. The position of EGFR on the blots is also indicated.
Starved HPAC, PANC-1, and MIA PaCa-2 cells exhibit robust ErbB2 expression but unstarved PANC-1 and MIA PaCa-2 cells exhibit markedly less ErbB2 expression (Figure 2). Starved and unstarved CaPan-1 cells exhibit little ErbB2 expression. In the absence of starvation, HPAC cells exhibit abundant ErbB2 tyrosine phosphorylation whereas CaPan-1 and MIA PaCa-2 cells exhibit little ErbB2 tyrosine phosphorylation and PANC-1 cells do not exhibit detectable ErbB2 tyrosine phosphorylation. ErbB2 tyrosine phosphorylation in the HPAC, CaPan-1, and MIA PaCa-2 cells is greatly reduced upon starvation of the cells.
Figure 2. ErbB2 expression and tyrosine phosphorylation vary in the four pancreatic tumor cell lines.

ErbB2 was immunoprecipitated from each pancreatic tumor cell line. C127 mouse fibroblasts engineered to express a constitutively-active rat ErbB2 mutant [85] (C127 ErbB2*) served as a control. Samples were resolved by SDS-PAGE and electroblotted onto nitrocellulose. The blot was probed with an anti-phosphotyrosine antibody and a parallel blot was probed with an anti-ErbB2 antibody. The mobility and the reported molecular weight of the protein markers are indicated. The position of ErbB2 on the blots is also indicated.
HPAC cells exhibit robust ErbB3 expression and tyrosine phosphorylation (Figure 3) PANC-1 and CaPan-1 cells do not exhibit detectable ErbB3 expression or tyrosine phosphorylation. MIA PaCa-2 cells exhibit ErbB3 expression, yet fail to exhibit detectable ErbB3 tyrosine phosphorylation (Figure 3).
Figure 3. ErbB3 expression and tyrosine phosphorylation vary in the four pancreatic tumor cell lines.

ErbB3 was immunoprecipitated from each pancreatic tumor cell line. BaF3 mouse lymphoid cells engineered to express ErbB2 and ErbB3 (BaF3/ErbB2+ErbB3 – [58]) served as a control. Samples were resolved by SDS-PAGE and electroblotted onto nitrocellulose. The blot was probed with an anti-phosphotyrosine antibody and a parallel blot was probed with an anti-ErbB3 antibody. The mobility and the reported molecular weight of the protein markers are indicated. The position of ErbB3 on the blots is also indicated.
None of the four cell lines tested exhibit ErbB4 expression or tyrosine phosphorylation, even when 1 mg of cell lysate was used for ErbB4 immunoprecipitations. In contrast, 10 μg of lysate from ψ2 fibroblasts engineered to express the constitutively-active ErbB4 Q646C mutant is sufficient to yield detectable ErbB4 expression and tyrosine phosphorylation (Figure 4).
Figure 4. ErbB4 expression and tyrosine phosphorylation are not detected in any of the four pancreatic tumor cell lines.

ErbB4 was immunoprecipitated from each pancreatic tumor cell line. Controls included ψ2 mouse fibroblasts engineered to express the constitutively-active ErbB4 Q646C mutant (Ψ2 ErbB4 Q646C) and ψ2 vector control cells (Ψ2 LXSN). Samples were resolved by SDS-PAGE and electroblotted onto nitrocellulose. The blot was probed with an anti-phosphotyrosine antibody and an analogous blot was probed with an anti-ErbB4 antibody. The mobility and the reported molecular weight of the protein markers are indicated. The position of ErbB4 on the blots is also indicated.
The constitutively-active ErbB4 Q646C mutant inhibits clonogenic proliferation (colony formation) by human pancreatic tumor cell lines in plastic culture dishes
One explanation for the apparent absence of ErbB4 expression in the four human pancreatic tumor cell lines is that the loss of ErbB4 signaling is a factor that contributes to prostate tumorigenesis. We tested this hypothesis by restoring ErbB4 expression and signaling to the four human pancreatic tumor cell lines.
The ErbB4 Q646C mutant [59] features substitution of a cysteine residue for a glutamine residue in the extracellular juxtamembrane region of ErbB4. This mutation results in constitutive ErbB4 tyrosine phosphorylation [59] and coupling to tumor suppressor activity in human prostate and breast tumor cell lines [25, 36], presumably as a consequence of disulfide-linked ErbB4 homodimerization. Indeed, MIA PaCa-2 cells infected with a recombinant adenovirus that encodes the ErbB4 Q646C mutant display a greater degree of ErbB4 multimerization and tyrosine phosphorylation than do cells infected with a recombinant adenovirus that encodes wild-type ErbB4. Indeed, the ErbB4 multimers present in cells that express Q646C display a high degree of tyrosine phosphorylation (Figure 5).
Figure 5. Cells that express the ErbB4 Q646C mutant display a higher degree of ErbB4 tyrosine phosphorylation and multimerization than do cells that express wild-type ErbB4.

MIA PaCa-2 cells were infected with recombinant adenoviruses that encode wild-type ErbB4 or the ErbB4 Q646C mutant. Controls included ψ2 mouse fibroblasts engineered to express the ErbB4 Q646C mutant or wild-type ErbB4. ErbB4 was immunoprecipitated and resolved using reducing or non-reducing polyacrylamide gels. The resolved samples were electroblotted onto PVDF. Blots were probed with an anti-phosphotyrosine antibody or an anti-ErbB4 antibody. The mobility and the reported molecular weight of the protein standards are indicated. The positions of ErbB4 monomers and multimers are also indicated.
Next we assayed the effects of the ErbB4 Q646C mutant on clonogenic proliferation (colony formation) by the four pancreatic tumor cell lines. These cells were infected with a recombinant retrovirus that carries the ErbB4 Q646C mutant along with the neomycin resistance gene, after which infected cells were selected using G418. As controls we infected these pancreatic tumor cell lines with a recombinant retrovirus that carries only the neomycin resistance gene (Vector) or the neomycin resistance gene and the wild-type ErbB4 gene (ErbB4 WT). To control for differences in absolute retroviral titers, we infected C127 mouse fibroblasts in parallel. C127 cells are readily infected by these recombinant retroviruses and the ErbB4 Q646C mutant does not affect clonogenic proliferation (drug-resistant colony formation) of these cells, making C127 cells ideal for comparing the absolute viral titers of these retroviruses. We have postulated that the failure of the C127 cells to respond to the ErbB4 Q646C mutant is due to the absence of appropriate ErbB4 effectors in this cell line [25, 26, 36].
As shown in Figure 6, human pancreatic tumor cells infected with the recombinant retrovirus that expresses the ErbB4 Q646C mutant exhibit less clonogenic proliferation (form fewer drug-resistant colonies) than do these cells infected with the other recombinant retroviruses. Indeed, the titer of the ErbB4 Q646C recombinant retrovirus in these pancreatic tumor cells is markedly less than the titers of the other recombinant retroviruses (Table 1). However, C127 cells infected with the ErbB4 Q646C retrovirus readily undergo clonogenic proliferation (form abundant drug-resistant colonies) and the titer of the ErbB4-Q646C retrovirus in C127 fibroblasts is not markedly less than the titer of the other recombinant retroviruses in C127 cells (Table 1). Thus, the low titer of the ErbB4 Q646C recombinant retrovirus in the human pancreatic tumor cell lines appears to reflect the ability of the ErbB4 Q646C mutant to inhibit clonogenic proliferation (colony formation) in these cells. This inhibition is specific for the ErbB4 Q646C mutant because in each of the pancreatic tumor cell lines the titer of the vector retrovirus and the titer of the ErbB4 WT retrovirus are similar.
Figure 6. The ErbB4 Q646C mutant inhibits clonogenic drug-resistant colony formation in plastic dishes by the four human pancreatic tumor cell lines.

These cells were infected with recombinant amphotropic retroviruses that carry the neomycin resistance gene (Vector) or with retroviruses that carry the neomycin resistance gene along with wild-type ErbB4 (ErbB4) or the constitutively-active ErbB4 Q646C mutant (Q646C). Infected cells were selected using G418. Following selection, the culture plates were stained using Giemsa and digitized using a flatbed scanner. Colonies were counted by hand from the actual culture plates.
Table 1.
The ErbB4 Q646C mutant exhibits tumor suppressor activity in the human pancreatic tumor cell lines
| Cell Lines | Viruses | Drug Resistant Colonies per mL of Virus |
Clonogenic Proliferation Efficiency (% of C127) |
Inhibition of Clonogenic Proliferation (%) |
|---|---|---|---|---|
| C127 | Vector | 2.6×105 | 100 | |
| ErbB4 WT | 2.9×105 | 100 | ||
| ErbB4 Q646C | 5.6×105 | 100 | ||
|
| ||||
| CaPan-1 | Vector | 1.3×102 | 0.043 (N=3) | |
| ErbB4 WT | 1.9×102 | 0.061 (N=4) | None (N=3) | |
| ErbB4 Q646C | 6.9×100 | 0.001 (N=4) | 99±1 (N=3) | |
|
| ||||
| HPAC | Vector | 1.6×105 | 65 (N=5) | |
| ErbB4 WT | 1.5×105 | 59 (N=5) | 7±11 (N=5) | |
| ErbB4 Q646C | 4.1×104 | 7.0 (N=5) | 88±4 (N=5) | |
|
| ||||
| PANC-1 | Vector | 1.0×104 | 4.3 (N=4) | |
| ErbB4 WT | 1.2×104 | 4.0 (N=4) | None (N=4) | |
| ErbB4 Q646C | 6.0×103 | 1.1 (N=4) | 71±14 (N=4) | |
|
| ||||
| MIA PaCa-2 | Vector | 4.7×104 | 20 (N=4) | |
| ErbB4 WT | 5.8×104 | 22 (N=4) | None (N=4) | |
| ErbB4 Q646C | 3.6×104 | 5.6 (N=4) | 71±8 (N=4) | |
These data indicate that the ErbB4 Q646C mutant displays tumor suppressor activity, presumably by inhibiting proliferation or by stimulating apoptosis. In Table 1 we have quantified this activity. The ErbB4 Q646C mutant inhibits clonogenic proliferation by the PANC-1 and MIA PaCa-2 cell lines by approximately 70%. The ErbB4 Q646C mutant inhibits clonogenic proliferation by the HPAC cell line by almost 90% and inhibits clonogenic proliferation by the CaPan-1 cell line by almost 100%.
We postulated that the failure of wild-type ErbB4 to display tumor suppressor activity was due to an absence of ErbB4 ligands. Thus, we compared the effects of wild-type ErbB4 and of the ErbB4 Q646C mutant on clonogenic proliferation (drug-resistant colony formation) by the MIA PaCa-2 cell line in the presence and absence of the ErbB4 ligand NRG1β. As a control we assayed clonogenic proliferation in the presence of the pro-apoptotic TRAIL peptide. The ErbB4 Q646C mutant inhibited clonogenic proliferation (colony formation) by MIA PaCa-2 cells to the same extent in the presence or absence of 10 nM NRG1β (Table 2). However, wild-type ErbB4 failed to inhibit clonogenic proliferation (colony formation) by MIA PaCa-2 cells in the presence or absence of 10 nM NRG1β. Yet, TRAIL markedly inhibited clonogenic proliferation (colony formation) both in the absence and presence of wild-type ErbB4. Moreover, 10 nM NRG1β stimulates ErbB4 tyrosine phosphorylation in MIA PaCa-2 cells infected with the wild-type ErbB4 retrovirus (data not shown). These data indicate that the constitutively-active ErbB4 mutant displays tumor suppressor activity, whereas ligand stimulation does not cause wild-type ErbB4 to display tumor suppressor activity.
Table 2.
NRG1β exerts no effect on the tumor suppressor activity of wild-type ErbB4 or of the ErbB4 Q646C mutant in the MIA PaCa-2 human pancreatic tumor cell line
| Cell Lines | Viruses | Treatments | Drug Resistant Colonies per mL of Virus |
Clonogenic Proliferation Efficiency (% of C127) |
Inhibition of Clonogenic Proliferation (%) |
|---|---|---|---|---|---|
| C127 | Vector | Mock | 5.7×105 | 100 | |
| ErbB4 WT | Mock | 4.6×105 | 100 | ||
| ErbB4 Q646C | Mock | 6.1×105 | 100 | ||
|
| |||||
| MIA PaCa-2 | Vector | Mock | 1.7×105 | 30 (N=3) | |
| Vector | 10 nM NRG1β | 1.7×105 | 31 (N=3) | None (N=3) | |
| Vector | 5 nM Trail | 3.7×103 | 0.70 (N=3) | 98±1 (N=3) | |
|
| |||||
| ErbB4 WT | Mock | 1.6×105 | 37 (N=3) | None (N=3) | |
| ErbB4 WT | 10 nM NRG1β | 1.7×105 | 39 (N=3) | None (N=3) | |
| ErbB4 WT | 5 nM Trail | 3.9×103 | 1.0 (N=3) | 97±2 (N=3) | |
|
| |||||
| ErbB4 Q646C | Mock | 5.2×104 | 8.4 (N=3) | 72±5 (N=3) | |
| ErbB4 Q646C | 10 nM NRG1β | 5.8×104 | 9.4 (N=3) | 69±6 (N=3) | |
An ErbB4 ligand stimulates anchorage-independent proliferation in cells that stably express wild-type ErbB4
Another explanation for the failure of wild-type ErbB4 to display tumor suppressor activity, even in the presence of an ErbB4 ligand, may be the failure to adequately stimulate coupling of wild-type ErbB4 to any biological response in MIA PaCa-2 cells. To address this possibility we have assessed ligand stimulation of coupling of wild-type ErbB4 to a biological response in MIA PaCa-2 cells. We have used recombinant lentiviruses to generate MIA PaCa-2 cells that ectopically express wild-type ErbB4 or the ErbB4 K751M mutant (which lacks tyrosine kinase activity) [25]. We were not able to include a cell line that expresses the ErbB4 Q646C mutant because we cannot generate a stable cell line that expresses this mutant, presumably due to the potent growth inhibitory effects of the mutant. Infected cells were seeded in a semi-solid medium consisting of 0.3% low melting point agarose supplemented with 10 nM NRG1β or the PBS diluent control. Cells were maintained for 15 days, after which anchorage-independent colonies of cells were photographed and measured. The colony size distributions for five independent trials are depicted and have been analyzed using Chi-square analysis (Figure 7).
Figure 7. Wild-type ErbB4 potentiates NRG1β stimulation of anchorage-independent growth of MIA PaCa-2 human pancreatic tumor cells.

These cells were infected with recombinant lentiviruses that carry the blasticidin resistance gene (Vector) or with lentiviruses that carry the blasticidin resistance gene along with wild-type ErbB4 (ErbB4) or the ErbB4 K751M mutant that lacks tyrosine kinase activity. Infected cells were selected using blasticidin and seeded in semisolid medium containing 10 nM NRG1β or PBS (diluent control). Following fifteen days of incubation, anchorage-independent colonies were photographed and colony diameter was measured by hand from the photographs. (A) Representative photomicrographs of cell lines cultured in the presence of NRG1β are depicted. (B) The effects of lentiviral infection and NRG1β stimulation on anchorage-independent proliferation were determined by using a Chi-square test to compare the colony size distribution for each experimental condition. The data represent five independent trials. P values are reported.
Expression of ectopic ErbB4 enhances NRG1β stimulation of anchorage-independent proliferation (P<0.05). In contrast, the ErbB4 mutant defective for tyrosine kinase activity (K751M) does not significantly enhance the effect of NRG1β (P>0.1). Thus, these data indicate that ligand stimulation of wild-type ErbB4 signaling is coupled to an oncogenic effect.
We have investigated a potential mechanism that may underlie the failure of the ErbB4 K751M mutant to enable increased NRG1β stimulation of anchorage-independent proliferation is due to inadequate NRG1β stimulation of ErbB4 tyrosine phosphorylation. Both ErbB4 constructs are expressed at relatively equal levels in MIA PaCa-2 cells infected with the two ErbB4 lentiviruses. However, only cells that ectopically express wild-type ErbB4 exhibit detectable levels of ErbB4 phosphorylation upon stimulation with NRG1β (Figure 8). This relative absence of ErbB4 tyrosine phosphorylation may account for the failure of the ErbB4 K751M mutant to enable NRG1β stimulation of anchorage-independent proliferation.
Figure 8. MIA PaCa-2 cells that ectopically express wild-type ErbB4, but not cells that express the ErbB4 K751M mutant, exhibit ErbB4 tyrosine phosphorylation following stimulation with NRG1β.
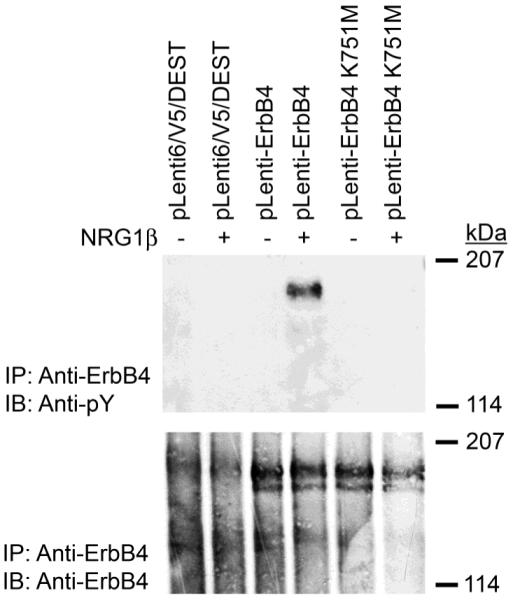
MIA PaCa-2 cells were infected with recombinant lentiviruses that carry the blasticidin resistance gene (Vector) or with lentiviruses that carry the blasticidin resistance gene along with wild-type ErbB4 (ErbB4) or the ErbB4 K751M mutant. Infected cells were selected using blasticidin and expanded into cell lines. These cell lines were starved and stimulated with NRG1β. ErbB4 expression and tyrosine phosphorylation were assessed by immunoprecipitation and immunoblotting. Data shown are representative of at least three independent experiments.
Discussion
Increased EGFR and ErbB2 expression or signaling may contribute to pancreatic tumorigenesis or tumor progression
Normal pancreatic cells exhibit little or no EGFR expression [64, 65]. In contrast, EGFR is abundantly expressed in three of the four pancreatic tumor cell lines tested here and is modestly expressed in the remaining pancreatic tumor cell line. These data suggest that EGFR signaling is important to pancreatic tumorigenesis or tumor progression. Our data are consistent with the observations that EGFR is deregulated in pancreatic tumors. Often, dysregulation of EGFR signaling is caused by EGFR overexpression [65, 66]. However, activating mutations of the EGFR tyrosine kinase domain are found in approximately 2% of pancreatic cancers [67-71], EGFR expression is correlated with a more aggressive phenotype in pancreatic cancer [54, 72]. Indeed, the EGFR tyrosine kinase inhibitor erlotinib has been approved for the treatment of pancreatic cancers [73].
Normal pancreatic cells exhibit little or no ErbB2 expression [64, 65, 74]. In contrast, ErbB2 is expressed in all four pancreatic tumor cell lines tested here and ErbB2 exhibits at least modest basal tyrosine phosphorylation in three of the four cell lines tested here. These data suggest that increased ErbB2 signaling contributes to pancreatic tumorigenesis or tumor progression. Our data are consistent with the observation that ErbB2 is overexpressed in 45-70% of pancreatic ductal carcinomas [64, 74] and with the observation that ErbB2 overexpression correlates with a poor prognosis in many malignancies and that ErbB2 is a validated drug target for these cancers [75].
ErbB4 may play dual roles in pancreatic tumorigenesis or tumor progression
ErbB4 is expressed in the normal pancreatic epithelia [53, 76]. In contrast, minimal ErbB4 expression is observed in the four human pancreatic tumor cell lines tested here. Moreover, the constitutively-dimerized ErbB4 Q646C mutant inhibits clonogenic proliferation in the four human pancreatic tumor cell lines. These data suggest that ErbB4 behaves as a tumor suppressor and that a loss of ErbB4 expression and signaling is critical for pancreatic tumorigenesis or tumor progression.
Indeed, the tumor suppressor activity displayed by the ErbB4 Q646C mutant in the four human pancreatic cell lines is similar to tumor suppressor activities displayed by other constitutively-active ErbB4 mutants in breast, prostate, and ovarian cancer cell lines [37-39]. Similarly, ErbB4 ligands can stimulate differentiation and growth arrest [58, 77, 78]. Finally, the hypothesis that ErbB4 functions as a tumor suppressor is consistent with the observation that some gastric, colorectal, NSCLC, and breast carcinomas harbor loss-of-function mutations in ErbB4 [79].
However, in the four human pancreatic tumor cell lines, an ErbB4 ligand does not inhibit clonogenic proliferation when accompanied by ectopic expression of wild-type ErbB4. Furthermore, ectopic expression of wild-type ErbB4 potentiates NRG1β stimulation of anchorage-independent growth in the MIA PaCa-2 pancreatic tumor cell line. These data suggest that ErbB4 behaves as an oncoprotein and may serve as a target for therapeutic intervention.
One possible explanation for this apparent dichotomy is that ErbB4 Q646C does not accurately model ligand-induced ErbB4 signaling and that the results generated using the ErbB4 Q646C mutant are artifacts of this mutant and cannot be used to predict ErbB4 function. Several observations would appear to argue against this explanation. The ErbB4 ligand Neuregulin 1β stimulates terminal differential and growth arrest of AU565 and MDA-MB-453 human breast tumor cell lines [31, 32]. Moreover, the constitutively-active ErbB4 I658E mutant is coupled to apoptosis in multiple cancer cell lines [37] and the constitutively-active s80 intracellular domain fragment of ErbB4 inhibits proliferation of multiple cancer cell lines [38, 39]. Consequently, the results obtained using the ErbB4 Q646C mutant do not appear to be artifacts of that mutant and suggest that wild-type ErbB4, in at least some contexts, possesses tumor suppressor activities.
Nonetheless, another possible explanation for our observation that wild-type ErbB4 and the ErbB4 Q646C mutant possess different signaling activities is that the ErbB4 Q646C mutant may be an oversimplified model for ligand-induced ErbB4 signaling. Note that ligands for ErbB4 can stimulate ErbB4 heterodimerization with EGFR and ErbB2, resulting in signaling by both ErbB4 and its dimerization partner. The presence of a heterodimerization partner for ErbB4 enables ErbB4 ligands to stimulate an expanded repertoire of signaling events, resulting in differences in the biological responses to ErbB4 ligands [77]. Indeed, ligand stimulation of ErbB4 homodimerization and signaling in the BaF3 lymphoid cell line fails to result in IL3-independent proliferation, whereas ErbB4 ligands stimulate IL3-independent proliferation in BaF3 cells that express both ErbB4 and EGFR or both ErbB4 and ErbB2 [58, 80]. Thus, we postulate that ErbB4 homodimers, which are best exemplified by the ErbB4 Q646C mutant, function as tumor suppressors. In contrast, ErbB4 heterodimers, which may arise in cells that co-express ErbB4 and ErbB2 or ErbB4 and EGFR following stimulation with ErbB4 ligands, appear to function as oncoproteins. Such a model is consistent with the observation that ErbB4 expression in breast tumors in the absence of EGFR and ErbB2 expression correlates with a more favorable prognosis, whereas ErbB4 co-expression with EGFR or ErbB2 correlates with a poorer prognosis [81]. Experiments to directly test this model are underway.
A confounding factor is that the ErbB4 transcript can undergo alternative splicing at two locations; thus, ErbB4 exists in four different splicing isoforms. The JM-a and JM-b isoforms differ in the extracellular juxtamembrane region and display differences in ligand-induced ErbB4 cleavage by proteases and differences in coupling to biological responses [82, 83]. The CT-a (Cyt-1) and CT-b (Cyt-2) isoforms differ in the carboxyl-terminal region of the cytoplasmic domain and appear to display differences in the sites of tyrosine phosphorylation as well as difference in coupling to downstream signaling effectors [16, 82, 84]. The experiments described herein were performed using the canonical JM-a/CT-a isoform and the results of these experiments may not be predictive of results obtained using other ErbB4 isoforms.
Finally, our model predicts that the changes in ErbB receptor expression that are characteristic of pancreatic tumor cells follow a defined sequence. Because ErbB4 heterodimers appear to function as oncogenes, we predict that the loss of ErbB4 expression would be selected against in cells that overexpress other ErbB family receptors. So, if ErbB4 homodimers do function as tumor suppressors and ErbB4 expression is indeed lost in pancreatic tumor cells, then the loss of ErbB4 expression must precede amplification and overexpression of other ErbB family receptors.
Acknowledgments
We thank Sarah Pitfield, Richard Gallo, Desi Penington, Steve Kaverman, and Ianthe Bryant for essential reagents. We acknowledge support from the National Cancer Institute (R01CA114209 to DJR), the U.S. Army Breast Cancer Research Program (DAMD17-00-1-0416), the American Cancer Society (IRG-58-006), the Indiana Elks Foundation, and the Purdue University Center for Cancer Research.
Grant Support:
US Army Medical Research and Materiel Command Prostate Cancer Research Program Grant DAMD17-02-1-0130
US Army Medical Research and Materiel Command Breast Cancer Research Program Grant DAMD17-00-1-0415
National Institutes of Health/National Cancer Institute Grant R01CA114209
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- [2].Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol. 2007;19:124–134. doi: 10.1016/j.ceb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- [3].Sanderson MP, Dempsey PJ, Dunbar AJ. Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors. 2006;24:121–136. doi: 10.1080/08977190600634373. [DOI] [PubMed] [Google Scholar]
- [4].Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and to cancer therapy. Mol Syst Biol. 2007;3:151. doi: 10.1038/msb4100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19:117–123. doi: 10.1016/j.ceb.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sarkar FH, Banerjee S, Li Y. Pancreatic cancer: pathogenesis, prevention and treatment. Toxicol Appl Pharmacol. 2007;224:326–336. doi: 10.1016/j.taap.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Henson ES, Gibson SB. Surviving cell death through epidermal growth factor (EGF) signal transduction pathways: implications for cancer therapy. Cell Signal. 2006;18:2089–2097. doi: 10.1016/j.cellsig.2006.05.015. [DOI] [PubMed] [Google Scholar]
- [8].Yeh JJ, Der CJ. Targeting signal transduction in pancreatic cancer treatment. Expert Opin Ther Targets. 2007;11:673–694. doi: 10.1517/14728222.11.5.673. [DOI] [PubMed] [Google Scholar]
- [9].Pedraza-Farina LG. Mechanisms of oncogenic cooperation in cancer initiation and metastasis. Yale J Biol Med. 2006;79:95–103. [PMC free article] [PubMed] [Google Scholar]
- [10].Bilanges B, Stokoe D. Mechanisms of translational deregulation in human tumors and therapeutic intervention strategies. Oncogene. 2007;26:5973–5990. doi: 10.1038/sj.onc.1210431. [DOI] [PubMed] [Google Scholar]
- [11].Reynolds AR, Kyprianou N. Growth factor signalling in prostatic growth: significance in tumour development and therapeutic targeting. Br J Pharmacol. 2006;147(Suppl 2):S144–152. doi: 10.1038/sj.bjp.0706635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- [13].Kim H, Muller WJ. The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp Cell Res. 1999;253:78–87. doi: 10.1006/excr.1999.4706. [DOI] [PubMed] [Google Scholar]
- [14].Hatakeyama M. System properties of ErbB receptor signaling for the understanding of cancer progression. Mol Biosyst. 2007;3:111–116. doi: 10.1039/b612800a. [DOI] [PubMed] [Google Scholar]
- [15].Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Maatta JA, Sundvall M, Junttila TT, Peri L, Laine VJ, Isola J, Egeblad M, Elenius K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol Biol Cell. 2006;17:67–79. doi: 10.1091/mbc.E05-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Starr A, Greif J, Vexler A, Ashkenazy-Voghera M, Gladesh V, Rubin C, Kerber G, Marmor S, Lev-Ari S, Inbar M, Yarden Y, Ben-Yosef R. ErbB4 increases the proliferation potential of human lung cancer cells and its blockage can be used as a target for anti-cancer therapy. Int J Cancer. 2006;119:269–274. doi: 10.1002/ijc.21818. [DOI] [PubMed] [Google Scholar]
- [18].Tang CK, Concepcion XZ, Milan M, Gong X, Montgomery E, Lippman ME. Ribozyme-mediated down-regulation of ErbB-4 in estrogen receptor-positive breast cancer cells inhibits proliferation both in vitro and in vivo. Cancer Res. 1999;59:5315–5322. [PubMed] [Google Scholar]
- [19].Zhu Y, Sullivan LL, Nair SS, Williams CC, Pandey AK, Marrero L, Vadlamudi RK, Jones FE. Coregulation of estrogen receptor by ERBB4/HER4 establishes a growth-promoting autocrine signal in breast tumor cells. Cancer Res. 2006;66:7991–7998. doi: 10.1158/0008-5472.CAN-05-4397. [DOI] [PubMed] [Google Scholar]
- [20].Lodge AJ, Anderson JJ, Gullick WJ, Haugk B, Leonard RC, Angus B. Type 1 growth factor receptor expression in node positive breast cancer: adverse prognostic significance of c-erbB-4. J Clin Pathol. 2003;56:300–304. doi: 10.1136/jcp.56.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gilbertson RJ, Clifford SC, MacMeekin W, Meekin W, Wright C, Perry RH, Kelly P, Pearson AD, Lunec J. Expression of the ErbB-neuregulin signaling network during human cerebellar development: implications for the biology of medulloblastoma. Cancer Res. 1998;58:3932–3941. [PubMed] [Google Scholar]
- [22].Jones FE, Jerry DJ, Guarino BC, Andrews GC, Stern DF. Heregulin induces in vivo proliferation and differentiation of mammary epithelium into secretory lobuloalveoli. Cell Growth Differ. 1996;7:1031–1038. [PubMed] [Google Scholar]
- [23].Aqeilan RI, Donati V, Gaudio E, Nicoloso MS, Sundvall M, Korhonen A, Lundin J, Isola J, Sudol M, Joensuu H, Croce CM, Elenius K. Association of Wwox with ErbB4 in breast cancer. Cancer Res. 2007;67:9330–9336. doi: 10.1158/0008-5472.CAN-07-2147. [DOI] [PubMed] [Google Scholar]
- [24].Naresh A, Long W, Vidal GA, Wimley WC, Marrero L, Sartor CI, Tovey S, Cooke TG, Bartlett JM, Jones FE. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006;66:6412–6420. doi: 10.1158/0008-5472.CAN-05-2368. [DOI] [PubMed] [Google Scholar]
- [25].Pitfield SE, Bryant I, Penington DJ, Park G, Riese DJ., 2nd Phosphorylation of ErbB4 on tyrosine 1056 is critical for ErbB4 coupling to inhibition of colony formation by human mammary cell lines. Oncol Res. 2006;16:179–193. doi: 10.3727/000000006783981134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Williams EE, Trout LJ, Gallo RM, Pitfield SE, Bryant I, Penington DJ, Riese DJ., 2nd A constitutively active ErbB4 mutant inhibits drug-resistant colony formation by the DU-145 and PC-3 human prostate tumor cell lines. Cancer Lett. 2003;192:67–74. doi: 10.1016/s0304-3835(02)00690-0. [DOI] [PubMed] [Google Scholar]
- [27].Kew TY, Bell JA, Pinder SE, Denley H, Srinivasan R, Gullick WJ, Nicholson RI, Blamey RW, Ellis IO. c-erbB-4 protein expression in human breast cancer. Br J Cancer. 2000;82:1163–1170. doi: 10.1054/bjoc.1999.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tovey SM, Witton CJ, Bartlett JM, Stanton PD, Reeves JR, Cooke TG. Outcome and human epidermal growth factor receptor (HER) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res. 2004;6:R246–251. doi: 10.1186/bcr783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sartor CI, Zhou H, Kozlowska E, Guttridge K, Kawata E, Caskey L, Harrelson J, Hynes N, Ethier S, Calvo B, Earp HS., 3rd Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol Cell Biol. 2001;21:4265–4275. doi: 10.1128/MCB.21.13.4265-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Culouscou JM, Plowman GD, Carlton GW, Green JM, Shoyab M. Characterization of a breast cancer cell differentiation factor that specifically activates the HER4/p180erbB4 receptor. J Biol Chem. 1993;268:18407–18410. [PubMed] [Google Scholar]
- [31].Peles E, Bacus SS, Koski RA, Lu HS, Wen D, Ogden SG, Levy RB, Yarden Y. Isolation of the neu/HER-2 stimulatory ligand: a 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell. 1992;69:205–216. doi: 10.1016/0092-8674(92)90131-u. [DOI] [PubMed] [Google Scholar]
- [32].Wen D, Peles E, Cupples R, Suggs SV, Bacus SS, Luo Y, Trail G, Hu S, Silbiger SM, Levy RB, Koski RA, Lu HS, Yarden Y. Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell. 1992;69:559–572. doi: 10.1016/0092-8674(92)90456-m. [DOI] [PubMed] [Google Scholar]
- [33].Li L, Seno M, Yamada H, Kojima I. Betacellulin improves glucose metabolism by promoting conversion of intraislet precursor cells to beta-cells in streptozotocin-treated mice. Am J Physiol Endocrinol Metab. 2003;285:E577–583. doi: 10.1152/ajpendo.00120.2003. [DOI] [PubMed] [Google Scholar]
- [34].Dunbar AJ, Goddard C. Structure-function and biological role of betacellulin. Int J Biochem Cell Biol. 2000;32:805–815. doi: 10.1016/s1357-2725(00)00028-5. [DOI] [PubMed] [Google Scholar]
- [35].Mashima H, Ohnishi H, Wakabayashi K, Mine T, Miyagawa J, Hanafusa T, Seno M, Yamada H, Kojima I. Betacellulin and activin A coordinately convert amylase-secreting pancreatic AR42J cells into insulin-secreting cells. J Clin Invest. 1996;97:1647–1654. doi: 10.1172/JCI118591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gallo RM, Bryant I, Fry R, Williams EE, Riese DJ., 2nd Phosphorylation of ErbB4 on Tyr1056 is critical for inhibition of colony formation by prostate tumor cell lines. Biochem Biophys Res Commun. 2006;349:372–382. doi: 10.1016/j.bbrc.2006.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vidal GA, Clark DE, Marrero L, Jones FE. A constitutively active ERBB4/HER4 allele with enhanced transcriptional coactivation and cell-killing activities. Oncogene. 2007;26:462–466. doi: 10.1038/sj.onc.1209794. [DOI] [PubMed] [Google Scholar]
- [38].Feng SM, Sartor CI, Hunter D, Zhou H, Yang X, Caskey LS, Dy R, Muraoka-Cook RS, Earp HS., 3rd The HER4 cytoplasmic domain, but not its C terminus, inhibits mammary cell proliferation. Mol Endocrinol. 2007;21:1861–1876. doi: 10.1210/me.2006-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Linggi B, Cheng QC, Rao AR, Carpenter G. The ErbB-4 s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene. 2006;25:160–163. doi: 10.1038/sj.onc.1209003. [DOI] [PubMed] [Google Scholar]
- [40].Michaud DS. Epidemiology of pancreatic cancer. Minerva Chir. 2004;59:99–111. [PubMed] [Google Scholar]
- [41].Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- [42].Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- [43].Baxter NN, Whitson BA, Tuttle TM. Trends in the treatment and outcome of pancreatic cancer in the United States. Ann Surg Oncol. 2007;14:1320–1326. doi: 10.1245/s10434-006-9249-8. [DOI] [PubMed] [Google Scholar]
- [44].Okusaka T, Kosuge T. Systemic chemotherapy for pancreatic cancer. Pancreas. 2004;28:301–304. doi: 10.1097/00006676-200404000-00017. [DOI] [PubMed] [Google Scholar]
- [45].Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- [46].Barton CM, Staddon SL, Hughes CM, Hall PA, O’Sullivan C, Kloppel G, Theis B, Russell RC, Neoptolemos J, Williamson RC, et al. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer. 1991;64:1076–1082. doi: 10.1038/bjc.1991.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cowgill SM, Muscarella P. The genetics of pancreatic cancer. Am J Surg. 2003;186:279–286. doi: 10.1016/s0002-9610(03)00226-5. [DOI] [PubMed] [Google Scholar]
- [48].Friess H, Yamanaka Y, Buchler M, Berger HG, Kobrin MS, Baldwin RL, Korc M. Enhanced expression of the type II transforming growth factor beta receptor in human pancreatic cancer cells without alteration of type III receptor expression. Cancer Res. 1993;53:2704–2707. [PubMed] [Google Scholar]
- [49].Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI, Korc M. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993;105:1846–1856. doi: 10.1016/0016-5085(93)91084-u. [DOI] [PubMed] [Google Scholar]
- [50].Korc M, Chandrasekar B, Yamanaka Y, Friess H, Buchier M, Beger HG. Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J Clin Invest. 1992;90:1352–1360. doi: 10.1172/JCI116001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yamanaka Y, Friess H, Kobrin MS, Buchler M, Kunz J, Beger HG, Korc M. Overexpression of HER2/neu oncogene in human pancreatic carcinoma. Hum Pathol. 1993;24:1127–1134. doi: 10.1016/0046-8177(93)90194-l. [DOI] [PubMed] [Google Scholar]
- [52].Friess H, Yamanaka Y, Kobrin MS, Do DA, Buchler MW, Korc M. Enhanced erbB-3 expression in human pancreatic cancer correlates with tumor progression. Clin Cancer Res. 1995;1:1413–1420. [PubMed] [Google Scholar]
- [53].Graber HU, Friess H, Kaufmann B, Willi D, Zimmermann A, Korc M, Buchler MW. ErbB-4 mRNA expression is decreased in non-metastatic pancreatic cancer. Int J Cancer. 1999;84:24–27. doi: 10.1002/(sici)1097-0215(19990219)84:1<24::aid-ijc5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- [54].Thybusch-Bernhardt A, Beckmann S, Juhl H. Comparative analysis of the EGF-receptor family in pancreatic cancer: expression of HER-4 correlates with a favourable tumor stage. Int J Surg Investig. 2001;2:393–400. [PubMed] [Google Scholar]
- [55].Leptak C, Ramon y Cajal S, Kulke R, Horwitz BH, Riese DJ, 2nd, Dotto GP, DiMaio D. Tumorigenic transformation of murine keratinocytes by the E5 genes of bovine papillomavirus type 1 and human papillomavirus type 16. J Virol. 1991;65:7078–7083. doi: 10.1128/jvi.65.12.7078-7083.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Riese DJ, 2nd, DiMaio D. An intact PDGF signaling pathway is required for efficient growth transformation of mouse C127 cells by the bovine papillomavirus E5 protein. Oncogene. 1995;10:1431–1439. [PubMed] [Google Scholar]
- [57].Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–982. 984–986, 989–990. [PMC free article] [PubMed] [Google Scholar]
- [58].Riese DJ, 2nd, van Raaij TM, Plowman GD, Andrews GC, Stern DF. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol. 1995;15:5770–5776. doi: 10.1128/mcb.15.10.5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Penington DJ, Bryant I, Riese DJ., 2nd Constitutively active ErbB4 and ErbB2 mutants exhibit distinct biological activities. Cell Growth Differ. 2002;13:247–256. [PubMed] [Google Scholar]
- [60].Mann R, Mulligan RC, Baltimore D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell. 1983;33:153–159. doi: 10.1016/0092-8674(83)90344-6. [DOI] [PubMed] [Google Scholar]
- [61].Miller AD, Buttimore C. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol Cell Biol. 1986;6:2895–2902. doi: 10.1128/mcb.6.8.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci U S A. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Riese DJ, 2nd, Komurasaki T, Plowman GD, Stern DF. Activation of ErbB4 by the bifunctional epidermal growth factor family hormone epiregulin is regulated by ErbB2. J Biol Chem. 1998;273:11288–11294. doi: 10.1074/jbc.273.18.11288. [DOI] [PubMed] [Google Scholar]
- [64].Liu N, Furukawa T, Kobari M, Tsao MS. Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol. 1998;153:263–269. doi: 10.1016/S0002-9440(10)65567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang L, Yuan SZ. Expression of c-erbB-2 oncogene protein, epidermal growth factor receptor, and TGF-beta1 in human pancreatic ductal adenocarcinoma. Hepatobiliary Pancreat Dis Int. 2002;1:620–623. [PubMed] [Google Scholar]
- [66].Ueda S, Ogata S, Tsuda H, Kawarabayashi N, Kimura M, Sugiura Y, Tamai S, Matsubara O, Hatsuse K, Mochizuki H. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2004;29:e1–8. doi: 10.1097/00006676-200407000-00061. [DOI] [PubMed] [Google Scholar]
- [67].Immervoll H, Hoem D, Kugarajh K, Steine SJ, Molven A. Molecular analysis of the EGFR-RAS-RAF pathway in pancreatic ductal adenocarcinomas: lack of mutations in the BRAF and EGFR genes. Virchows Arch. 2006;448:788–796. doi: 10.1007/s00428-006-0191-8. [DOI] [PubMed] [Google Scholar]
- [68].Kwak EL, Jankowski J, Thayer SP, Lauwers GY, Brannigan BW, Harris PL, Okimoto RA, Haserlat SM, Driscoll DR, Ferry D, Muir B, Settleman J, Fuchs CS, Kulke MH, Ryan DP, Clark JW, Sgroi DC, Haber DA, Bell DW. Epidermal growth factor receptor kinase domain mutations in esophageal and pancreatic adenocarcinomas. Clin Cancer Res. 2006;12:4283–4287. doi: 10.1158/1078-0432.CCR-06-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee J, Jang KT, Ki CS, Lim T, Park YS, Lim HY, Choi DW, Kang WK, Park K, Park JO. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer. 2007;109:1561–1569. doi: 10.1002/cncr.22559. [DOI] [PubMed] [Google Scholar]
- [70].Tzeng CW, Frolov A, Frolova N, Jhala NC, Howard JH, Buchsbaum DJ, Vickers SM, Heslin MJ, Arnoletti JP. Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancer. Surgery. 2007;141:464–469. doi: 10.1016/j.surg.2006.09.009. [DOI] [PubMed] [Google Scholar]
- [71].Jimeno A, Hidalgo M. Pharmacogenomics of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Biochimica et biophysica acta. 2006;1766:217–229. doi: 10.1016/j.bbcan.2006.08.008. [DOI] [PubMed] [Google Scholar]
- [72].Tobita K, Kijima H, Dowaki S, Kashiwagi H, Ohtani Y, Oida Y, Yamazaki H, Nakamura M, Ueyama Y, Tanaka M, Inokuchi S, Makuuchi H. Epidermal growth factor receptor expression in human pancreatic cancer: Significance for liver metastasis. Int J Mol Med. 2003;11:305–309. [PubMed] [Google Scholar]
- [73].Heeger S. Targeted therapy of the epidermal growth factor receptor in the treatment of pancreatic cancer. Recent Results Cancer Res. 2008;177:131–136. doi: 10.1007/978-3-540-71279-4_15. [DOI] [PubMed] [Google Scholar]
- [74].Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–1825. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wang SC, Hung MC. HER2 overexpression and cancer targeting. Semin Oncol. 2001;28:115–124. doi: 10.1016/s0093-7754(01)90289-1. [DOI] [PubMed] [Google Scholar]
- [76].Kritzik MR, Krahl T, Good A, Gu D, Lai C, Fox H, Sarvetnick N. Expression of ErbB receptors during pancreatic islet development and regrowth. J Endocrinol. 2000;165:67–77. doi: 10.1677/joe.0.1650067. [DOI] [PubMed] [Google Scholar]
- [77].Riese DJ, 2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. Bioessays. 1998;20:41–48. doi: 10.1002/(SICI)1521-1878(199801)20:1<41::AID-BIES7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- [78].Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ., 2nd Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacol Ther. 2009;122:1–8. doi: 10.1016/j.pharmthera.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tvorogov D, Sundvall M, Kurppa K, Hollmen M, Repo S, Johnson MS, Elenius K. Somatic mutations of ErbB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. J Biol Chem. 2009;284:5582–5591. doi: 10.1074/jbc.M805438200. [DOI] [PubMed] [Google Scholar]
- [80].Riese DJ, 2nd, Bermingham Y, van Raaij TM, Buckley S, Plowman GD, Stern DF. Betacellulin activates the epidermal growth factor receptor and erbB-4, and induces cellular response patterns distinct from those stimulated by epidermal growth factor or neuregulin-beta. Oncogene. 1996;12:345–353. [PubMed] [Google Scholar]
- [81].Abd El-Rehim DM, Pinder SE, Paish CE, Bell JA, Rampaul RS, Blamey RW, Robertson JF, Nicholson RI, Ellis IO. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer. 2004;91:1532–1542. doi: 10.1038/sj.bjc.6602184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- [83].Junttila TT, Sundvall M, Lundin M, Lundin J, Tanner M, Harkonen P, Joensuu H, Isola J, Elenius K. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005;65:1384–1393. doi: 10.1158/0008-5472.CAN-04-3150. [DOI] [PubMed] [Google Scholar]
- [84].Kainulainen V, Sundvall M, Maatta JA, Santiestevan E, Klagsbrun M, Elenius K. A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J Biol Chem. 2000;275:8641–8649. doi: 10.1074/jbc.275.12.8641. [DOI] [PubMed] [Google Scholar]
- [85].Petti LM, Ray FA. Transformation of mortal human fibroblasts and activation of a growth inhibitory pathway by the bovine papillomavirus E5 oncoprotein. Cell Growth Differ. 2000;11:395–408. [PubMed] [Google Scholar]