

Abstract
The protective effects of high-density lipoprotein (HDL) under lipopolysaccharide (LPS) conditions have been well documented. Here, we investigated whether an effect of HDL on Toll-like receptor 4 (TLR4) expression and signalling may contribute to its endothelial-protective effects and to improved survival in a mouse model of LPS-induced inflammation and lethality. HDL cholesterol increased 1.7-fold (p < 0.005) and lung endothelial TLR4 expression decreased 8.4-fold (p < 0.005) 2 weeks after apolipoprotein (apo) A-I gene transfer. Following LPS administration in apo A-I gene transfer mice, lung TLR4 and lung MyD88 mRNA expression, reflecting TLR4 signalling, were 3.0-fold (p < 0.05) and 2.1-fold (p < 0.05) lower, respectively, than in LPS control mice. Concomitantly, LPS-induced lung neutrophil infiltration, lung oedema and mortality were significantly attenuated following apo A–I transfer. In vitro, supplementation of HDL or apo A–I to human microvascular endothelial cells-1 24 h before LPS administration reduced TLR4 expression, as assessed by fluorescent-activated cell sorting, and decreased the LPS-induced MyD88 mRNA expression and NF-κB activity, independently of LPS binding. In conclusion, HDL reduces TLR4 expression and signalling in endothelial cells, which may contribute significantly to the protective effects of HDL in LPS-induced inflammation and lethality.
Electronic supplementary material
The online version of this article (doi:10.1007/s00109-010-0690-6) contains supplementary material, which is available to authorized users.
Keywords: Toll-like receptor 4, High-density lipoprotein, Endothelial cells, Lipopolysaccharide, Gene transfer
Introduction
Epidemiological studies have shown that high-density lipoprotein (HDL) cholesterol levels inversely correlate with the outcome of endotoxic shock [1–3]. Moreover, systemic administration of reconstituted HDL in human volunteers down-regulated CD14 on monocytes and attenuated pro-inflammatory mediators caused by small doses of intravenous LPS [4]. HDL also protects against LPS-induced inflammation and lethality in experimental animal models [5–8]. It has been shown that a twofold increase of plasma HDL in human apolipoprotein (apo) A–I transgenic mice enhances the binding of intraperitoneally administered LPS to HDL, reduces plasma tumour necrosis factor (TNF)-α levels and improves the survival rate compared to control mice [5]. Similarly, intravenous injection of reconstituted HDL increased survival in mice challenged with LPS [5], as well as in rabbits challenged with live Escherichia coli [9]. Several in vitro studies demonstrated that apo A–I, the main apo of HDL, and sphingosine-1-phosphate (S1P), a sphingolipid associated with lipoproteins, especially with HDL, inhibit LPS-induced inflammatory responses. Reconstituted HDL and an apo A–I mimetic peptide reduced LPS-induced expression of endothelial cell adhesion molecules in vitro [6, 7]. S1P significantly reduced pulmonary vascular leakage and inflammation in a murine model of LPS-induced acute lung injury [8].
Notwithstanding the well-documented anti-inflammatory effects of HDL and its components during LPS-induced endotoxemia, a potential direct role of HDL or apo A–I in innate immunity regulation under LPS-induced inflammation has not yet been investigated. Innate immunity is characterized by a natural selection of germline-encoded receptors, which focus the host response to highly conserved pathogen-associated molecular patterns (PAMPs) shared by many microorganisms [10, 11]. Toll-like receptors (TLRs), a group of PAMP recognition receptors, play an important role in innate immune signalling in response to microbial infection. TLR4 is the main protein involved in recognizing LPS [12–14] and is of importance in the pathogenesis of different cardiovascular disorders [15, 16]. Recently, it has been demonstrated that endothelial TLR4 rather than leukocyte TLR4 is the key player in LPS-induced neutrophil sequestration into lungs and subsequent lung oedema [17].
Given the well-known endothelial-protective [18–20] and anti-inflammatory [21, 22] properties of HDL, we hypothesized in the current study that HDL may attenuate endothelial TLR4 expression and TLR4 signalling and improve hereby inflammation and the outcome of LPS-induced lethality. To explore this hypothesis, we investigated the effect of HDL on TLR4 regulation and TLR4-mediated signalling in vivo, following human apo A–I gene transfer in an experimental model of LPS-induced inflammation and lethality. We show that human apo A–I gene transfer decreases TLR4 expression in vivo in the absence of LPS and attenuates TLR4 signalling following LPS administration. Similar effects on TLR4 expression and TLR4-mediated signalling were induced by HDL in human microvascular endothelial cells (HMEC)-1 in vitro. These effects could be mimicked by apo A–I and were observed independently of LPS binding. These novel endothelial protective effects of HDL may contribute to increased survival in this murine model of LPS-induced inflammation and lethality.
Materials and methods
For detailed methodology, please see the Electronic supplementary material (ESM) ESM 1 (available online at http://www.springer.com/biomed/molecular/journal/109). In brief, 8-week-old male C57BL/6 mice were intravenously injected with 5 × 1010 particles of the E1E3E4-deleted adenoviral vector Ad.hapoA–I, expressing human apo A–I [23]. As controls, age-matched C57BL/6 mice were injected with the same dose of Ad.Null, containing no expression cassette [23]. Fourteen days hereafter, LPS from E. coli, serotype 055:B5 (Sigma, Steinheim, Germany), was intraperitoneally injected at a dose of 80 mg/kg. Mice were killed 20 h after LPS injection. Lung TLR4 expression as well as localisation of TLR4 in lung endothelial cells and neutrophils was determined by immunohistology. In addition, mRNA expression of lung TLR4, myeloid differentiation factor 88 (MyD88), Toll/IL-1R-containing adaptor inducing interferon β (TRIF) and ribosomal protein L32 was analysed and lung myeloperoxidase activity was quantified. In vitro, the effect of pre-incubation with HDL or its component apo A–I on TLR4 expression and signalling was analysed in HMEC-1 cells. Determinations were performed in the absence or presence of LPS supplementation following the pre-incubation period. In the case of LPS, all LPS supplementations were performed in the absence of HDL or apo A–I to ensure that effects were independent of LPS binding. The LPS binding capacity of HDL and apo A–I was also analysed.
Results
Apo A–I transfer selectively increases HDL cholesterol both before and after LPS administration
Human apo A–I gene transfer resulted in human apo A–I levels of 85 ± 4.8 mg/dl at day 14 (ESM Fig. S1A), leading to 1.7-fold (p < 0.005) higher HDL cholesterol levels compared to Ad.Null controls (Table 1). In agreement with other reports [24–26], human apo A–I gene transfer reduced murine apo A–I concentrations 2.8-fold (p < 0.0001) and 2.8-fold (p = 0.0005) versus saline-control and Ad.Null-control mice, respectively (ESM Fig. S1B). Ad.Null gene transfer did not affect murine apo A–I plasma levels. Twenty hours after LPS injection, HDL cholesterol significantly declined in both Ad.Null-LPS and Ad.hapoA-I-LPS mice, but HDL cholesterol levels in the Ad.hapoA-I-LPS group were 2.4-fold (p < 0.005) higher compared to the Ad.Null-LPS group (Table 1). Human apo A–I levels decreased 1.9-fold (p < 0.005) after LPS administration (ESM Fig. S1A). Furthermore, LPS administration resulted in a 1.3-fold (p < 0.05) and 1.5-fold (p < 0.05) drop in murine apo A–I plasma levels in saline and Ad.Null-injected mice, respectively (ESM Fig. S1B), whereas murine apo A–I concentrations were not significantly altered by LPS in Ad.hapoA–I mice. Human apo A–I gene transfer did not affect non-HDL cholesterol levels (Table 1). Moreover, the relative content of S1P in HDL was not influenced by human apo A–I gene transfer (ESM Fig. S2), which is in agreement with a recent study indicating that apo A–I is not the determinant protein in HDL responsible for S1P levels [27]. Since Ad.Null gene transfer did not alter murine apo A–I plasma levels, nor the cholesterol lipoprotein profile (ESM Fig. S1C) under control and LPS conditions, comparisons were consistently performed between Ad.Null mice and Ad.hapoA–I mice in the majority of subsequent experiments.
Table 1.
HDL, non-HDL and total cholesterol
| Ad.Null-Co | Ad.hapoA-I-Co | Ad.Null-LPS | Ad.hapoA-I-LPS | |
|---|---|---|---|---|
| HDL-C (mg/dl) | 42 ± 2 | 73 ± 4** | 23 ± 1** | 55 ± 2*,§ |
| Non-HDL-C (mg/dl) | 25 ± 2 | 24 ± 2 | 40 ± 4** | 38 ± 2** |
| Total-C (mg/dl) | 66 ± 3 | 91 ± 4** | 63 ± 5 | 93 ± 2**,§ |
Mouse lipoproteins were separated by density gradient ultracentrifugation as described previously [39]. Fractions were stored at −20°C until analysis. Cholesterol in lipoprotein fractions was determined with commercially available enzymes (Roche Diagnostics, Basel, Switzerland). Precipath L (Roche Diagnostics) was used as a standard. Data are represented as mean ± SEM
Co control, C cholesterol
*p < 0.05; ** p < 0.005 (versus Ad.Null-Co); § p < 0.005 (versus Ad.Null-LPS)
Apo A–I transfer reduces lung TLR4 expression in LPS-injected mice
Immunohistochemistry identified increased TLR4 expression 20 h after LPS injection in the lungs of Ad.Null-LPS mice (Fig. 1a). Human apo A–I transfer did not alter lung TLR4 mRNA expression in control mice, but TLR4 mRNA was 3.0-fold (p < 0.05) lower in Ad.hapoA-I-LPS mice compared to Ad.Null-LPS mice. This Ad.hapoA-I-induced reduction of TLR4 mRNA expression under LPS conditions was associated with a 2.1-fold (p < 0.05) decrease in MyD88 mRNA expression, whereas TRIF mRNA expression declined 1.5-fold (p = NS, Fig. 1b). The percentage of neutrophils expressing TLR4 increased 1.9-fold (p < 0.05) after LPS administration in control mice, but this induction was abrogated in Ad.hapoA-I-LPS mice (Fig. 2).
Fig. 1.
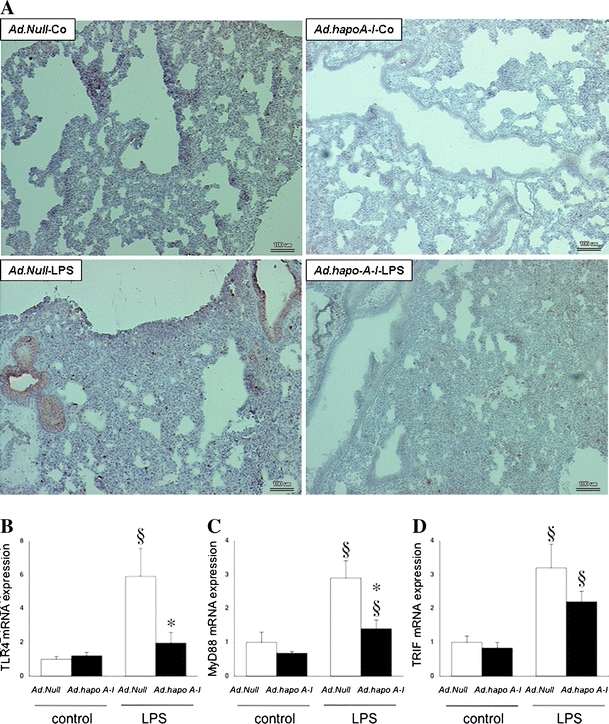
Apo A–I transfer reduces LPS-induced lung TLR4 expression and signalling. a Representative photomicrographs of Toll-like receptor 4-stained cryosections (magnification ×200; bar corresponds to 100 μm). Top Baseline TLR4 expression in a lung of a non-septic C57BL/6 mouse 14 days after intravenous injection with Ad.Null (Ad.Null-Co) or Ad.hapoA-I (Ad.hapoA-I-Co) and 20 h after intraperitoneally injected saline (Ad.Null-Co and Ad.hapoA-I-Co, respectively). Bottom TLR4 abundance and less pronounced TLR4 expression in a lung of a septic C57BL/6 mouse 14 days after intravenous injection with Ad.Null or Ad.hapoA-I and 20 h after intraperitoneally injected LPS (Ad.Null-LPS and Ad.hapoA-I-LPS, respectively). Bar graphs representing lung TLR4 (b), MyD88 (c) and TRIF (d) mRNA expression, relative to the Ad.Null-saline group set as 1. Bar graphs indicate mean ± SEM (n = 6), with Ad.Null (open bars) and Ad.hapoA-I (black bars). *p < 0.05 versus Ad.Null-LPS, § p < 0.05 versus controls
Fig. 2.
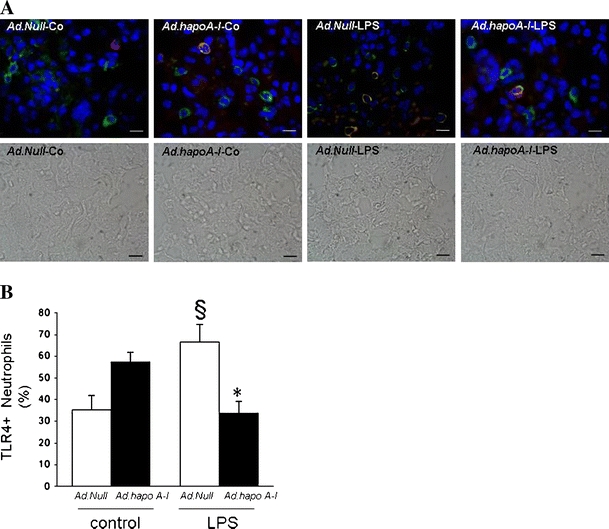
Apo A–I transfer reduces LPS-induced lung neutrophil TLR4 expression. Neutrophil TLR4 expression in the lung of C57BL/6 mice 14 days after intravenous injection with Ad.Null or Ad.hapoA-I and 20 h after intraperitoneal injection with saline or LPS. a Representative fluorescent microscopy pictures of double-stained TLR4 (green)/myeloperoxidase (red) cryosections of (from left to right) Ad.Null-control, Ad.hapoA-I-control, Ad.Null-LPS and Ad.hapoA-I-LPS mice with below respective phase contrast pictures (magnification ×1,000; bar corresponds to 10 μm). b Bar graph representing percentage of neutrophils expressing TLR4 represented as mean ± SEM (n = 5–7), with Ad.Null (open bars) and Ad.hapoA-I (black bars). *p < 0.05 versus Ad.Null-LPS, § p < 0.05 versus controls
HDL and apo A–I bind LPS
The effects of human apo A–I transfer on TLR4 expression after LPS administration may be due to the direct effects of HDL on TLR4 expression or to LPS binding by HDL or one of its components. Since the relative content of S1P in HDL was unchanged after apo A–I gene transfer, suggesting no major S1P-mediated contribution to the effects of apo A–I transfer on TLR4 signalling, further in vitro analyses evaluating the effect of S1P were not performed. We compared the in vitro LPS binding capacity of HDL and apo A–I using LPS FITC and by evaluating the fluorescence intensity. ESM Fig. S3 illustrates that both HDL and apo A–I bind LPS.
HDL reduce endothelial TLR4 expression and signalling in vivo and in vitro
Considering the critical role of endothelial TLR4 in neutrophil sequestration in the lungs after LPS administration, we investigated the effect of apo A–I transfer on the expression of TLR4 on endothelial cells. Human apo A–I transfer reduced the percentage of endothelial cells expressing TLR4 by 8.4-fold (p < 0.005) in control mice (Fig. 3).
Fig. 3.
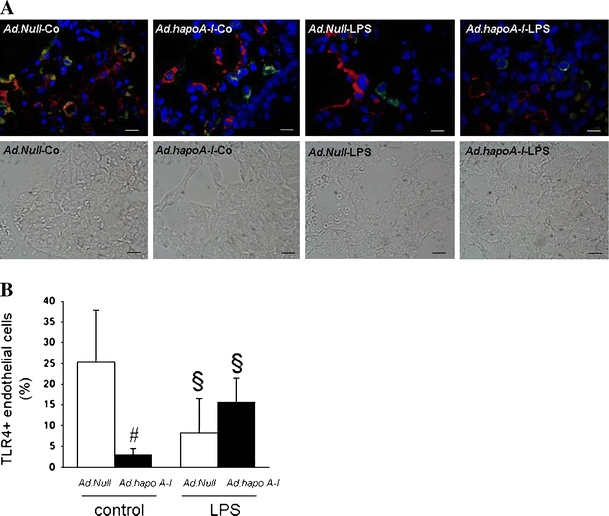
Apo A–I transfer reduces endothelial TLR4 expression. Endothelial TLR4 expression in the lung of C57BL/6 mice 14 days after intravenous injection with Ad.Null or Ad.hapoA–I and 20 h after intraperitoneal injection with saline or LPS. a Representative fluorescent microscopy pictures of double-stained TLR4 (green)/von Willebrand factor (red) cryosections of (from left to right) Ad.Null-control, Ad.hapoA-I-control, Ad.Null-LPS and Ad.hapoA-I-LPS mice with below respective phase contrast pictures (magnification ×1,000; bar corresponds to 10 μm). b Bar graph representing per cent of endothelial cells expressing TLR4 represented as mean ± SEM (n = 5–7), with Ad.Null (open bars) and Ad.hapoA-I (black bars). # p < 0.05 versus Ad.Null-control, *p < 0.05 versus Ad.Null-LPS, § p < 0.05 versus controls
To further analyse the effect of HDL and apo A–I on endothelial TLR4 expression and TLR4-mediated signalling in vitro, HMEC-1 cells were pre-incubated with HDL or apo A–I. In the absence of LPS supplementation, TLR4 expression quantified by fluorescent-activated cell sorting (FACS) was reduced by 2.6-fold (p < 0.05) and 2.5-fold (p < 0.01) by pre-incubation with HDL and apo A–I, respectively (Fig. 4a). MyD88 and TRIF mRNA expressions were not altered (Fig. 4b, c). Following the pre-incubation period, all LPS supplementations were performed in the absence of HDL or apo A–I to ensure that effects were independent of LPS binding. LPS supplementation without pre-incubation with HDL or any of its components resulted in decreased TLR4 expression (3.5-fold, p < 0.001), whereas MyD88 mRNA expression increased. Under conditions of pre-incubation with HDL or apo A–I, the LPS-up-regulated MyD88 mRNA expression was decreased (Fig. 4b). Concomitantly, pre-incubation with HDL or apo A–I reduced the LPS-induced p65 NF-κB activity by 1.6-fold (p < 0.05) and 2.0-fold (p < 0.05), respectively (Fig. 4d). Taken together, these results indicate that HDL may exert direct effects on TLR4 signalling independent of LPS binding.
Fig. 4.
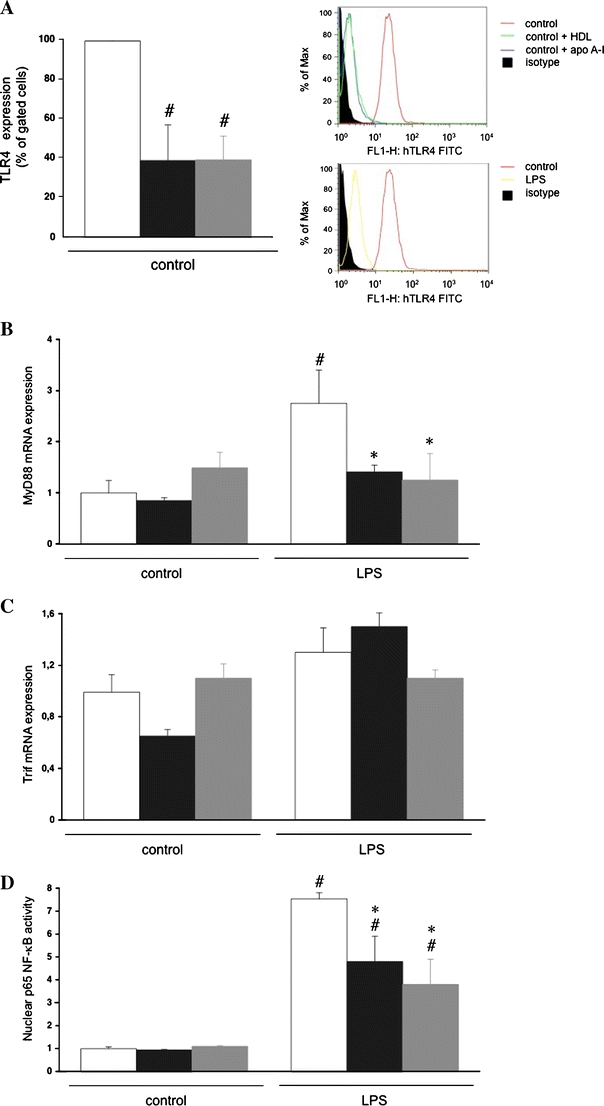
HDL and its component apo A–I reduce TLR4 expression and underlying signalling in human microvascular endothelial cells-1. Human microvascular endothelial cells-1 (HMEC-1) were incubated in the presence or absence of HDL (50 μg/ml) or apo A–I (35 μg/ml) for 24 h. Next, LPS (100 ng/ml) was supplemented in the absence of HDL or apo A–I for 2 h for FACS and mRNA expression or for 4 h for NF-κB activity analysis, respectively. a Bar graph representing per cent gated cells of n = 3 independent experiments. Right upper panel Histogram representing counts of human TLR4 expression in control, control+apo A–I, control+HDL and isotype control (see legend on the right). Right lower panel Histogram representing counts of human TLR4 expression in control, LPS and isotype control (see legend on the right). Bar graphs representing MyD88 mRNA expression (b), TRIF mRNA expression (c) and p65 NF-κB activity (d), relative to the normal control group set as 1. Bar graphs indicate mean ± SEM (n = 4), with untreated (open bars), HDL (black bars) and apo A-I (grey bars). # p < 0.01 versus untreated control, *p < 0.05 versus untreated LPS
Apo A–I gene transfer attenuates lipopolysaccharide-induced lung oedema and endothelial damage
Haematoxylin and eosin staining on paraffin sections demonstrated interstitial oedema, infiltration of inflammatory cells and increased alveolar area in Ad.Null-LPS mice, which was less pronounced in Ad.hapoA-I-LPS mice (Fig. 5a). The decrease in pulmonary oedema in Ad.hapoA-I-LPS mice was also shown by a 1.7-fold (p < 0.05) lower wet lung weight/body weight ratio compared to Ad.Null-LPS mice (Fig. 5b). In addition, human apo A–I gene transfer significantly reduced neutrophil infiltration in LPS-treated mice, as indicated by 1.9-fold (p < 0.05) reduced myeloperoxidase activity and 2.4-fold (p = 0.05) decreased density of myeloperoxidase-positive cells compared to Ad.Null-LPS mice (Fig. 5c, d). The LPS-induced decrease of endothelial cell density was 2.0-fold (p < 0.05) less pronounced in Ad.hapoA-I-LPS mice than in Ad.Null mice (Fig. 5e).
Fig. 5.
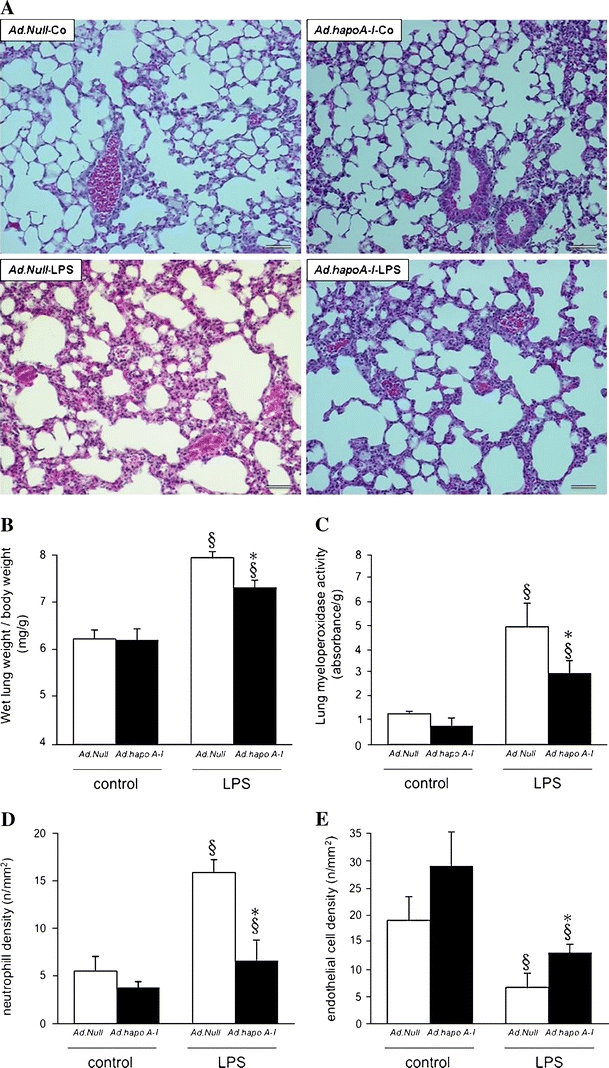
Apo A–I transfer attenuates LPS-induced lung damage. a Representative photomicrographs of haematoxylin and eosin-stained paraffin sections (magnification ×200; bar corresponds to 100 μm) from lungs of control C57BL/6 mice 14 days after intravenous injection with Ad.Null (Ad.Null-Co) or Ad.hapoA-I (Ad.hapoA-I-Co, top) and C57BL/6 mice 20 h after LPS treatment (Ad.Null-LPS and Ad.hapoA-I-LPS, bottom). Extensive interstitial oedema, infiltration of inflammatory cells and haemorrhage is present in Ad.Null-LPS mice, whereas lung damage is markedly reduced in mice overexpressing human apo A–I compared with Ad.Null-LPS mice. Bar graphs representing wet lung weight-to-body weight ratio (b), lung myeloperoxidase activity (c), as marker of neutrophil infiltration, neutrophil cell density (n/mm2) (d) and endothelial cell density (n/mm2) (e). Bar graphs indicate mean ± SEM (n = 6), with Ad.Null (open bars) and Ad.hapoA-I (black bars). *p < 0.05 versus Ad.Null-LPS, § p < 0.05 versus controls
Apo A–I transfer improves survival in lipopolysaccharide-induced lethality
To investigate whether human apo A–I gene transfer may improve LPS-induced lethality, we followed survival for 60 h. Kaplan–Meier survival curves are shown in ESM Fig. S4. Comparison of survival curves by logrank test showed significantly higher survival (p < 0.005) in Ad.hapoA-I-LPS mice compared to saline-LPS and Ad.Null-LPS mice. No significant difference was found between the survival rate of Ad.Null-LPS and saline-LPS mice.
Discussion
This study reveals the reduction of endothelial TLR4 signalling after human apo A–I gene transfer or HDL supplementation on endothelial cells as a novel endothelial-protective feature of HDL contributing to the decreased infiltration of neutrophils and improved survival rate in an experimental model of LPS-induced inflammation.
Epidemiological studies demonstrate an inverse correlation between HDL cholesterol concentrations and the outcome of septic shock [1–3]. In septic patients, the concentrations of HDL are markedly reduced and the observed changes are closely related to the severity of the infection [1]. Low serum concentrations of apo A–I are an independent predictor of mortality in human sepsis [2, 3]. In contrast to other (apo)lipoproteins, apo A–I and HDL further decrease in non-survivors during intensive care unit stay [2]. The current study shows that an intervention that increases apo A–I levels improves survival in a mouse model of LPS-induced inflammation and lethality. This is in agreement with a prior report which showed that human apo A–I transgenic mice with elevated apo A–I and twofold higher HDL levels as control mice are protected against LPS-induced mortality [5]. These effects may be the direct consequence of increased plasma apo A–I levels or may be the result of the quantitative or qualitative changes of HDL. Although LPS neutralisation by HDL or human apo A–I [7] may have contributed to the in vivo effects of human apo A–I transfer, direct effects of HDL or apo A–I can be postulated. Pre-incubation of HMEC-1 cells in vitro with HDL or apo A–I resulted in profound effects on TLR4 signalling after LPS supplementation in the absence of HDL or apo A–I, indicating LPS binding-independent effects.
Stimulation or attenuation of inflammation may be beneficial or detrimental depending on the stage of sepsis. In this respect, the current model of LPS-induced inflammation and lethality differs from models of sepsis. Notwithstanding this, intravenous injection of reconstituted HDL has previously been shown to increase survival in rabbits challenged with live E. coli [9]. LPS-induced inflammation and lethality likely corresponds to a stage of sepsis where reduced inflammation is beneficial. HDL has generally anti-inflammatory effects, e.g. by inhibiting LPS-induced CD14 expression on monocytes [5, 9, 28]. Interestingly, Berbée et al. [29] have demonstrated that plasma levels of apo C–I, an apolipoprotein partially associated with HDL, are markedly decreased in patients with severe sepsis. Apo C–I levels were higher in survivors, even after adjustment for lipid levels [29, 30]. In an experimental murine study, apo C–I dose-dependently increased the early inflammatory response to Klebsiella pneumoniae-induced pneumonia and protected mice against fatal sepsis [31].
The endothelium plays a crucial role in the pathogenesis of sepsis: Endothelial cells are the key stromal cell type involved in innate immune responses to LPS, which recruit and activate leukocytes to the inflammatory site. Recently, it has been demonstrated that endothelium-derived rather than neutrophil-derived TLR4 is the key molecule in LPS-induced neutrophil sequestration into lungs [17]. This is of importance since the rapid and selective accumulation of neutrophils into the lungs is thought to underlie the pulmonary failure that leads to sepsis-related death. Given the importance of the endothelium in the pathogenesis of sepsis and given the endothelial-protective effects of HDL, we further investigated the regulation of endothelial TLR4 after apo A–I gene transfer in the lung as well as after HDL supplementation in HMEC-1. Apo A–I transfer reduced endothelial TLR4 expression in mice before LPS administration, indicating an effect of HDL, independent of its LPS binding capacities. In agreement, supplementation of HDL or its component apo A–I on HMEC-1 cells reduced TLR4 expression under control conditions, i.e. in the absence of LPS. Like other cell types, endothelial cells respond to LPS by the activation of NF-κB [32], which in turn induces the transcription of genes encoding for cytokines and adhesion molecules [33, 34]. Recent studies have demonstrated that this effect is mediated through TLR4 [35]. The TLR4-dependent response to LPS in endothelial cells leads to induced ICAM-1 and VCAM-1 expression [35]. Under control conditions, HDL or apo A–I decreased TLR4 expression in the absence of a reduction in NF-κB activity or a decrease in MyD88 or TRIF mRNA expression. In contrast, after LPS challenge, a reduction in LPS-induced NF-κB activity was observed, which was paralleled by a decrease in MyD88 expression, with MyD88 being the specific TLR4 signalling pathway in vascular endothelial cells [36]. HDL did not reduce TRIF mRNA expression, suggesting a MyD88-specific down-regulation. These endothelial-protective effects of HDL took place in the absence of LPS sequestration, underscoring direct effects of HDL on endothelial TLR4 expression and TLR4 signalling. Reduced TLR4 signalling likely contributes to a reduction in ICAM-1 and VCAM-1 expression, leading to decreased neutrophil infiltration and subsequent less pulmonary failure in vivo.
Interestingly, despite the LPS-induced abundance of TLR4 in the lung, a decrease in endothelial TLR4 expression was observed in the lung after LPS challenge. Also in vitro, LPS reduced endothelial TLR4 expression on protein level, whereas underlying signalling, the expression of MyD88 and NF-κB activity, was induced. This is in line with Yvan-Charvet et al. [37] who recently demonstrated LPS-mediated down-regulation of TLR4 in macrophages in vivo. LPS is rapidly internalized and transported to the Golgi apparatus [38], suggesting that the down-regulation of TLR4 can be attributed to co-localisation and co-internalisation with LPS. Recently, it has been demonstrated that cholesterol accumulation in the plasma membrane of ATP-binding cassette transporter (ABC) G1−/− and ABCA1−/−ABCG1−/− macrophages, associated with abrogated cholesterol efflux to HDL, leads to increased levels and signalling of TLR4 and an increased inflammatory response after exposure to LPS [37]. Our study, illustrating HDL-mediated down-regulation of TLR4 expression and signalling, further supports that there exists a strong link between cellular cholesterol metabolism and TLR4 signalling.
In summary, our study demonstrates the down-regulation of endothelial TLR4 as a novel endothelial-protective effect of HDL, which takes place independently of the LPS binding capacities of HDL and contributes to the reduced accumulation of neutrophils in the septic lung, leading to an improved mortality rate.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 161 kb)
Acknowledgments
This study was supported by the SFB Transregio 19, Project Z3 to CT. The Center for Molecular and Vascular Biology (BDG, EVC) is supported by the Excellentiefinanciering KU Leuven (EF/05/013). Eline Van Craeyveld is a Research Assistant of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen. We thank Annika Koschel and Beatrice Testa for excellent technical support.
Disclosure
There is no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- Apo
Apolipoprotein
- C
Cholesterol
- IFN-γ
Interferon-γ
- LPS
Lipopolysaccharide
- MyD88
Myeloid differentiation factor 88
- PAMP
Pathogen-associated molecular pattern
- S1P
Sphingosine-1-phosphate
- TNF-α
Tumour necrosis factor-α
- TLR
Toll-like receptor
- TRIF
Toll/IL-1R-containing adaptor inducing interferon β
References
- 1.van Leeuwen HJ, Heezius EC, Dallinga GM, van Strijp JA, Verhoef J, van Kessel KP. Lipoprotein metabolism in patients with severe sepsis. Crit Care Med. 2003;31:1359–1366. doi: 10.1097/01.CCM.0000059724.08290.51. [DOI] [PubMed] [Google Scholar]
- 2.Barlage S, Gnewuch C, Liebisch G, Wolf Z, Audebert FX, Gluck T, Frohlich D, Kramer BK, Rothe G, Schmitz G. Changes in HDL-associated apolipoproteins relate to mortality in human sepsis and correlate to monocyte and platelet activation. Intensive Care Med. 2009;35:1877–1885. doi: 10.1007/s00134-009-1609-y. [DOI] [PubMed] [Google Scholar]
- 3.Tsai MH, Peng YS, Chen YC, Lien JM, Tian YC, Fang JT, Weng HH, Chen PC, Yang CW, Wu CS. Low serum concentration of apolipoprotein A–I is an indicator of poor prognosis in cirrhotic patients with severe sepsis. J Hepatol. 2009;50:906–915. doi: 10.1016/j.jhep.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 4.Pajkrt D, Doran JE, Koster F, Lerch PG, Arnet B, van der Poll T, ten Cate JW, van Deventer SJ. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J Exp Med. 1996;184:1601–1608. doi: 10.1084/jem.184.5.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine DM, Parker TS, Donnelly TM, Walsh A, Rubin AL. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci USA. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calabresi L, Franceschini G, Sirtori CR, De Palma A, Saresella M, Ferrante P, Taramelli D. Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochem Biophys Res Commun. 1997;238:61–65. doi: 10.1006/bbrc.1997.7236. [DOI] [PubMed] [Google Scholar]
- 7.Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra V, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005;97:236–243. doi: 10.1161/01.RES.0000176530.66400.48. [DOI] [PubMed] [Google Scholar]
- 8.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 9.Hubsch AP, Casas AT, Doran JE. Protective effects of reconstituted high-density lipoprotein in rabbit Gram-negative bacteremia models. J Lab Clin Med. 1995;126:548–558. [PubMed] [Google Scholar]
- 10.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/S0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- 12.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 13.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elson G, Dunn-Siegrist I, Daubeuf B, Pugin J. Contribution of Toll-like receptors to the innate immune response to Gram-negative and Gram-positive bacteria. Blood. 2007;109:1574–1583. doi: 10.1182/blood-2006-06-032961. [DOI] [PubMed] [Google Scholar]
- 15.Riad A, Bien S, Gratz M, Escher F, Westermann D, Heimesaat MM, Bereswill S, Krieg T, Felix SB, Schultheiss HP, Kroemer HK, Tschope C. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur J Heart Fail. 2008;10:233–243. doi: 10.1016/j.ejheart.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, Pauschinger M, Schultheiss HP, Tschope C. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- 17.Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M, Escher F, Filenberg E, Demir O, Li J, Shakibaei M, Schimke I, Staudt A, Felix SB, Schultheiss HP, De Geest B, Tschope C. Human apolipoprotein A–I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation. 2008;117:1563–1573. doi: 10.1161/CIRCULATIONAHA.107.710830. [DOI] [PubMed] [Google Scholar]
- 19.Van Linthout S, Spillmann F, Lorenz M, Meloni M, Jacobs F, Egorova M, Stangl V, De Geest B, Schultheiss HP, Tschope C. Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension. 2009;53:682–687. doi: 10.1161/HYPERTENSIONAHA.108.118919. [DOI] [PubMed] [Google Scholar]
- 20.Van Linthout S, Spillmann F, Schultheiss HP, Tschope C. High-density lipoprotein at the interface of type 2 diabetes mellitus and cardiovascular disorders. Curr Pharm Des. 2010;16:1504–1516. doi: 10.2174/138161210791051031. [DOI] [PubMed] [Google Scholar]
- 21.Van Linthout S, Foryst-Ludwig A, Spillmann F, Peng J, Feng Y, Meloni M, Van Craeyveld E, Kintscher U, Schultheiss HP, De Geest B, Tschope C. Impact of HDL on adipose tissue metabolism and adiponectin expression. Atherosclerosis. 2010;210:438–444. doi: 10.1016/j.atherosclerosis.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Spillmann F, Schultheiss HP, Tschope C, Van Linthout S. High-density lipoprotein-raising strategies: update 2010. Curr Pharm Des. 2010;16:1517–1530. doi: 10.2174/138161210791050988. [DOI] [PubMed] [Google Scholar]
- 23.Van Linthout S, Lusky M, Collen D, De Geest B. Persistent hepatic expression of human apo A–I after transfer with a helper-virus independent adenoviral vector. Gene Ther. 2002;9:1520–1528. doi: 10.1038/sj.gt.3301824. [DOI] [PubMed] [Google Scholar]
- 24.Belalcazar LM, Merched A, Carr B, Oka K, Chen KH, Pastore L, Beaudet A, Chan L. Long-term stable expression of human apolipoprotein A–I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 25.Feng Y, Jacobs F, Van Craeyveld E, Brunaud C, Snoeys J, Tjwa M, Van Linthout S, De Geest B. Human ApoA-I transfer attenuates transplant arteriosclerosis via enhanced incorporation of bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:278–283. doi: 10.1161/ATVBAHA.107.158741. [DOI] [PubMed] [Google Scholar]
- 26.Feng Y, Van Craeyveld E, Jacobs F, Lievens J, Snoeys J, De Geest B. Wild-type apo A–I and apo A–I(Milano) gene transfer reduce native and transplant arteriosclerosis to a similar extent. J Mol Med. 2009;87:287–297. doi: 10.1007/s00109-008-0427-y. [DOI] [PubMed] [Google Scholar]
- 27.Bode C, Sensken SC, Peest U, Beutel G, Thol F, Levkau B, Li Z, Bittman R, Huang T, Tolle M, van der Giet M, Graler MH. Erythrocytes serve as a reservoir for cellular and extracellular sphingosine 1-phosphate. J Cell Biochem. 2010;109:1232–1243. doi: 10.1002/jcb.22507. [DOI] [PubMed] [Google Scholar]
- 28.McDonald MC, Dhadly P, Cockerill GW, Cuzzocrea S, Mota-Filipe H, Hinds CJ, Miller NE, Thiemermann C. Reconstituted high-density lipoprotein attenuates organ injury and adhesion molecule expression in a rodent model of endotoxic shock. Shock. 2003;20:551–557. doi: 10.1097/01.shk.0000097249.97298.a3. [DOI] [PubMed] [Google Scholar]
- 29.Berbee JF, van der Hoogt CC, de Haas CJ, van Kessel KP, Dallinga-Thie GM, Romijn JA, Havekes LM, van Leeuwen HJ, Rensen PC. Plasma apolipoprotein CI correlates with increased survival in patients with severe sepsis. Intensive Care Med. 2008;34:907–911. doi: 10.1007/s00134-008-1006-y. [DOI] [PubMed] [Google Scholar]
- 30.Berbee JF, Mooijaart SP, de Craen AJ, Havekes LM, van Heemst D, Rensen PC, Westendorp RG. Plasma apolipoprotein CI protects against mortality from infection in old age. J Gerontol A Biol Sci Med Sci. 2008;63:122–126. doi: 10.1093/gerona/63.2.122. [DOI] [PubMed] [Google Scholar]
- 31.Berbee JF, van der Hoogt CC, Kleemann R, Schippers EF, Kitchens RL, van Dissel JT, Bakker-Woudenberg IA, Havekes LM, Rensen PC. Apolipoprotein CI stimulates the response to lipopolysaccharide and reduces mortality in Gram-negative sepsis. FASEB J. 2006;20:2162–2164. doi: 10.1096/fj.05-5639fje. [DOI] [PubMed] [Google Scholar]
- 32.Wrighton CJ, Hofer-Warbinek R, Moll T, Eytner R, Bach FH, de Martin R. Inhibition of endothelial cell activation by adenovirus-mediated expression of I kappa B alpha, an inhibitor of the transcription factor NF-kappa B. J Exp Med. 1996;183:1013–1022. doi: 10.1084/jem.183.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 34.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/S0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 35.Zeuke S, Ulmer AJ, Kusumoto S, Katus HA, Heine H. TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc Res. 2002;56:126–134. doi: 10.1016/S0008-6363(02)00512-6. [DOI] [PubMed] [Google Scholar]
- 36.Harari OA, Alcaide P, Ahl D, Luscinskas FW, Liao JK. Absence of TRAM restricts Toll-like receptor 4 signaling in vascular endothelial cells to the MyD88 pathway. Circ Res. 2006;98:1134–1140. doi: 10.1161/01.RES.0000220105.85182.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via Toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thieblemont N, Wright SD. Transport of bacterial lipopolysaccharide to the Golgi apparatus. J Exp Med. 1999;190:523–534. doi: 10.1084/jem.190.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobs F, Van Craeyveld E, Feng Y, Snoeys J, De Geest B. Adenoviral low density lipoprotein receptor attenuates progression of atherosclerosis and decreases tissue cholesterol levels in a murine model of familial hypercholesterolemia. Atherosclerosis. 2008;201:289–297. doi: 10.1016/j.atherosclerosis.2008.02.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 161 kb)