

Abstract
Familial transthyretin (TTR) amyloidosis is caused by a mutation in the TTR gene, although wild-type (wt) TTR is also incorporated into the amyloid fibrils. Liver transplantation (LT) is the prevailing treatment of the disease and is performed in order to eliminate the mutant TTR from plasma. The outcome of the procedure is varied; especially problematic is a progressive cardiomyopathy seen in some patients, presumably caused by continued incorporation of wtTTR. What determines the discrepancy in outcome is not clear. We have previously shown that two structurally distinct amyloid fibrils (with or without fragmented ATTR) are found among ATTRV30M patients. In this study, we investigated the proportion of wtATTR in cardiac and adipose amyloid from patients having either fibril type. It was found that cardiac amyloid more easily incorporates wtTTR than adipose amyloid, offering a potential explanation for the vulnerability of cardiac tissue for continued amyloidosis after LT. In cardiac tissue, fibrils with fragmented ATTR contained a higher wt proportion than fibrils without, suggesting that continued incorporation of wtTTR after LT, perhaps, can take place more easily in these patients. In adipose tissue, a rapid increase in wt proportion after LT indicates that a rather fast turnover of the deposits must occur. A difference in wt proportion between the fibril types was seen post-LT but not pre-LT, possibly caused by differences in turnover rate. Conclusively, this study further establishes the basic dissimilarities between the two fibril types and demonstrates that their role in LT outcome needs to be further investigated.
Keywords: Amyloid, Transthyretin, Familial amyloidotic polyneuropathy, Liver transplantation, Wild-type
Introduction
Amyloidoses are a group of diseases characterized by aggregation of proteins into a stable fibrillar structure that accumulates in tissues. Familial transthyretin (TTR) amyloidosis is a lethal systemic disease caused by a mutation in the gene coding for the plasma transporter TTR. Around 100 amyloidogenic mutations have been found to date [1]. The mutation renders the protein more prone to form amyloid, but the wild-type (wt) protein is also amyloidogenic and is incorporated into the fibrils of heterozygotes [2–4]. Moreover, in senile systemic amyloidosis, wtTTR is the only fibrillar protein [5]. The main features of the familial forms are polyneuropathy and/or cardiomyopathy, but the clinical picture is highly varied between mutations and also between patients with the same mutation [6, 7].
The reason(s) behind the clinicopathological differences between patients is/are far from understood, not only in ATTR amyloidosis (nomenclature according to [8]), but in all systemic variants of amyloid disease. In a previous study on Swedish ATTRV30M patients, we found that in some of the individuals, the amyloid fibrils contain a mixture of full-length and fragmented ATTR (fibril type A) while in other patients the fibrils are composed of only full-length ATTR (fibril type B)[9]. The vast majority of the fragmented ATTR species found in fibril type A has been found to be N-terminally truncated and starts at positions around amino acid residue 50 while only very small amounts of C-terminally truncated fragments are found [9–11]. The fibril type seems to be consistent between different organs within an individual [12]. We further discovered that the fibril type was correlated with the clinical phenotype of the patients; individuals with fragmented ATTR present in the fibrils had a late onset of disease and an enlarged heart due to heavy amyloid deposition while patients having fibrils without fragments had an early disease onset and much smaller amyloid amounts in cardiac tissue [12].
Liver transplantation (LT) is performed on ATTR amyloidosis patients, as this removes the main production site of the mutant protein [13, 14]. In many cases, the progress of amyloid deposition and neurological symptoms seems to halt or slow down after transplantation, and some patients also show an improvement; however, for other patients the symptoms have worsened [14, 15]. Especially problematic is the fact that in some transplanted individuals, a rapid continued deposition is taking place in cardiac tissue, presumably caused by the addition of wtTTR to the amyloid [16, 17]. Mostly, this has been seen in patients with other mutations than V30M [18, 19], but progression of cardiac amyloidosis has been reported to occur also in some V30M patients [20–23]. The special vulnerability of heart tissue for continued deposition after transplantation and the reason why some patients benefit from the procedure, whereas amyloid progression is triggered in others, are not understood. In order to be able to select patients that are suitable for the operation, it is highly important to define the factor(s) determining the outcome of the transplantation.
We hypothesize that amyloid in cardiac tissue more easily incorporates wtTTR than amyloid in other organs and that the cardiac deposits therefore are more prone to continue growing after liver transplantation. We also theorize that the two fibril composition types found in ATTRV30M patients (presence of fragmented ATTR or not) differ in their propensity to incorporate wtTTR molecules and that this is a factor that could affect the outcome of transplantation. In this study, we therefore investigated the proportion of wild-type ATTR in cardiac and adipose amyloid from ATTRV30M patients, having fibrils of type A or B.
Material and methods
Tissue material and patients
All patients in the study have been diagnosed with familial ATTR amyloidosis and were heterozygous for the mutation ATTRV30M. The patients originated from the same geographical area in northern Sweden. Duration of disease was estimated anamnestically.
Heart tissue was obtained from 12 patients at autopsy, of whom one had undergone liver transplantation. Abdominal subcutaneous adipose tissue biopsy was taken from 32 patients, of whom 20 were liver transplanted. These 32 patients were not the same individuals from whom the heart tissue was obtained, except for two cases. Patient data are displayed in Tables 1 and 2. The study was approved by the ethical committees of Umeå University Hospital and Uppsala University Hospital and was performed in accordance with the 1964 Declaration of Helsinki. Informed consent was obtained from all subjects.
Table 1.
Clinical data of patients and proportion of wt to total ATTR in cardiac tissue
| Patient no. | Sex | Age at onset of symptomatic disease (years) | Duration of symptomatic disease (years) | Duration after LT (years) | wtATTR proportion [mean (range), %] | |||
|---|---|---|---|---|---|---|---|---|
| Homogenized | Paraffin-embedded | |||||||
| Intact ATTR | Fragmented ATTR | Intact ATTR | Fragmented ATTR | |||||
| Fibril type A (fragmented ATTR present) | ||||||||
| Pre-LT | ||||||||
| 1 | M | 69 | 5.5 | – | 73 (68–77) | 78 (74–82) | 74 (65–80) | 79 (77–81) |
| 2 | M | NK | NK | – | 69 (65–75) | 77 (77–78) | NA | NA |
| 3 | F | NK | NK | – | 72 (69–74) | 76 (73–78) | 72 (68–76) | 77 (68–85) |
| 4 | M | NK | 13 | – | 73 (71–75) | 79 | 70 | 81 |
| 5 | M | 62 | 13 | – | 75 (74–76) | 85 (82–88) | 75 (69–84) | 81 (76–89) |
| 6 | M | NK | NK | – | 74 (71–79) | 79 (73–84) | 74 (71–77) | 79 (78–79) |
| Mean ± SD | 65 ± 3.5 | 10 ± 3.5 | – | 73 ± 2.1 | 79 ± 3.5 | 73 ± 2.0 | 79 ± 1.7 | |
| Post-LT | ||||||||
| 7 | M | 65 | 10 | 7 | 96 (95–97) | 97 (96–98) | NA | NA |
| Fibril type B (fragmented ATTR not present) | ||||||||
| Pre-LT | ||||||||
| 8 | F | 52 | 10 | – | 43 (40–45) | – | NA | – |
| 9 | F | 46 | 11 | – | 52 (51–54) | – | 53 (50–57) | – |
| 10 | F | NK | NK | – | 51 (49–52) | – | 59 (54–63) | – |
| 11 | NK | NK | NK | – | 57 (54–60) | – | NA | – |
| 12 | M | NK | NK | – | 57 (55–59) | – | 52 | – |
| Mean ± SD | 49 ± 4.2 | 10.5 ± 0.7 | – | 52 ± 5.7 | – | 55 ± 3.8 | – | |
NK not known, NA not available
Table 2.
Clinical data of patients and proportion of wt to total ATTR in adipose tissue
| Patient no. | Sex | Age at onset of symptomatic disease (years) | Duration of symptomatic disease (years) | Duration after LT (years) | wtATTR proportion [mean (range), %] | |||
|---|---|---|---|---|---|---|---|---|
| Not guanidine-treated | Guanidine-treated | |||||||
| Intact ATTR | Fragmented ATTR | Intact ATTR | Fragmented ATTR | |||||
| Fibril type A (fragmented ATTR present) | ||||||||
| Pre-LT | ||||||||
| 1 | M | 69 | 5.6 | − | 48 (42–52) | 67 (58–74) | 60 (59–61) | 63 (62–64) |
| 13 | M | 67 | 3.9 | − | 28 (27–29) | 39 (38–39) | NA | NA |
| 14 | M | 65 | 6.4 | − | 35 (31–39) | 50 (48–51) | NA | NA |
| 15 | F | 80 | 4.1 | − | 34 | 59 | 55 (54–56) | 64 (55–73) |
| 16 | M | 63 | 0.9 | − | 14 | 20 | NA | NA |
| 17 | M | 68 | 4.7 | − | 18 | Signal too weak | NA | NA |
| 18 | M | 49 | 9.5 | − | 36 (30–41) | 32 (26–37) | NA | NA |
| 19 | M | 68 | 2.1 | − | 55 | 58 | 52 | 52 |
| 20 | M | 67 | 3.2 | − | 79 (73–85) | 79 (70–87) | 80 (75–85) | 85 (83–87) |
| 21 | M | 59 | 3.4 | − | 18 | 24 | 17 (14–21) | 22 (18–25) |
| Mean ± SD | 66 ± 7.9 | 4.4 ± 2.4 | − | 37 ± 20 | 48 ± 20 | 53 ± 23 | 57 ± 23 | |
| Post-LT | ||||||||
| 7 | M | 65 | 7.9 | 4.9 | 99 (98–100) | 99 (99–99) | 100 (99–100) | 96 (95–96) |
| 18 | M | 49 | 12.5 | 2.4 | 97 (96–98) | 85 | 95 (94–96) | 83 (83–83) |
| 21 | M | 59 | 5.4 | 0.6 | 77 | 72 | NA | NA |
| 22 | M | 63 | 4.6 | 3.1 | 98 (97–98) | 94 (90–97) | 99 (98–99) | 92 (91–94) |
| 23 | M | 53 | 6.5 | 4.0 | 99 (99–99) | 98 (98–98) | NA | NA |
| 24 | F | 50 | 14.4 | 5.1 | 100 (99–100) | 96 (96–96) | 98 (98–98)a | 95 (92–97) |
| Mean ± SD | 57 ± 6.8 | 8.6 ± 4.0 | 3.4 ± 1.7 | 95 ± 8.9 | 91 ± 10 | 98 ± 2.2 | 92 ± 5.9 | |
| Fibril type B (fragmented ATTR not present) | ||||||||
| Pre-LT | ||||||||
| 25 | M | 30 | 3.3 | – | 47 | – | NA | – |
| 26 | M | 63 | 1.6 | – | 28 (26–30) | – | NA | – |
| 27 | F | 45 | 4.2 | – | 35 (31–38) | – | 31 (20–36) | – |
| 28 | F | 40 | 1.6 | – | 36 | – | NA | – |
| 29 | M | 36 | 1.7 | – | 24 | – | 31 (31–31) | – |
| 30 | M | 30 | 1.1 | – | 24 | – | 34 (29–37) | – |
| 31 | M | 52 | 2.4 | – | 42 | – | 43 (40–45) | – |
| 32 | F | 62 | 2.4 | – | 40 (34–48) | – | NA | – |
| 33 | F | 60 | 4.9 | – | 37 | – | NA | – |
| Mean ± SD | 46 ± 13 | 2.6 ± 1.3 | – | 35 ± 8.0 | – | 35 ± 5.7 | ||
| Post-LT | ||||||||
| 26 | M | 63 | 5.4 | 3.4 | 76 (69–82) | – | NA | – |
| 27 | F | 45 | 6.8 | 2.0 | 81 (79–83) | – | 74 (68–78) | – |
| 28 | F | 40 | 5.4 | 3.1 | 73 (71–74) | – | 70 (66–73) | – |
| 29 | M | 36 | 5.2 | 3.3 | 71 (66–75) | – | 65 | – |
| 31 | M | 52 | 5.9 | 2.7 | 77 (74–80) | – | 84 | − |
| 34 | F | 31 | 15.3 | 6.3 | 63 (56–70) | – | 46 (43–51) | – |
| 35 | F | 60 | 9.6 | 6.7 | 97 (95–99) | – | 76 (73–78) | – |
| 36 | M | 60 | 5.6 | 3.1 | 81 (72–89) | – | NA | – |
| 37 | M | 37 | 6.1 | 2.2 | 66 (62–69) | – | NA | – |
| 38 | M | 26 | 12.6 | 10.6 | 81 (73–88) | – | 64 (61–68) | – |
| 39 | M | 48 | 17.6 | 13.6 | 99 (98–99) | – | NA | – |
| 40 | M | 66 | 6.9 | 4.5 | 78 (75–80) | – | NA | – |
| 41 | F | 43 | 9.5 | 6.9 | 95 (94–95) | – | 89 (89–89) | – |
| 42 | F | 60 | 10.6 | 6.1 | 95 | – | 79 (72–83) | – |
| Mean ± SD | 48 ± 13 | 8.8 ± 4.0 | 5.3 ± 3.4 | 81 ± 12 | – | 72 ± 13 | – | |
NA not available
aThe material treated with guanidine was obtained from a biopsy taken 6 months earlier than the biopsy from where the not guanidine-treated tissue was obtained.
Tissue preparation
Heart tissue was either stored at −20°C or fixed in formalin and embedded in paraffin. The amyloid was extracted from the frozen tissue as described [9]. In short, the tissue was repeatedly homogenized in 0.15 M NaCl and 0.05 M sodium citrate and centrifuged before washing in distilled water several times. The resulting pellet was lyophilized, defatted in acetone, and left to dry. The samples were dissolved in a solution of 6 M guanidine–HCl, 0.1 M Tris–HCl, pH 8.0, and 0.1 M dithiothreitol (DTT) for 4 days, followed by centrifugation. The supernatant was dialyzed against saturated ammonium sulfate followed by distilled water and the sample was thereafter lyophilized. To extract amyloid from paraffin-embedded tissue, 10-μm sections were deparaffinized, scraped off the slides, and pooled [24]. The material was dissolved in guanidine–HCl with DTT, dialyzed, and lyophilized as described for the frozen tissue.
Subcutaneous adipose tissue obtained through biopsy was cut into smaller pieces, rinsed in 0.15 M NaCl with 0.02% sodium azide several times, and incubated in a 0.88% ammonium chloride solution for lysis of erythrocytes. The material was thereafter washed in distilled water and defatted in acetone for several changes and allowed to dry [25]. Where enough biopsy tissue was available, some of the dry material was dissolved in guanidine–HCl with DTT, dialyzed, and lyophilized as described above.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blot analysis
The tissue materials, either heart or adipose tissue, were dissolved in a sample buffer containing 4% sodium dodecyl sulfate (SDS), 0.02 M of DTT, and 0.06 M of iodoacetamide and separated in a 16% gel using a Tris–tricine system [26]. The protein bands were visualized by staining with Coomassie Brilliant Blue.
For Western blot, the samples were transferred to a Hybond-ECL nitrocellulose membrane (GE Healthcare, Uppsala, Sweden). Polyclonal antisera produced in rabbit against TTR50–127 and TTR24–35 were used as primary antibodies (characterized in [27] and [28]). Swine anti-rabbit antibodies conjugated with horseradish peroxidase (Dako, Glostrup, Denmark) were used as secondary antibody. The reaction was visualized with an enhanced chemiluminescence system (GE Healthcare).
Analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
Bands shown by Western blot to correspond to full-length monomeric ATTR, at approximately 15 kDa, and fragmented ATTR, at approximately 5–10 kDa, were cut out from the Coomassie Brilliant Blue-stained gels. Trypsin was used to digest the proteins into peptides as described [29], and these were analyzed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Ultraflex III TOF/TOF; Bruker Daltonics, Bremen, Germany). The spectrum peaks corresponding to the tryptic peptides consisting of amino acid residues 22–34 were investigated. Other tryptic peptides containing amino acid residue 30, which would have resulted from incomplete cleavage, were searched for but were not seen. The height of the peaks was taken as a measure of relative quantity. As the methionine in the mutant variant may become oxidized, the height of the peaks corresponding to the mutant peptide in non-oxidized (MH+ = 1,398.732) and mono-oxidized (sulfoxide) state (MH+ = 1,414.726) was combined to achieve a quantitative value of the mutant peptide. The peak corresponding to the di-oxidized (sulfone) state (MH+ = 1,430.722) was either absent or negligible in size. The wild-type peptide contains no methionine and gives rise to only one peak (MH+ = 1,366.759). The proportion of wt to total ATTR was calculated.
For samples where enough material was present, gel electrophoresis and mass spectrometry analysis was repeated, up to three times if possible, and a mean value was calculated.
Statistical analysis
Data are reported as mean ± SD. Statistical analyses were performed using non-parametrical methods. The Mann–Whitney test was used to compare patient groups or tissue types. The Wilcoxon matched pairs test was used when comparing results obtained by different methods for the same patients [guanidine–HCl-treated versus guanidine–HCl-untreated adipose tissue as well as frozen and homogenized versus formalin-fixed and paraffin-embedded (FFPE) heart tissue]. Spearman’s correlation was used to calculate correlations. P ≤ 0.05 was considered significant. All tests were performed with the use of the GraphPad Prism 5 software.
Results
Heart tissue
The fibril composition type was determined for each sample by Western blot using an antiserum against TTR50–127 (see Fig. 1). The proportion of wt to total ATTR was determined by MALDI-TOF mass spectrometry (Figs. 2 and 3). The results from the protein bands on SDS-polyacrylamide gels corresponding to monomeric full-length ATTR are described here while the results of the fragmented ATTR species are presented separately below.
Fig. 1.
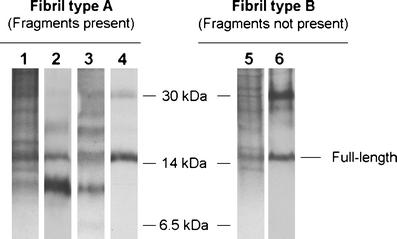
The two fibril composition types, shown by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blot analyses on adipose tissue. Lanes 1–4 show a fibril composition of both full-length and fragmented ATTR (fibril type A) while lanes 5–6 show a fibril composition of only full-length ATTR (fibril type B). Lanes 1 and 5 = Coomassie Blue-stained SDS-PAGE gels; lanes 2 and 6 = Western blot using an in-house produced antiserum against TTR50–127 that detects C-terminal fragments; lane 3 = Western blot using an in-house produced antiserum against TTR24–35 that detects fragments containing position 30; lane 4 = Western blot using commercial α-TTR antibodies (Dako), which do not recognize the fragmented ATTR species [12]
Fig. 2.
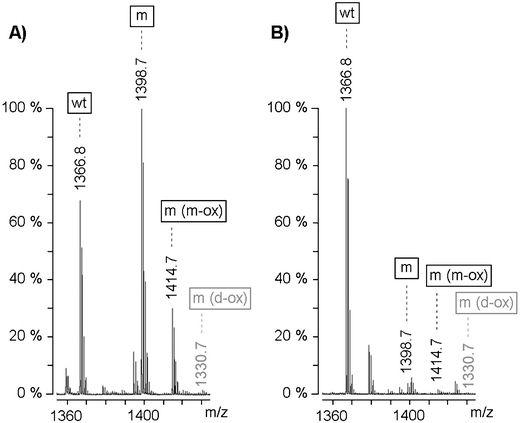
Typical examples of mass spectra obtained from MALDI-TOF mass spectrometry analysis of adipose tissue amyloid fibrils from a non-transplanted (a) and transplanted (b) patient. The tryptic peptides ATTR22–34 containing valine (wt) or methionine in non-oxidized (m), mono-oxidized (m (m-ox)), or di-oxidized (m (d-ox)) state in position 30 are marked. The height of the peaks was used as a relative measure of quantity. Peaks corresponding to non-oxidized and mono-oxidized mutant peptides were combined to obtain the quantity of mutant protein. The peak corresponding to the di-oxidized peptide was either absent or negligible in size. The proportion of wt to total ATTR was determined to be 34% in A and 95% in B
Fig. 3.
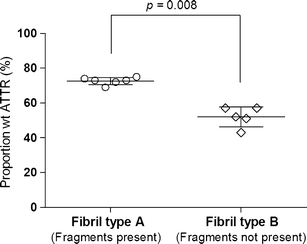
Proportion of wild-type molecules among full-length ATTR species in cardiac amyloid fibrils, grouped according to fibril composition type. Each measure point represents one individual. The horizontal lines represent the mean value ± SD for each group. Fibrils composed of a mixture of full-length and fragmented ATTR consisted of a higher proportion wild-type ATTR than fibrils composed of only full-length ATTR. The difference between the groups is highly significant, p = 0.008
Frozen heart tissue obtained at autopsy from 12 Swedish patients with the V30M mutation, who had not undergone liver transplantation, was homogenized and analyzed (see Table 1 for details). Individuals with fibril type A had a clearly higher proportion of wild-type molecules than patients with type B (73 ± 2.1% to 52 ± 5.7%, p = 0.008). Unfortunately, we only had access to cardiac tissue from one transplanted patient. This patient had a fibril composition of type A and the wt proportion was determined to be 96%, which is noticeably higher than in any of the non-transplanted patients.
To confirm that the wtATTR proportions obtained was not affected by the storage or extraction method used, FFPE tissue was also analyzed for patients where such material was available (see Table 1). No significant difference in wild-type proportion was seen for the FFPE tissue compared to the frozen and homogenized tissue, regardless if the fibril type groups were analyzed separately (p = 1.0 for type A and p = 0.75 for type B) or combined (p = 1.0).
Adipose tissue
Subcutaneous fat biopsies were obtained from 32 patients (see Table 2 for details). For seven individuals, biopsies were available from both before and after transplantation. For the other patients, the tissue material was either from pre or posttransplantation.
Comparison of wild-type ATTR proportion between fibril types
The wild-type proportion of the monomeric full-length ATTR molecules was determined as described for the cardiac amyloid. The results of the fragmented species are presented separately below. In both fibril composition groups, the proportion of wtATTR was significantly higher among the LT patients than the non-LT patients (95 ± 8.9% compared to 37 ± 20%, p = 0.002 for type A and 81 ± 12% compared to 35 ± 8.0%, p = 0.0001 for type B) (Fig. 4). For the seven individuals where biopsies were available both pre and posttransplantation, a large increase in wt proportion was seen after LT in each case. Among non-LT patients, there was no difference in mean value of wt proportion between the two fibril types (37 ± 20% compared to 35 ± 8.0%, p = 0.74), although the values were more spread among those with type A; however, among the LT patients, individuals with type A had a higher wt proportion than patients with type B (95 ± 8.9% compared to 81 ± 12%, p = 0.02). In fact, fibrils of type A showed an almost complete dominance of wtATTR (in all cases but one patient, who had been transplanted very recently) while the patients lacking fragments had a more varied distribution of wt proportion.
Fig. 4.
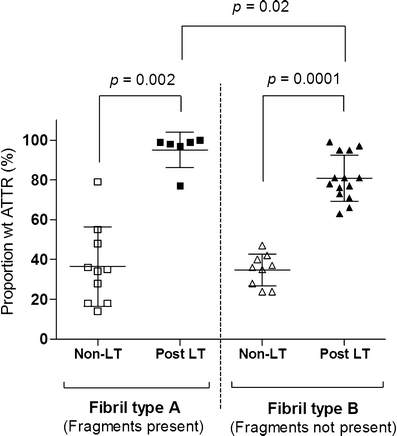
Proportion of wild-type molecules among full-length ATTR species in adipose amyloid fibrils. The samples are grouped according to fibril composition type and whether the patients have undergone liver transplantation or not. Each measure point represents one individual. The horizontal lines represent the mean value ± SD for each group. Fibrils from transplanted patients contained a higher proportion of wild-type ATTR than samples from non-transplanted patients regardless of fibril composition (p = 0.002 and 0.0001). A significant difference between the two fibril types was only seen among transplanted patients (p = 0.02)
Because of the limited size of many of the adipose biopsies, the tissue was not dissolved in any amyloid breaking agent besides SDS. To exclude the possibility that only parts of the amyloid was dissolved and the results therefore unrepresentative of the amyloid mass as a whole, a portion of the tissue was dissolved in guanidine–HCl, for patients where enough material was available (see Table 2). There was no statistically significant difference between the results obtained with or without guanidine–HCl treatment when all patient groups were combined (p = 0.31). Neither was a difference seen when each patient group was compared separately, except for the group of transplanted patients with fibril type B (p = 0.50 for pre-LT type A, p = 0.38 for pre-LT type B, p = 0.58 for post-LT type A and p = 0.04 for post-LT type B). In this group, the values of the guanidine–HCl-treated samples were slightly lower than those not treated with guanidine–HCl (mean difference = −10% units).
Wild-type ATTR proportion in relation to other factors
As it seems likely that the time elapsed between transplantation and time of biopsy can affect the proportion of wtATTR, it was investigated if patients with fibril type A in general had a longer duration after transplantation than patients with type B. Such association was not seen (3.4 ± 1.7 years after LT among type A patients compared to 5.3 ± 3.4 years among type B patients, p = 0.30). When patients with a similar duration but different fibril type are compared (Fig. 5), it is even clearer that duration after LT is not the reason for the higher wt proportion among fibril type A patients. A possible correlation between wt proportion and duration after LT is seen within each fibril type group (r = 0.99, p = 0.03 for type A and r = 0.52, p = 0.05 for type B), but not when both groups are combined (r = 0.36, p = 0.12).
Fig. 5.
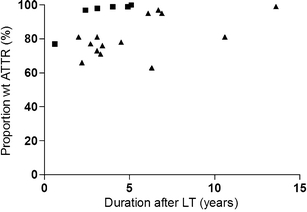
Correlation between time elapsed after liver transplantation and proportion of wild-type molecules among full-length ATTR species in adipose amyloid fibrils. Each measure point represents one individual. Black square, fibrils with fragmented ATTR (type A); black triangle, fibrils without fragmented ATTR (type B). The figure shows that the higher percentage of wild-type ATTR found among fragment-containing fibrils is not due to longer duration after transplantation among these patients compared to the patients lacking fragments. A possible correlation between wt proportion and duration after LT is seen within each fibril type group (r = 0.99, p = 0.003 for type A and r = 0.52, p = 0.05 for type B), but not when both groups are combined (r = 0.36, p = 0.1)
Other factors, such as age, sex, and duration before transplantation were also investigated for potential correlations to wild-type proportion, but none was found (data not shown).
Comparison of adipose and cardiac amyloid
Among non-LT patients, heart amyloid contained a higher proportion of wtATTR than adipose amyloid (73 ± 2.1% compared to 37 ± 20%, p = 0.01 among fibril type A and 52 ± 5.7% compared to 35 ± 8.0%, p = 0.005 among type B).
Proportion of wild-type ATTR among fragmented species
The wild-type proportion among the truncated ATTR molecules was investigated for the patients where such species were present (fibril type A). Unfortunately, the vast majority of the fragmented species starts at positions around amino acid residue 50 while the fragments containing position 30 seem to be present in the fibrils in only very small amounts [9, 10]. Western blot with an antiserum recognizing TTR24–35 was used to find fragments containing amino acid residue 30. Weak reactions could be seen at sizes of around 10 and 5 kDa (see Fig. 1). The whole area of approximately 5–10 kDa on the SDS gels was cut out and analyzed with mass spectrometry.
Overall, there was a rather good agreement between the wild-type proportion among the full-length and the fragmented ATTR molecules in both cardiac and adipose amyloid (r = 0.94, p = 0.0001, Fig. 6). A tendency for a slightly higher wild-type proportion among the fragmented than among the full-length ATTR can be noted in samples from non-transplanted patients, whereas the opposite seems to be true for specimens from transplanted patients.
Fig. 6.
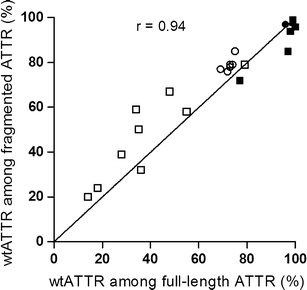
Correlation between wild-type proportion among full-length and fragmented ATTR species. Each measure point represents one individual. White square, adipose tissue from non-transplanted patients; black square, adipose tissue from transplanted patients; white circle, cardiac tissue from non-transplanted patients; black circle, cardiac tissue from transplanted patient (r = 0.94, p = 0.0001)
Discussion
It has been proven difficult to halt amyloid progression by liver transplantation, as the wtTTR also is amyloidogenic [5] and apparently continues to be incorporated into the fibrils even when virtually all variant TTR has been eliminated from the plasma. Such development was first discovered in heart tissue [2, 16, 17], as some patients showed a troublesome increase in cardiac amyloid load after the procedure [18]. Lately, an increase in wtATTR proportion after liver transplantation has been found also in amyloid from kidney [16], nerve [4], and adipose tissue [3] but, at least for the adipose tissue, the higher wild-type proportion was said not to be accompanied by a progression of the deposits. In the present study, we also saw an increase in wtATTR proportion after transplantation in adipose amyloid. These observations show that continued incorporation of wtTTR is not restricted to cardiac tissue; however, why the heart is especially prone to develop a progressive disease and why this is not a problem in all patients is far from understood.
Cardiac versus adipose amyloid
It is not possible to discern from earlier studies if there is a variation in wild-type proportion in amyloid from different tissue types, as these studies are rare and most often are based on only one type of tissue, a single individual, different mutations, or various purifying and analyzing techniques. The reported values are also quite diverse; 30–65% for cardiac amyloid [2, 11, 17, 30, 31], 0–35% for kidney [16, 32], 35–40% for nerve [4], and 30–50% for subcutaneous fat tissue [3].
In an attempt to learn if cardiac tissue diverges from other organs regarding the propensity to incorporate wtTTR into the amyloid fibrils, we compared the proportion wtATTR in cardiac to adipose amyloid in non-transplanted patients. The result clearly showed a higher wtATTR percentage in heart than adipose tissue, thus suggesting that cardiac amyloid is especially prone to incorporate wtTTR molecules. This could be a reason for why the heart is especially vulnerable to progression of amyloid deposits after transplantation, when only wt molecules are available.
Impact of fibril type
We have earlier reported that two compositionally and morphologically distinct fibrils are seen in patients with ATTRV30M amyloidosis [9, 12]. Interestingly, a difference in wtATTR proportion was found between the two types when heart tissue from non-transplanted patients was examined, as the fraction of wtATTR consistently was higher in fibril type A. In fact, the wild-type molecules even dominated in amount over the mutant protein in this type of fibrils, suggesting that the former molecules actually are more easily incorporated than the latter. This renders into speculations that the rate of fibril incorporation actually might be accelerated after LT, as the supply of the most easily added species thereby is increased. Even if this is not the case, wt molecules are still more easily incorporated into fibrils of type A than type B. It, therefore, seems possible that patients of type A are more susceptible than those of type B for progressive amyloid deposition in cardiac tissue after LT.
A higher content of wtATTR among patients having fibrils with fragmented ATTR was probably also found by Koike et al. [31]. They reported that fibrils with a weak and non-glittering kind of birefringence after Congo red staining had a higher wild-type percentage than fibrils with a strong and glittering birefringence, and it has been shown before that there is a strong relationship between a fibril composition with fragmented ATTR and a non-glittering appearance of the fibrils [9].
It should be noted that the relative amount of wtATTR found in the cardiac amyloid of the present study and especially the values obtained for the fragment-containing fibrils are unusually high compared to previous studies by others on ATTRV30M [2, 17] as well as other mutations [2, 11, 16, 33]. On the other hand, two of the samples used in the present study have been analyzed earlier by Edman degradation, and this technique also gave a similarly high wild-type content [30].
Amyloid turnover
Another issue raised by the results of the present study is the occurrence of turnover of the amyloid deposits. The fact that the proportion of wtATTR in transplanted patients was very high even after a fairly short time after LT, suggests that a quite rapid turnover of the deposits occurs. The amyloid amount was not assessed in this study, but as the increase in amyloid load would have to be immense to account for the massive increase in wild-type percentage, it is not likely to be the explanation. Also, follow-up reports demonstrate that the amyloid mass in adipose tissue does not seem to increase after LT, at least not to such extent [3, 22].
It can of course not be excluded that the latest added subunits also most easily detach from the amyloid, thereby producing a higher wild-type proportion than what is representative for the whole material. The lower values of wild-type proportion seen among transplanted type B patients when the tissue was treated with guanidine–HCl compared to non-treated specimens might be in line with this. On the other hand, such effect of guanidine–HCl treatment was not seen in amyloid from type A patients, which still showed an almost complete dominance of wild-type molecules.
Regardless if guanidine–HCl was used or not, a difference in proportion of wtATTR was present between the two fibril type groups among transplanted patients. The fact that a similar divergence was not seen among non-transplanted individuals is not easily interpreted, but a higher turnover rate among type A patients can perhaps be an explanation.
Fragmented versus full-length ATTR species
The V30M mutation is situated outside the C-terminal fragments, which in many cases are the main constituent of the fibrils. Therefore, a majority of the fibril components can, unfortunately, not be included in the analysis. The higher wild-type proportion seen among the fragment-containing fibrils could thus in reality be an effect of selective cleavage of the mutant molecules over the wild-type molecules. The few existing studies where the wild-type proportion is measured in both full-length ATTR and C-terminal fragments separately imply, however, that the wt proportion is similar in both species [11, 33]; however, these investigations are based on other mutations and mutational differences in cleavage propensity might exist.
In this study, we found that the wild-type proportion among the investigated fragments approximately followed the values of the full-length molecules. This implies that wt and mutant protein are equally likely to be truncated, but it should be kept in mind that it is not known if the wild-type proportion among the investigated fragments (those that contain position 30) reflects the wild-type proportion among the much more abundant C-terminal fragments.
Time point and importance of TTR fragmentation
A correlation between fibril composition and clinicopathological features has also been reported in AA-amyloidosis patients [34]. Why different types of amyloid fibrils are seen in AA and ATTRV30M amyloidosis is far from understood. Issues such as time of precursor cleavage (before or after fibril incorporation) and potential importance of fragmentation in triggering fibrillogenesis are still to be answered.
When the wt proportion was compared between full-length and fragmented ATTR in the present study, a slightly higher value among the former than the latter species was found in transplanted patients, whereas the opposite was true for non-transplanted patients. The implication of this is uncertain, but interestingly, this pattern is consistent with what would be expected if cleavage occurs after fibril incorporation. In such case, the wt proportion in transplanted patients would always be somewhat higher among full-length than fragmented species, as the most newly incorporated subunits (which after LT always will be wt molecules) would not have been cleaved yet.
However, the fact that several patients in this study had been ill for 10–17 years and still showed no sign of fragmented ATTR strongly suggests, that cleavage is not occurring after fibril incorporation in all individuals with time. Instead, variations in, for example, enzyme repertoire or amyloid fibril associated molecules (serum amyloid P component, heparan sulfate, etc.) could explain the differences between patients. Another possibility is that full-length TTR could form two structurally and morphologically distinct fibrils, where exposure of the cleavage site occurs in one of the structures but not in the other. Perhaps age-related conditions of the extracellular environment render the formation of one or the other form, which then is sustained throughout the lifetime of the individual. Structural polymorphism of amyloid-like fibrils formed from a single precursor molecule has been reported for several different proteins in vitro, for example Aß1–40 and IAPP [35], and the awareness of this phenomenon is increasing. Whether it is commonly occurring also in vivo deserves further attention.
Acknowledgements
Supported by the Swedish Research Council, the European Framework 6 program (EURAMY), and the patient associations FAMY, FAMY Norrbotten, and AMYL.
Disclosure statement
The authors declare that no competing interests exist.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- 2.Yazaki M, Tokuda T, Nakamura A, Higashikata T, Koyama J, Higuchi K, Harihara Y, Baba S, Kametani F, Ikeda S. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild-type transthyretin. Biochem Biophys Res Commun. 2000;274:702–706. doi: 10.1006/bbrc.2000.3203. [DOI] [PubMed] [Google Scholar]
- 3.Tsuchiya A, Yazaki M, Kametani F, Takei Y, Ikeda S. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl. 2008;14:563–570. doi: 10.1002/lt.21395. [DOI] [PubMed] [Google Scholar]
- 4.Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75:324–327. doi: 10.1212/WNL.0b013e3181ea15d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westermark P, Sletten K, Johansson B, Cornwell GG., III Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87:2843–2845. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranlöv PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254:225–235. doi: 10.1046/j.1365-2796.2003.01173.x. [DOI] [PubMed] [Google Scholar]
- 7.Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, Hattori N, Mukai E, Ando Y, Ikeda S, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63:129–138. doi: 10.1212/01.wnl.0000132966.36437.12. [DOI] [PubMed] [Google Scholar]
- 8.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. A primer of amyloid nomenclature. Amyloid. 2007;14:179–183. doi: 10.1080/13506120701460923. [DOI] [PubMed] [Google Scholar]
- 9.Bergström J, Gustavsson Å, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson B-O, Westermark P. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206:224–232. doi: 10.1002/path.1759. [DOI] [PubMed] [Google Scholar]
- 10.Pras M, Prelli F, Franklin EC, Frangione B. Primary structure of an amyloid prealbumin variant in familial polyneuropathy of Jewish origin. Proc Natl Acad Sci USA. 1983;80:539–542. doi: 10.1073/pnas.80.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liepnieks JJ, Wilson DL, Benson MD. Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid. 2006;13:170–177. doi: 10.1080/13506120600877003. [DOI] [PubMed] [Google Scholar]
- 12.Ihse E, Ybo A, Suhr O, Lindqvist P, Backman C, Westermark P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol. 2008;216:253–261. doi: 10.1002/path.2411. [DOI] [PubMed] [Google Scholar]
- 13.Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, Andersen O, Karlberg I, Norden G, Nakazato M, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30) Clin Genet. 1991;40:242–246. doi: 10.1111/j.1399-0004.1991.tb03085.x. [DOI] [PubMed] [Google Scholar]
- 14.Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 2004;77:64–71. doi: 10.1097/01.TP.0000092307.98347.CB. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto S, Wilczek HE, Nowak G, Larsson M, Oksanen A, Iwata T, Gjertsen H, Soderdahl G, Wikstrom L, Ando Y, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant. 2007;7:2597–2604. doi: 10.1111/j.1600-6143.2007.01969.x. [DOI] [PubMed] [Google Scholar]
- 16.Yazaki M, Mitsuhashi S, Tokuda T, Kametani F, Takei Y-I, Koyama J, Kawamorita A, Kanno H, Ikeda S-I. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant. 2006;7:235–242. doi: 10.1111/j.1600-6143.2006.01585.x. [DOI] [PubMed] [Google Scholar]
- 17.Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14:277–282. doi: 10.1080/13506120701614032. [DOI] [PubMed] [Google Scholar]
- 18.Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D, Falk RH. Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation. 1997;64:74–80. doi: 10.1097/00007890-199707150-00014. [DOI] [PubMed] [Google Scholar]
- 19.Stangou AJ, Hawkins PN, Heaton ND, Rela M, Monaghan M, Nihoyannopoulos P, O’Grady J, Pepys MB, Williams R. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation. 1998;66:229–233. doi: 10.1097/00007890-199807270-00016. [DOI] [PubMed] [Google Scholar]
- 20.Olofsson BO, Backman C, Karp K, Suhr OB. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002;73:745–751. doi: 10.1097/00007890-200203150-00015. [DOI] [PubMed] [Google Scholar]
- 21.Delahaye N, Rouzet F, Sarda L, Tamas C, Dinanian S, Plante-Bordeneuve V, Adams D, Samuel D, Merlet P, Syrota A, et al. Impact of liver transplantation on cardiac autonomic denervation in familial amyloid polyneuropathy. Medicine. 2006;85:229–238. doi: 10.1097/01.md.0000232559.22098.c3. [DOI] [PubMed] [Google Scholar]
- 22.Haagsma EB, Van G, II, Bijzet J, Posthumus MD, Hazenberg BP. Familial amyloidotic polyneuropathy: long-term follow-up of abdominal fat tissue aspirate in patients with and without liver transplantation. Amyloid. 2007;14:221–226. doi: 10.1080/13506120701461368. [DOI] [PubMed] [Google Scholar]
- 23.Okamoto S, Yamashita T, Ando Y, Ueda M, Misumi Y, Obayashi K, Horibata Y, Uchino M. Evaluation of myocardial changes in familial amyloid polyneuropathy after liver transplantation. Intern Med. 2008;47:2133–2137. doi: 10.2169/internalmedicine.47.1399. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan B, Hrncic R, Murphy CL, Gallo G, Weiss DT, Solomon A. Microextraction and purification techniques applicable to chemical characterization of amyloid proteins in minute amounts in tissue. Meth Enzymol. 1999;309:67–81. doi: 10.1016/S0076-6879(99)09007-2. [DOI] [PubMed] [Google Scholar]
- 25.Westermark P, Davey E, Lindbom K, Enqvist S. Subcutaneous fat tissue for diagnosis and studies of systemic amyloidosis. Acta Histochem. 2006;108:209–213. doi: 10.1016/j.acthis.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 26.Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 27.Gustavsson Å, Engström U, Westermark P. Mechanisms of transthyretin (TTR) amyloidogenesis: antigenic mapping of transthyretin purified from plasma and amyloid fibrils and within in situ tissue localizations. Am J Pathol. 1994;144:1301–1311. [PMC free article] [PubMed] [Google Scholar]
- 28.Bergström J, Murphy CL, Weiss DT, Solomon A, Sletten K, Hellman U, Westermark P. Two different types of amyloid deposits—apolipoprotein A-IV and transthyretin—in a patient with systemic amyloidosis. Lab Invest. 2004;84:981–988. doi: 10.1038/labinvest.3700124. [DOI] [PubMed] [Google Scholar]
- 29.Hellman U. Sample preparation by SDS/PAGE and in-gel digestion. In: Jollés P, Jörnvall H, editors. Proteomics in functional genomics. Basel, Switzerland: Birkhäuser Verlag; 2000. pp. 43–54. [Google Scholar]
- 30.Westermark P, Sletten K, Olofsson B-O. Prealbumin variants in the amyloid fibrils of Swedish familial amyloidotic polyneuropathy. Clin Exp Immunol. 1987;69:695–701. [PMC free article] [PubMed] [Google Scholar]
- 31.Koike H, Ando Y, Ueda M, Kawagashira Y, Iijima M, Fujitake J, Hayashi M, Yamamoto M, Mukai E, Nakamura T, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287:178–184. doi: 10.1016/j.jns.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 32.Dwulet FE, Benson MD. Primary structure of an amyloid prealbumin and its plasma precursor in a heredofamilial polyneuropathy of Swedish origin. Proc Natl Acad Sci USA. 1984;81:694–698. doi: 10.1073/pnas.81.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ihse E, Stangou AJ, Heaton ND, O’Grady J, Ybo A, Hellman U, Edvinsson A, Westermark P. Proportion between wild-type and mutant protein in truncated compared to full-length ATTR: an analysis on transplanted transthyretin T60A amyloidosis patients. Biochem Biophys Res Commun. 2009;379:846–850. doi: 10.1016/j.bbrc.2008.12.095. [DOI] [PubMed] [Google Scholar]
- 34.Westermark GT, Sletten K, Westermark P. Massive vascular AA-amyloidosis: a histologically and biochemically distinctive subtype of reactive systemic amyloidosis. Scand J Immunol. 1989;30:605–613. doi: 10.1111/j.1365-3083.1989.tb02468.x. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen JS, Dikov D, Flink JL, Hjuler HA, Christiansen G, Otzen DE. The changing face of glucagon fibrillation: structural polymorphism and conformational imprinting. J Mol Biol. 2006;355:501–523. doi: 10.1016/j.jmb.2005.09.100. [DOI] [PubMed] [Google Scholar]