

Abstract
Background and Aim. EGFR is commonly expressed in cancers of the head and neck (H and N), and anti-EGFR agents have demonstrated improvements in outcomes (TTP and OS). The aim of this study was to determine EGFR gene status in H and N cancer patients treated with gefitinib and to correlate mutational status with clinico-pathological data and response. Patients and Methods. Patients with histologically confirmed H and N cancer having failed prior treatment for advanced disease entered this compassionate-use-program. Nineteen patients received gefitinib. EGFR expression was assessed by IHC, gene copy number by FISH, and mutation analysis was conducted for EGFR (18-21), KRAS, BRAF (V600E), and HER-2 exon 20. An additional TKI naive cohort of 73 patients was also screened. Results. Mutations were detected in 6/19 patients (3× EGFR, 1× KRAS, and 2× HER2-exon 20). There were no significant differences in TTP or OS for patients with somatic EGFR mutations. No BRAF mutations were detected. Conclusions. The incidence of EGFR mutations in H and N cancer in this study was 5.3%. No statistically relevant correlations between mutation or gene gain and response or survival were observed. Due to the limited number of patients and low incidence of genetic aberrations in the genes analyzed, additional studies are warranted.
1. Introduction
Cancer of the head and neck (H and N) is the fifth most common cancer in the United States, and despite significant progress in therapeutic modalities, almost half of patients with this diagnosis will relapse with local or distant disease, indicating the need for novel therapeutic interventions [1, 2]. Treatment of advanced disease usually involves combinations of chemotherapeutic agents, such as cisplatin, with radiation, while new agents are being studied in platinum-refractory metastatic disease.
The epidermal growth factor receptor (EGFR) along with its ligands epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α) are important in many aspects of cell survival, differentiation, proliferation, and invasion [3–5]. EGFR is almost universally expressed in H and N cancers, and high levels of expression have been correlated with poor outcome [6, 7]. Studies have shown that blockade of EGFR signaling using antibody-based approaches (cetuximab) can offer improved outcomes. Recent randomized studies have demonstrated that the combination of radiation with the anti-EGFR monoclonal antibody cetuximab results in improvements of response and overall survival over radiation alone in patients with locally advanced H and N cancer [8]. Combinations of EGFR inhibition with chemotherapy have also been studied with promising results in the metastatic and recurrent settings [9, 10]. Although other EGFR inhibitors, such as small tyrosine kinase inhibitors (TKIs; gefitinib and erlotinib) are available for the treatment of other tumor types; only modest response rates of up to 11% as monotherapy have been obtained in H and N cancers [11, 12]. Considering that the oral TKI erlotinib is licensed for second line NSCLC, and the TKI gefitinib has recently been approved for first line advanced NSCLC carrying somatic EGFR mutations, a new level of clinical interpretation may be necessary in this apparently EGFR sensitive disease.
Gefitinib is an orally active and selective inhibitor of the EGFR tyrosine kinase, which has principally been studied in NSCLC. Clinical responses to gefitinib differ among NSCLC patients, and several studies have aimed at identifying prognostic and/or predictive markers of response to these agents. Clinical studies indicated that EGFR TKIs were more effective in women, of Asian origin, individuals with adenocarcinoma, and never smokers [13, 14]. The impact of EGFR expression levels on drug sensitivity is still an open issue, since preclinical and clinical data show no obvious correlation between EGFR immunohistochemical expression and response although no thorough analysis has been performed. EGFR gene copy number on the other hand has been shown to be associated with improved response rates and survival outcomes to TKI treatment compared to WT patients, albeit in NSCLC [15, 16]. However, the defining molecular event appears to be the presence of activating sensitizing mutations in EGFR [17–19]. These are virtually exclusive to NSCLC but have been reported in numerous other cancers [20]. Patients harboring such mutations have a much higher response rate that is translated into improved survival times compared to WT patients treated with EGFR TKIs, at least in NSCLC [20, 21]. The molecular signatures of NSCLC have also highlighted that the presence of somatic mutations in KRAS, occurring mutually exclusively to EGFR mutations, earmarks tumors that are essentially resistant to TKIs [22]. Similar molecular events are being unearthed in colorectal cancers with respect to treatment outcomes with the anti-EGFR monoclonal antibodies cetuximab and panitumumab. Here, mutations in key signaling molecules KRAS [22], BRAF [23], PIK3CA [24], and loss of expression of PTEN [25] have been correlated with a lack of response [26].
Many recent studies have consolidated our understanding on the functional blockade of EGFR with various agents. Investigations by numerous groups have now broadened the scope of TKIs by the discovery of similar somatic mutations in other cancer types; however, their correlation with response to TKI treatment is as yet not conclusive. These insights have raised questions as to the effect and incidence of such mechanisms and also as to their prognostic significance.
Although the response rates of H and N cancer to gefitinib are similar to those seen in NSCLC, as yet there appear to be no clinicopathological predictors so far identified for the responsive cases. From limited literature studies in H and N cancer, there are suggestions that EGFR mutations similar to those in NSCLC exist, while there are other reports that have failed to detect mutations [27–30]. However, recently there have been reports of the presence of EGFR somatic mutations in H and N cancers, albeit at a low incidence (1–14%) [31–37]. Unlike NSCLC where there are multiple studies investigating the predictive nature of gene copy number analysis to TKIs, the data in H and N cancer remains scant and inconclusive [27, 38, 39].
Based on the above, we hypothesized that if somatic mutations and gene copy gain of EGFR occur in H and N cancer, then treatment with a TKI such as gefitinib could be a potentially beneficial treatment option for many of these patients. The objective of this study was to determine whether the molecular mechanisms seen in NSCLC regarding EGFR mutations and gene copy number and correlation with TKI response extend to H and N cancer. Furthermore, we extended this analysis to examine the incidence of additional molecular events that have been proposed as candidate biomarkers for response to anti-EGFR agents in NSCLC and colorectal cancer.
2. Materials and Methods
2.1. Patients
Patients with histologically confirmed H and N cancer, who had failed prior treatment for advanced or metastatic disease and were not amenable to further chemotherapy or chemotherapy-naïve patients due to contraindication, were eligible for the study. Eligibility criteria included an ECOG PS of 0–2, estimated life expectancy of at least 3 months, adequate bone marrow, and hepatic and renal function, indicated by an absolute neutrophil count of ≥1,500/μl, platelets ≥75,000/μl, total bilirubin ≤2× the upper limit of normal; serum AST or ALT levels ≤3× the upper limit of normal; serum creatinine ≤2 mg/dl; serum albumin ≥2.5 g/dl. Previous chemotherapy and/or radiotherapy were allowed. All histological subtypes were allowed. Patients with a history of serious cardiac disease, other serious medical illness, or inability to comply with the treatment plan and followup visits were excluded from the study.
An additional cohort of 37 patients, previously reported [40], were also included in this analysis for comparative purposes.
A third cohort of anti-EGFR naive patients was also included to aid in the study of the prognostic significance of the biomarkers analysed. These patients were randomly selected from the School of Medicine, University of Thessaloniki, with the only entry criteria being adequate biological material access and patient's informed consent for biomarker analysis.
All patients signed informed consent as a requirement for study inclusion. They similarly signed consent for the use of biological materials for research purposes. The study was conducted according to the Declaration of Helsinki and the guidelines for Good Clinical Practice. The Local Ethics Committees approved the study and the collection of biological material.
2.2. TKI Treatment
Patients received gefitinib at a dose of 250 mg per day orally. Gefitinib was supplied free of charge by AstraZeneca as part of a compassionate use program. Treatment was administered daily with a treatment cycle constituting 28 days. Treatment was discontinued for up to 7 days for grade 3 or 4 toxicity, until resolution of toxicity to ≤1. For non-resolving toxicities of more than 15 days, patients were taken off study. Treatment was continued until disease progression, serious adverse toxicity, at the direction of the treating physician, or following patient withdrawal. Patients were eligible for response evaluation after completion of at least 2 months of treatment. All patients have been routinely followedup at 6 monthly intervals from their last treatment (data on file, HeCOG Data Office). Complete clinical data including smoking history, clinical stage, pathological diagnosis, and response data for all patients was available.
2.3. Determination of EGFR Expression
Paraffin blocks of tumor were collected retrospectively, and peripheral blood samples were collected during treatment or followup. Immunohistochemical detection of EGFR was performed as previously described [41] to semiquantify EGFR expression levels. Tumor tissue sections showing 2+ or 3+ were considered as positive.
Assessment of EGFR gene copy number was conducted by FISH and scored as previously described [40, 41].
2.4. Mutation Analyses
Genomic DNA was extracted from paraffin embedded tumors as previously described [42]. All paraffin blocks were examined on H and E for histological verification according to WHO [43], tumors consisting of >75% tumor cell content (% TCC) were considered as eligible for DNA extraction and sequence analysis. For those biopsies where the % TCC was inadequate, macrodissection on 5 μm sections was performed to increase the content to >75%.
Mutational analysis for all genes was conducted as previously described [40].
Additional genes for which analysis was conducted included KRAS mutation analysis of codons 12 and 13. PCR was performed using the same conditions as for EGFR, using KRAS-specific primers amplifying Exon 2 as previously described [40]. BRAF exons 14 and 15 were analyzed as previously described [40], and the 3′ and 5′ intron-exon splice sites of MET exon 14 were also screened. The primer sequences for all reactions are available upon request.
All studied exons were confirmed as previously described above for EGFR [40]. All PCR products were purified by solid-phase reversible immobilization chemistry followed by bidirectional dye-terminator fluorescent sequencing. Sequences were analyzed by BLAST and chromatograms by manual review and compared to the following representative gene accession numbers: EGFR, NM_005228, and/or the EGFR gene sequence accession number: AF288738; KRAS, gi: 14277199; HER2 exon 20, gi: 23462913; MET, gi: 212720875 (http://www.ncbi.nlm.nci/).
The EGFR exon 21 mutation L858R which represents approximately 40% of all reported mutations in NSCLC [44] was also analyzed by PCR-RFLP based on the presence of a new Sau96I restriction site created by the mutation [40]. KRAS mutations of codons 12 and 13 were also analyzed by PCR-RFLP based on modified versions of the protocols of Boldrini et al. [45] and Kislitsin et al. [46]. MET exon 14 5′ and 3′ intron-exon deletions first reported in NSCLC [47] were also analyzed by a mutant allele-specific PCR method, which only amplifies in the presence of the given deletions, while BRAF V600E was also analyzed by PCR-RFLP based on a modified version of Salvesen et al. [48].
2.5. Statistical Analysis
Endpoints included TTP (time to progression) and survival in association with the candidate biomarkers. Survival was defined as the time from first day of treatment until death from any cause. TTP was computed as the time from initiation of treatment until recurrence of tumor or death from any cause. The Fisher's exact test was used for comparing groups of categorical data, while for continuous data the Mann-Whitney test. P-values of at least .05 were considered statistically significant. Kaplan-Meier curves and log-rank test were used for comparing time to event distributions. All analyses were performed using SPSS version 18, in the HeCOG data office.
3. Results
3.1. Clinicopathological Charateristics of Whole Cohort
Characteristics of the 73 TKI naive patients are listed in Table 1. A subcohort of these patients received the anti-EGFR agent cetuximab, and details of these patients have been previously presented [49]. In the TKI treatment group, a total of 19 patients were enrolled into a compassionate-use program between 7/2002 and 11/2005. Gefitinib-treated patients were predominantly white males, median age 66 years, with median PS 1. The majority of patients had a strong smoking history (1 unknown), and the majority had received prior chemotherapy (79%). Four patients had never received chemotherapy (i.e., treatment naïve) entering the compassionate use study due to contraindication to standard chemotherapy. Since this study was not designed to determine the response rate, TTP, or survival to treatment with gefitinib, it was not deemed necessary to analyze untreated patients in this investigational/translational study.
Table 1.
Patient and tumor characteristics by group.
| TKI treated | Naive | |||
|---|---|---|---|---|
| Number | % | Number | % | |
| Total | 19 | 100 | 73 | 100 |
| Gender | ||||
| Male | 19 | 100 | 60 | 82.2 |
| Female | 13 | 17.8 | ||
| Age (years) | ||||
| Median | 66 | 60 | ||
| Range | 38–75 | 36–82 | ||
| Histological type | ||||
| SCC | 18 | 94.7 | 69 | 94.5 |
| Other | 1 | 5.3 | 2 | 2.7 |
| Unknown | 2 | 2.7 | ||
| Smoking status | ||||
| Yes | 18 | 94.7 | 64 | 87.7 |
| No | 8 | 11.0 | ||
| Unknown | 1 | 5.3 | 1 | 1.3 |
| Alcohol | ||||
| Yes | 11 | 57.9 | 56 | 76.7 |
| No | 7 | 36.8 | 15 | 20.5 |
| Unknown | 1 | 5.3 | 2 | 2.7 |
The second cohort of untreated (TKI naive) patients had similar patient and tumor characteristics.
3.2. TKI-Treated Cohort
3.2.1. Treatment Characteristics
Patients received a total of 98 cycles of treatment (median 4 cycles, range 1–16 cycles). At the time of analysis, all patients had died. Reasons for treatment discontinuation included progression (5/19), death (1/19), and patient refusal (2/19).
3.2.2. Response, TTP, and Survival
A total of 4 (21.1%) of patients achieved an overall objective response (CR + PR) and further 6 (31.6%) achieved disease stabilization. Interestingly only one of the responding patients had gene copy number gain; the 3 patients with EGFR mutations demonstrated disease stabilization. There were 19 disease progressions for a median TTP of 3.6 months (95% CI: 1.5–5.6) and a total of 19 deaths for a median survival of 6.5 months (95% CI: 2.4–10.6).
3.2.3. Somatic Mutation Analysis
We performed mutational analysis of exons 18, 19, 20, and 21 of biopsy material from 19 patients with H and N cancer treated with Gefitinib. In this cohort, a total of 4 different somatic mutations located within the exons of the TK domain of EGFR were observed in 3 patients. Mutational status of all mutations was confirmed using germline DNA extracted from peripheral blood or from macrodissected normal tissue available from the paraffin embedded biopsy. Two patients harbored delL747-P753insS (one with associated gene amplification), Figure 1, and another patient harbored V843I with a secondary silent R846R somatic mutation (also in the presence of EGFR gene amplification), Table 4 and Figure 2.
Figure 1.
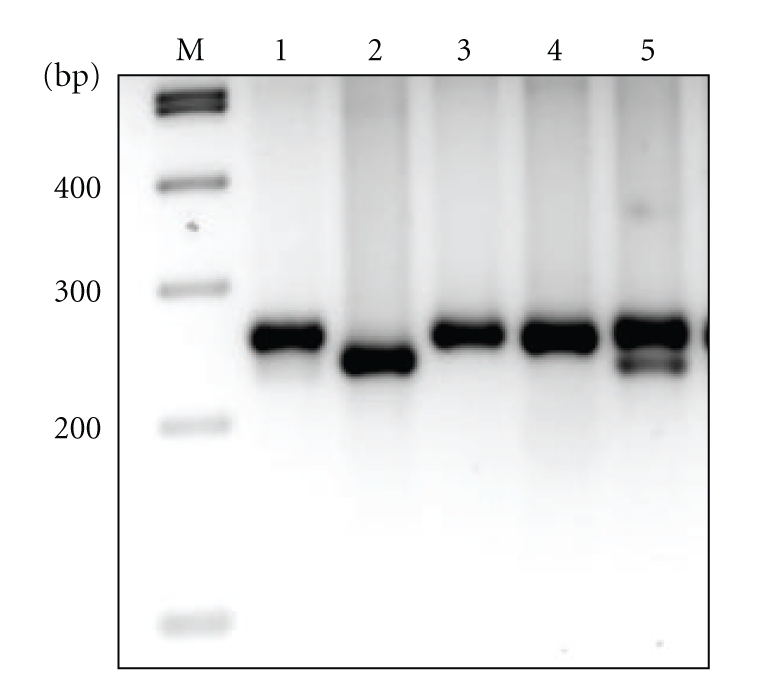
EGFR Exon 19 deletions. M: molecular weight markers; 1, 3, 4: WT Exon 19 EGFR; 2: delL747-P753insS Exon 19 and EGFR gene amplification; 5: delL747-P753insS Exon 19 EGFR.
Table 4.
Response correlations. Patients, tumor characteristics and mutations by response for TKI treated cohort.
| CR or PR (N = 4) | SD or PD (N = 13) | ||||
|---|---|---|---|---|---|
| N | % | N | % | P | |
| Alcohol | .999 | ||||
| No | 2 | 50.0 | 5 | 41.7 | |
| Yes | 2 | 50.0 | 7 | 58.3 | |
| RAS | .235 | ||||
| MUT | 1 | 25.0 | |||
| WT | 3 | 75.0 | 13 | 100.0 | |
| EGFR | .541 | ||||
| MUT | 3 | 23.1 | |||
| WT | 4 | 100.0 | 10 | 76.9 | |
| HER2 | .999 | ||||
| MUT | 1 | 7.7 | |||
| WT | 4 | 100.0 | 12 | 92.3 | |
| EGFR (IHC) | .516 | ||||
| Negative | 4 | 33.3 | |||
| Positive | 3 | 100.0 | 8 | 66.7 | |
| EGFR (FISH) | .999 | ||||
| Negative | 3 | 100.0 | 8 | 72.7 | |
| Positive | 3 | 27.3 | |||
Figure 2.

Representative chromatograms of somatic mutations (sense 5′) of (a): HER2 Exon 20 Q795R; (b): HER2 Exon 20 W825X (Stop); (c): HER2 Exon 20 Y781C; (d) EGFR Exon 21, R836R, and V843I.
No patient was found to harbor the common exon 21 mutation L858R either on sequencing or by RFLP. KRAS mutations were observed in 1 patient, occurring at codon 12. EGFR and KRAS mutations were mutually exclusive. No patient was found to harbor a BRAF V600E point mutation, nor was there any other mutations observed in exon 15 of the BRAF gene. Similarly no analyzed patient was found to harbor an inton-exon 14 deletion of the MET gene. Two patients were found to have somatic mutations within exon 20 of HER-2, one harboring two independent point mutations, Table 5, Figure 2.
Table 5.
Characteristics of patients receiving Gefitinib.
| Somatic Mutational Spectrum | EGFR | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Age | Previous Chemo-therapy | Response | TTP (months) |
Survival (months) |
RAS 12/13 |
EGFR Exons 18–21 | BRAF exon 15 | MET 5′–3′ exon 14 intro-exon boundaries | Her2 Exon20 |
FISH | IHC |
| 1 | 38 | Y | PD | 1.25 | 8.85 | WT | WT | WT | WT | Y781C | ND | 0 |
| 2 | 50 | Y | PR | 2.20 | 5.70 | WT | WT | WT | WT | WT | ND | ND |
| 3 | 75 | Y | SD | 5.31 | 6.46 | WT | WT | WT | WT | WT | WT | 0 |
| 4 | 59 | Y | NE | 2.66 | 2.66 | WT | WT | WT | WT | Q795R | WT | 3 |
| W825X | ||||||||||||
| 5 | 63 | Y | SD | 12.13 | 12.13 | WT | delL747-P753insS | WT | WT | WT | Amp | ND |
| 6 | 66 | Y | PD | 2.07 | 2.43 | WT | WT | WT | WT | WT | WT | 3 |
| 7 | 63 | Y | SD | 7.74 | 9.54 | WT | delL747-P753insS | WT | WT | WT | WT | 0 |
| 8 | 72 | Y | SD | 8.46 | 11.70 | WT | R836R | WT | WT | WT | Amp | 3 |
| V843I | ||||||||||||
| 9 | 62 | Y | SD | 7.84 | 8.03 | WT | WT | WT | WT | WT | ND | 3 |
| 10 | 72 | Y | PR | 3.11 | 3.11 | WT | WT | WT | WT | WT | WT | 3 |
| 11 | 75 | Y | PD | 5.15 | 5.15 | WT | WT | WT | WT | WT | Amp | 2 |
| 12 | 66 | N | PR | 4.10 | 11.64 | WT | WT | WT | WT | WT | Gain | 2 |
| 13 | 74 | N | PD | 3.57 | 9.15 | WT | WT | WT | WT | WT | WT | 3 |
| 14 | 61 | N | PD | 1.64 | 3.74 | WT | WT | WT | WT | WT | WT | 3 |
| 15 | 70 | N | CR | 15.05 | 17.57 | G12D | WT | WT | WT | WT | WT | 3 |
| 16 | 69 | Y | PD | 0.82 | 10.75 | WT | WT | WT | WT | WT | WT | 0 |
| 17 | 63 | Y | PD | 0.75 | 2.89 | WT | WT | WT | WT | WT | WT | 3 |
| 18 | 72 | Y | NE | 4.59 | 4.59 | WT | WT | WT | WT | WT | WT | 3 |
| 19 | 59 | Y | SD | 1.48 | 3.80 | WT | WT | WT | WT | WT | WT | 2 |
Abbreviations: Y: Yes; N: No; IHC: immunohistochemistry; FISH: fluorescent in situ hybridization; ND: not determined; gain: aneuploidy; CR: complete response; PR: partial response; SD: stable disease; PR: progressive disease; NE: not evaluable.
Somatic mutations were detected in 6/19 patients (3×EGFR, 1×KRAS, and 2×HER2-exon 20). The incidence of EGFR mutations in this population was 15.8% (3/19). Patients with somatic EGFR Exon19 deletions and point mutations in Exon21 had a longer TTP and survival compared to patients without mutations (not statistically significant), as shown in Table 4. For patients without mutations, TTP was 2.2 m versus 7.7 m for patients with mutations (Log Rank P = .145), and survival was 4.6 m for patients without mutations versus 11.6 m for mutation carriers (Log Rank P = .125). Similarly no statistical significant difference was found with respect to any other biomarker analyzed in this study.
Immunohistochemical analysis for EGFR expression identified positive tumors in 76.5% (13/17) of cases. Three patients had gene amplification of EGFR (two with concomitant somatic mutations in EGFR), and one additional patient had gene copy number gain (aneuploidy), Table 5. Although EGFR copy gain was not associated with a prolonged survival (survival was 11.64 m), when we combined patients with a somatic EGFR mutation and/or gene copy number gain, they performed better (but not statistical significant) compared to the wild-type (WT) group, 4.6 to 11.6 months, for survival; 2.2 to 7.7 months for TTP (WT versus mutation/gene gain, resp.).
3.3. Naive Cohort
The TKI naive cohort consisted of additional 73 patients, Tables 1 and 2. Of these patients, 37 have previously been analyzed for EGFR and KRAS mutational status [49]. These patients were included to obtain more accurate estimation of the frequency of gene alterations present in H and N cancers. The overall incidence per aberration analyzed is indicated in Table 3.
Table 2.
Molecular characteristics per group.
| TKI treated | Naive | |||
|---|---|---|---|---|
| (N = 19) | (N = 73) | |||
| N | % | N | % | |
| RAS | ||||
| MUT | 1 | 5.3 | ||
| WT | 18 | 94.7 | 73 | 100.0 |
| EGFR | ||||
| MUT | 3 | 15.8 | 1 | 1.4 |
| WT | 16 | 84.2 | 56 | 76.7 |
| Unknown | 16 | 21.9 | ||
| MET | ||||
| WT | 19 | 100.0 | 36 | 49.3 |
| Unknown | 37 | 50.7 | ||
| BRAF | ||||
| WT | 19 | 100.0 | ||
| Unknown | 73 | 100.0 | ||
| HER2 exon 20 | ||||
| MUT | 2 | 10.5 | ||
| WT | 17 | 89.5 | ||
| Unknown | 73 | 100.0 | ||
| EGFR (IHC) | ||||
| 0 | 4 | 21.1 | ||
| 1 | 2 | 2.7 | ||
| 2 | 3 | 15.8 | 8 | 11.0 |
| 3 | 10 | 52.6 | 22 | 30.1 |
| NE | 2 | 10.5 | 3 | 4.1 |
| Unknown | 38 | 52.1 | ||
| EGFR (FISH) | ||||
| Amplified | 3 | 15.8 | 1 | 1.4 |
| Nonamplified | 13 | 68.4 | 32 | 43.8 |
| NE | 3 | 15.8 | ||
| Unknown | 40 | 54.8 | ||
Table 3.
Survival according to molecular markers for TKI-treated cohort.
| Survival | Log rank P | TTP | Log rank P | |
|---|---|---|---|---|
| EGFR | ||||
| MUT | 11.7 | .131 | 8.5 | .106 |
| WT | 5.1 | 2.7 | ||
| HER2 | ||||
| MUT | 2.7 | .340 | 1.2 | .100 |
| WT | 6.5 | 4.1 | ||
| EGFR (IHC) | ||||
| Negative | 8.9 | .700 | 1.2 | .641 |
| Positive | 4.6 | 3.6 | ||
| EGFR (FISH) | ||||
| Negative | 4.6 | .271 | 3.1 | .185 |
| Positive | 11.7 | 8.5 | ||
| EGFR combination: (Mutation and/or Gene gain) | ||||
| Negative | 4.6 | .125 | 2.2 | .145 |
| Positive | 11.6 | 7.7 |
4. Discussion
We have analyzed three differently selected populations of H and N cancer in order to further understand the incidence and potential predictive ability of a subset of specific anti-EGFR candidate biomarkers. Although the representative population of TKI-treated patients was small, the incidence of both EGFR somatic mutations and gene gain/amplification was relatively high (3 and 4, resp., (2 coexistent)) leading us to incorporate additional 73 patients to more closely reflect their respective incidence in a more balanced population.
With the inclusion of 92 patients, the overall incidence of EGFR mutations indicates that at 4% (4/92) they are rare among cases of H and N cancer within the Greek population. A number of other authors have reported on the presence of EGFR kinase domain mutations in H and N cancers. These have ranged from not being detected from a total of 221 patients [27–30], through to between 1 and 14% in other studies (n = 15/425) [31–37]. These differences suggest that such mutations are a rare event, but that ethnicity or other cofactors may play a role. Indeed, differences in incidence have been well characterized with respect to EGFR mutations in NSCLC, wherein the incidence is twice that in Asians compared to Whites [20]. Bearing in mind that the TKI gefitinib has recently gained license approval for the treatment of first line NSCLC for patients harboring EGFR mutations [50], the search for similar biomarkers of response to anti-EGFR agents has become a priority in virtually all cancer types.
Head and neck cancers are not without their fair share of responses to anti-EGFR therapies. There are a number of studies that have reported similar response rates of H and N cancer to TKIs as to the response rates observed with single agent TKIs in unselected NSCLC [11, 12]. Further to this is the clinical utilization of anti-EGFR monoclonal antibodies including cetuximab in the treatment of H and N cancers [8]. Given such utility and with the knowledge that none of the biomarkers analyzed herein were found to correlate with outcomes in the FLEX [51] study, each of these biomarkers will need to be more thoroughly investigated in H and N cancer. We also investigated the incidence of EGFR gene copy gain finding 5.3% (1/19) of such patients. This matches previous data indicating that between 8 and 30% of esophageal cancers have EGFR gene gain [52, 53]. As with EGFR somatic mutations, there is currently little evidence to speculate on the overall predictive nature of these aberrations in H and N cancers receiving anti-EGFR-based agents. There are of course some limitations to interpretations being derived from this study, the major of which is the small sample size respective to the TKI-treated population. Although there were differences between the EGFR mutation positive and negative groups, their numbers were small and any difference could simply be attributable to chance. The relatively low incidences of such mutations in H and N cancer, as well as mutations in KRAS, BRAF, and HER2, indicate the necessity for analysis of much larger patient populations together with investigation of alternative molecular pathways and mechanisms to identify predictive or prognostic markers.
In the continued search for additional biomarkers that may be predictive of response to anti-EGFR agents, we and others [12] have extrapolated data from breast cancer [54], colorectal cancer [22, 26], and NSCLC [20] in order to address additional candidate genes/biomarkers to receptor tyrosine kinases in general. As indicated in our cohorts, no somatic mutations were identified in BRAF or MET. Only one other report has analyzed HER2 mutations, suggesting that they may be a potential biomarker of EGFR-TKI sensitivity [12]. Although our investigation did not expand into the other 2 cohorts, the Erbitux-treated cohort previously investigated [49] was also analyzed for the presence of PTEN loss. Both PTEN and PIK3CA remain potential candidate biomarkers of response to anti-EGFR agents. Further analyses should incorporate analysis of these two molecular events, as preliminary data suggests that PIK3CA mutations may be as common as 20% [55] and PTEN expression may be of predictive/prognostic significance [56]. The limited data that currently exists not only to anti-EGFR agents but also to chemotherapy in general in H and N cancers beckons further concerted efforts into additional molecular taxinometry in order to start to substratify patient populations for treatment individualization.
In conclusion, from a relatively small cohort of TKI-treated H and N cancers, there is little evidence of any single biomarker or biomarker algorithm from the genes investigated as capable of subclassifying H and N cancer into two distinct responsive subgroups. Although differences in survival endpoints did not reach statistical significance between these groups, there are many contributing factors to small retrospective analyses, such as inappropriate patient selection and/or reporter bias compounding the analysis. Considering that there are (to date) no effective biomarkers in H and N cancer, additional studies are welcome in order to identify and further clarify if any underlying mechanisms of response or resistance to anti-EGFR agents exist in H and N cancer.
Conflict of Interests
Consultant or Advisory Role. Dr. S. Murray, Merck KGaA, Darmstadt, Germany. Merck distributes the MoAb Cetuximab (Erbitux); AstraZeneca, Maccelsfield, United Kingdom. AstraZeneca are proprietors of gefitinib (Iressa); Amgen Thousand Oaks, Ca, USA. Amgen distributes the MoAb Panitumumab (Vectibix). Dr. S. Murray is an employee of Biomarker Solutions; in this role, he has no conflict of interests with proprietary agents mentioned in this paper. No other author has a conflict of interests.
Funding Source
This study was solely funded by HeCOG.
Acknowledgment
The authors would like to thank Miss I.Grimani for her assistance with the statistical analysis.
References
- 1.Forastiere AA, Metch B, Schuller DE, et al. Randomized comparison of cisplatin plus fluorouracil and carboplatin plus fluorouracil versus methotrexate in advanced squamous-cell carcinoma of the head and neck: a Southwest Oncology Group study. Journal of Clinical Oncology. 1992;10(8):1245–1251. doi: 10.1200/JCO.1992.10.8.1245. [DOI] [PubMed] [Google Scholar]
- 2.Cohen EEW, Lingen MW, Vokes EE. The expanding role of systemic therapy in head and neck cancer. Journal of Clinical Oncology. 2004;22(9):1743–1752. doi: 10.1200/JCO.2004.06.147. [DOI] [PubMed] [Google Scholar]
- 3.Grandis JR, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor- α and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78(6):1284–1292. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 4.Grandis JR, Melhem MF, Gooding WE, et al. Levels of TGF-α and EGFR protein in head and neck squamous cell carcinoma and patient survival. Journal of the National Cancer Institute. 1998;90(11):824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 5.Santini J, Formento JL, Francoual M, et al. Characterization, quantification, and potential clinical value of the epidermal growth factor receptor in head and neck squamous cell carcinomas. Head and Neck. 1991;13(2):132–139. doi: 10.1002/hed.2880130209. [DOI] [PubMed] [Google Scholar]
- 6.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Research. 2002;62(24):7350–7356. [PubMed] [Google Scholar]
- 7.Hitt R, Ciruelos E, Amador ML, et al. Prognostic value of the epidermal growth factor receptor (EGRF) and p53 in advanced head and neck squamous cell carcinoma patients treated with induction chemotherapy. European Journal of Cancer. 2005;41(3):453–460. doi: 10.1016/j.ejca.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncology. 2010;11(1):21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 9.Rivera F, García-Castaño A, Vega N, Vega-Villegas ME, Gutiérrez-Sanz L. Cetuximab in metastatic or recurrent head and neck cancer: the EXTREME trial. Expert Review of Anticancer Therapy. 2009;9(10):1421–1428. doi: 10.1586/era.09.113. [DOI] [PubMed] [Google Scholar]
- 10.William WN, Jr., Kim ES, Herbst RS. Cetuximab therapy for patients with advanced squamous cell carcinomas of the head and neck. Nature Clinical Practice Oncology. 2009;6(3):132–133. doi: 10.1038/ncponc1321. [DOI] [PubMed] [Google Scholar]
- 11.Cohen EEW, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. Journal of Clinical Oncology. 2003;21(10):1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 12.Cohen EEW, Kane MA, List MA, et al. Phase II trial of gefitinib 250 mg daily in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. Clinical Cancer Research. 2005;11(23):8418–8424. doi: 10.1158/1078-0432.CCR-05-1247. [DOI] [PubMed] [Google Scholar]
- 13.Jänne PA, Gurubhagavatula S, Yeap BY, et al. Outcomes of patients with advanced non-small cell lung cancer treated with gefitinib (ZD1839, ’Iressa’) on an expanded access study. Lung Cancer. 2004;44(2):221–230. doi: 10.1016/j.lungcan.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Wu MF, Fahn HJ, Wu TC, et al. Experience of gefitinib (Iressa) for previously treated patients with advanced non-small cell lung cancer. Thoracic Medicine. 2003;18, article 203 [Google Scholar]
- 15.Dahabreh IJ, Linardou H, Siannis F, Paris Kosmidis P, Bafaloukos D, Murray S. Somatic EGFR mutation and amplification as predictive biomarkers for response to tyrosine kinase inhibitors in non-small-cell lung cancer: a systematic review and meta-analysis. Clinical Cancer Research. 2010;16:291–303. doi: 10.1158/1078-0432.CCR-09-1660. [DOI] [PubMed] [Google Scholar]
- 16.Dahabreh IJ, Linardou H, Kosmidis P, Bafaloukos D, Murray S. EGFR gene copy number as a predictive biomarker for patients receiving tyrosine kinase inhibitor treatment: a systematic review and meta-analysis in non-small-cell lung cancer. Annals of Oncology. 2010 doi: 10.1093/annonc/mdq432. [DOI] [PubMed] [Google Scholar]
- 17.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 18.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung, cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 19.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nature Reviews Clinical Oncology. 2009;6(6):352–366. doi: 10.1038/nrclinonc.2009.62. [DOI] [PubMed] [Google Scholar]
- 21.Mok TSK, Wu YL, Yu CJ, et al. Randomized, placebo-controlled, phase II study of sequential erlotinib and chemotherapy as first-line treatment for advanced non-small-cell lung cancer. Journal of Clinical Oncology. 2009;27(30):5080–5087. doi: 10.1200/JCO.2008.21.5541. [DOI] [PubMed] [Google Scholar]
- 22.Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncology. 2008;9(10):962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 23.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Research. 2007;67(6):2643–2648. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 24.Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Annals of Oncology. 2009;20(1):84–90. doi: 10.1093/annonc/mdn541. [DOI] [PubMed] [Google Scholar]
- 25.Frattini M, Saletti P, Romagnani E, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. British Journal of Cancer. 2007;97(8):1139–1145. doi: 10.1038/sj.bjc.6604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sartore-Bianchi A, Di Nicolantonio F, Nichelatti M, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS ONE. 2009;4(10, article e7287) doi: 10.1371/journal.pone.0007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung CH, Ely K, McGavran L, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. Journal of Clinical Oncology. 2006;24(25):4170–4176. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 28.Lemos-González Y, Páez de la Cadena M, Rodríguez-Berrocal FJ, Rodríguez-Piñeiro AM, Pallas E, Valverde D. Absence of activating mutations in the EGFR kinase domain in spanish head and neck cancer patients. Tumor Biology. 2007;28(5):273–279. doi: 10.1159/000110425. [DOI] [PubMed] [Google Scholar]
- 29.Janmaat ML, Gallegos-Ruiz MI, Rodriguez JA, et al. Predictive factors for outcome in a phase II study of gefitinib in second-line treatment of advanced esophageal cancer patients. Journal of Clinical Oncology. 2006;24(10):1612–1619. doi: 10.1200/JCO.2005.03.4900. [DOI] [PubMed] [Google Scholar]
- 30.Cohen EEW, Lingen MW, Martin LE, et al. Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR . Clinical Cancer Research. 2005;11(22):8105–8108. doi: 10.1158/1078-0432.CCR-05-0926. [DOI] [PubMed] [Google Scholar]
- 31.Pühringer-Oppermann FA, Stein HJ, Sarbia M. Lack of EGFR gene mutations in exons 19 and 21 in esophageal (Barrett’s) adenocarcinomas. Diseases of the Esophagus. 2007;20(1):9–11. doi: 10.1111/j.1442-2050.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- 32.Willmore-Payne C, Holden JA, Layfield LJ. Detection of EGFR- and HER2-activating mutations in squamous cell carcinoma involving the head and neck. Modern Pathology. 2006;19(5):634–640. doi: 10.1038/modpathol.3800552. [DOI] [PubMed] [Google Scholar]
- 33.Kwak EL, Jankowski J, Thayer SP, et al. Epidermal growth factor receptor kinase domain mutations in esophageal and pancreatic adenocarcinomas. Clinical Cancer Research. 2006;12(14):4283–4287. doi: 10.1158/1078-0432.CCR-06-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sudo T, Mimori K, Nagahara H, et al. Identification of EGFR mutations in esophageal cancer. European Journal of Surgical Oncology. 2007;33(1):44–48. doi: 10.1016/j.ejso.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 35.Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clinical Cancer Research. 2005;11(8):2879–2882. doi: 10.1158/1078-0432.CCR-04-2029. [DOI] [PubMed] [Google Scholar]
- 36.Loeffler-Ragg J, Witsch-Baumgartner M, Tzankov A, et al. Low incidence of mutations in EGFR kinase domain in Caucasian patients with head and neck squamous cell carcinoma. European Journal of Cancer. 2006;42(1):109–111. doi: 10.1016/j.ejca.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 37.Guo M, Liu S, Lu F. Gefitinib-sensitizing mutations in esophageal carcinoma. New England Journal of Medicine. 2006;354(20):2193–2194. doi: 10.1056/NEJMc052698. [DOI] [PubMed] [Google Scholar]
- 38.Thomas F, Rochaix P, Benlyazid A, et al. Pilot study of neoadjuvant treatment with erlotinib in nonmetastatic head and neck squamous cell carcinoma. Clinical Cancer Research. 2007;13(23):7086–7092. doi: 10.1158/1078-0432.CCR-07-1370. [DOI] [PubMed] [Google Scholar]
- 39.Agulnik M, Santos GDC, Hedley D, et al. Predictive and pharmacodynamic biomarker studies in tumor and skin tissue samples of patients with recurrent or metastatic squamous cell carcinoma of the head and neck treated with erlotinib. Journal of Clinical Oncology. 2007;25(16):2184–2190. doi: 10.1200/JCO.2006.07.6554. [DOI] [PubMed] [Google Scholar]
- 40.Murray S, Timotheadou E, Linardou H, et al. Mutations of the epidermal growth factor receptor tyrosine kinase domain and associations with clinicopathological features in non-small cell lung cancer patients. Lung Cancer. 2006;52(2):225–233. doi: 10.1016/j.lungcan.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 41.Fountzilas G, Bobos M, Kalogera-Fountzila A, et al. Gemcitabine combined with gefitinib in patients with inoperable or metastatic pancreatic cancer: a phase II study of the Hellenic Cooperative Oncology Group with biomarker evaluation. Cancer Investigation. 2008;26(8):784–793. doi: 10.1080/07357900801918611. [DOI] [PubMed] [Google Scholar]
- 42.Goelz SE, Hamilton SR, Vogelstein B. Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochemical and Biophysical Research Communications. 1985;130(1):118–126. doi: 10.1016/0006-291x(85)90390-0. [DOI] [PubMed] [Google Scholar]
- 43.Travis WD, Colby TV, Corrin B, Shimosato Y, Brambilla E. Histological Typing of Lung and Pleural Tumors. 3rd edition. Berlin, Germany: Springer; 1999. [Google Scholar]
- 44.Murray S, Dahabreh IJ, Linardou H, Manoloukos M, Bafaloukos D, Kosmidis P. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. Journal of Thoracic Oncology. 2008;3(8):832–839. doi: 10.1097/JTO.0b013e31818071f3. [DOI] [PubMed] [Google Scholar]
- 45.Boldrini L, Gisfredi S, Ursino S, et al. Mutational analysis in cytological specimens of advanced lung adenocarcinoma: a sensitive method for molecular diagnosis. Journal of Thoracic Oncology. 2007;2(12):1086–1090. doi: 10.1097/JTO.0b013e31815ba1fa. [DOI] [PubMed] [Google Scholar]
- 46.Kislitsin D, Lerner A, Rennert G, Lev Z. K-ras mutations in sporadic colorectal tumors in Israel: unusual high frequency of codon 13 mutations and evidence for nonhomogeneous representation of mutation subtypes. Digestive Diseases and Sciences. 2002;47(5):1073–1079. doi: 10.1023/a:1015090124153. [DOI] [PubMed] [Google Scholar]
- 47.Kong-Beltran M, Seshagiri S, Zha J, et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Research. 2006;66(1):283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 48.Salvesen HB, Kumar R, Stefansson I, et al. Low frequency of BRAF and CDKN2A mutations in endometrial cancer. International Journal of Cancer. 2005;115(6):930–934. doi: 10.1002/ijc.20702. [DOI] [PubMed] [Google Scholar]
- 49.Fountzilas G, Kalogera-Fountzila A, Lambaki S, et al. MMP9 but Not EGFR, MET, ERCC1, P16, and P-53 is associated with response to concomitant radiotherapy, cetuximab, and weekly cisplatin in patients with locally advanced head and neck cancer. Journal of Oncology. 2009;2009:17 pages. doi: 10.1155/2009/305908. Article ID 305908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. New England Journal of Medicine. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 51.Pirker R, Pereira JR, Szczesna A, et al. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. The Lancet. 2009;373(9674):1525–1531. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 52.Miller CT, Moy JR, Lin L, et al. Gene amplification in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clinical Cancer Research. 2003;9(13):4819–4825. [PubMed] [Google Scholar]
- 53.Al-Kasspooles M, Moore JH, Orringer MB, Beer DG. Amplification and over-expression of the EGFR and erbB-2 genes in human esophageal adenocarcinomas. International Journal of Cancer. 1993;54(2):213–219. doi: 10.1002/ijc.2910540209. [DOI] [PubMed] [Google Scholar]
- 54.Calabrich A, Fernandes GDS, Katz A. Trastuzumab: mechanisms of resistance and therapeutic opportunities. Oncology. 2008;22(11):1250–1258. [PubMed] [Google Scholar]
- 55.Qiu W, Tong GX, Manolidis S, Close LG, Assaad AM, Su GH. Novel mutant-enriched sequencing identified high frequency of PIK3CA mutations in pharyngeal cancer. International Journal of Cancer. 2008;122(5):1189–1194. doi: 10.1002/ijc.23217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang D, Wang TY, Li HC, Wei JC, Song JX. Prognostic significance of PTEN expression in esophageal squamous cell carcinoma from Linzhou City, a high incidence area of northern China. Diseases of the Esophagus. 2007;20(6):491–496. doi: 10.1111/j.1442-2050.2007.00695.x. [DOI] [PubMed] [Google Scholar]