

Abstract
The ribonucleoprotein complex ribonuclease (RNase) MRP is a site-specific endoribonuclease essential for the survival of the eukaryotic cell. RNase MRP closely resembles RNase P (a universal endoribonuclease responsible for the maturation of the 5′ ends of tRNA) but recognizes distinct substrates including pre-rRNA and mRNA. Here we report the results of an in vitro selection of Saccharomyces cerevisiae RNase MRP substrates starting from a pool of random sequences. The results indicate that RNase MRP cleaves single-stranded RNA and is sensitive to sequences in the immediate vicinity of the cleavage site requiring a cytosine at the position +4 relative to the cleavage site. Structural implications of the differences in substrate recognition by RNases P and MRP are discussed.
Keywords: ribonuclease MRP, RNase MRP, Saccharomyces cerevisiae, in vitro selection, ribonucleoprotein
INTRODUCTION
Ribonuclease (RNase) MRP is a universal eukaryotic site-specific endoribonuclease (Chang and Clayton 1987a,b; Karwan et al. 1991) closely related to RNase P, an RNA-based enzyme responsible for the maturation of 5′ ends of tRNA (Altman and Kirsebom 1999). RNase MRP is an essential enzyme (Schmitt and Clayton 1992) found in practically all eukaryotes analyzed (Piccinelli et al. 2005; Rosenblad et al. 2006). RNase MRP is involved in the maturation of rRNA (Schmitt and Clayton 1993; Chu et al. 1994; Lygerou et al. 1994, 1996) and the degradation of a specific mRNA involved in the regulation of the cell cycle (Cai et al. 2002; Gill et al. 2004). A very small fraction of RNase MRP is found in the mitochondria (Chang and Clayton 1987a; Kiss and Filipowicz 1992); however, mitochondrial RNase MRP has a distinct composition and substrate specificity (Lu et al. 2010) and is beyond the scope of this work. Mutations in the RNA component of human RNase MRP result in a variety of pleiotropic diseases (Ridanpaa et al. 2001; Martin and Li 2007 and references therein).
RNase MRP is a ribonucleoprotein with a composition closely resembling that of eukaryotic RNase P (for review, see Esakova and Krasilnikov 2010). In Saccharomyces cerevisiae, RNase MRP contains 10 essential proteins, eight of which are also found in RNase P (Chamberlain et al. 1998, Salinas et al. 2005). The RNA component of RNase P (Fig. 1A) is the enzyme's catalytic moiety (Guerrier-Takada et al. 1983; Pannucci et al. 1999; Thomas et al. 2000; Kikovska et al. 2007); the RNA component of RNase MRP (Fig. 1B) shows clear similarity to that of RNase P. In particular, the catalytic (C-) domain of RNase P (a generally phylogenetically conserved domain that contains the active site) appears to have the same overall architecture as the corresponding part of RNase MRP (Domain 1, Fig. 1A,B; Esakova et al. 2008 and references therein). Several nucleotides that are universally conserved in RNase P are also found in RNase MRP (Lopez et al. 2009). Moreover, the P3 subdomains (Perederina et al. 2010) of the two enzymes (Fig. 1A,B) can be interchanged (Lindahl et al. 2000). The similarity of the C-domain of RNase P to the corresponding part of RNase MRP strongly suggests that the two enzymes use a common mechanism of catalysis.
FIGURE 1.

Secondary structure diagrams. (A) S. cerevisiae RNase P. The catalytic (C-) and specificity (S-) domains are separated by a solid line. (B) S. cerevisiae RNase MRP. Domains 1 and 2 are separated by a solid line. The phylogenetically conserved sequence 5′-GARAR-3′ (R is G or A) (Lopez et al. 2009) is outlined in gray. (C) A putative secondary structure of yeast pre-rRNA internal transcribed spacer 1 near the RNase MRP cleavage site A3. (Arrow) The A3 site. The diagrams are based on Esakova and Krasilnikov (2010).
The specificity (S-) domain of RNase P (Fig. 1A) is responsible for pre-tRNA substrate recognition by this enzyme (for review, see Esakova and Krasilnikov 2010). This domain contains a structurally conserved region that is involved in substrate recognition (Krasilnikov et al. 2003, 2004; Reiter et al. 2010). In general, RNase P does not recognize specific sequences but appears to use recognizable structural features in its substrates (for review, see Kirsebom 2007).
RNase MRP does not have a structural element resembling the specificity domain of RNase P but possesses a distinct element, Domain 2, instead (Fig. 1B). The divergence between the specificity domain in RNase P and the corresponding part in RNase MRP is consistent with distinct specificities of the two closely related enzymes. RNase MRP Domains 2 can vary significantly when enzymes from different organisms are compared; the only phylogenetically conserved part of Domain 2 is a GARAR (where R is a purine nucleotide) sequence element (Fig. 1B; Lopez et al. 2009). The function of this conserved segment has not yet been established.
RNase MRP and RNase P represent a case of two closely related RNA-based enzymes that have developed distinct specificities. While the substrate specificity of RNase P has been well characterized, little was known about substrate selection by nuclear RNase MRP. To help fill this gap, we used a library of random oligonucleotides to perform an in vitro selection of cleavable RNase MRP substrates and identified key substrate elements located in the vicinity of the cleavage site.
RESULTS
In vitro selection of RNase MRP substrates
The RNase MRP holoenzyme was purified from S. cerevisiae using an affinity tag attached to the C terminus of the protein component Rmp1 (see Materials and Methods). The purified holoenzyme contained all known RNase MRP protein components; the RNA component of the purified complex was essentially pure (Supplemental Fig. 1). To select RNase MRP substrates, we used a procedure generally based on the approach previously described in Pan and Uhlenbeck (1992) and Pan (1995). Briefly, a library of deoxyribonucleotides was used to create a pool of circularized RNA molecules that contained a stretch of 68 random nucleotides. The starting number of RNA molecules (1.8 × 1013) was significantly smaller than the number of possible sequences in the 68-nt-long random stretch (8.7 × 1040); thus, practically each RNA molecule was expected to be unique. The pool of these circular RNA molecules was subjected to a cleavage with RNase MRP; the cleavage resulted in the linearization of RNA. The linearized molecules were separated from the remaining circular ones using electrophoresis in a denaturing polyacrylamide gel, and then extracted from the gel and self-ligated using T4 RNA ligase 1. The ligation resulted in a pool of circular RNA molecules similar to the starting pool but enriched in sequences cleavable by RNase MRP. Due to the specificity of T4 RNA ligase 1, only RNA molecules containing 5′-phosphate and 3′-hydroxyl termini (such as the ones produced after RNase P/MRP cleavage but not after typical nonspecific RNA degradation) were re-circularized. Re-circularized RNA was used as a template for reverse transcription, followed by PCR to produce template DNA for the subsequent round of selection. After six rounds of selection, most of the RNA in the pool was cleavable by RNase MRP.
After the sixth round of selection, the pool of final DNAs was inserted into a plasmid vector, which was used to transform Escherichia coli cells. Plasmid DNA from individual colonies was isolated, sequenced, and used to produce RNAs by run-off transcription with T7 RNA polymerase. The resultant individual RNAs were labeled with 32P and subjected to RNase MRP cleavage, followed by the analysis of the cleavage products on nucleotide-resolution polyacrylamide gels. In total, 144 cleavage sites were identified and analyzed (Supplemental Table 1).
Analysis of identified RNase MRP substrates
The analysis of the results of the substrate selection shows that all cleavage sites are located in single-stranded regions of RNA (as judged by the secondary structure modeling with RNAstructure) (Mathews et al. 2004).
More than 80% of all substrates have one of these five sequences in the positions +2 to +4: CUC, UUC, CGC, AUC, or CAC (Fig. 2A,B). When only the strongest 40% of all sites are analyzed (Supplemental Table 1), the distribution of the sequences in these positions changes considerably, with CUC dominating in the strongest substrates (Fig. 2C). All identified substrates have a cytosine located in the position +4 from the cleavage site (Fig. 2A). The position −1 is typically occupied by a pyrimidine, while guanines were selected against at both +1 and −1 (Supplemental Table 1). Positions from −2 to −7 are enriched in uridines (Fig. 2A). An analysis of the sequences of individual substrates shows that the majority of them have U-rich stretches located in this area (Supplemental Table 1).
FIGURE 2.

Sequences of in vitro selected RNase MRP substrates. (A) Consensus sequence logo. (Arrow) RNase MRP cleavage site. (B) Frequencies of the occurrence of different sequences at positions +2 to +4 in all RNase MRP substrates. (C) Frequencies of the occurrence of different sequences at positions +2 to +4 in 40% of the strongest RNase MRP substrates.
To test if the features that are typical to the identified substrates (CUC/UUC/CGC/AUC/CAC in the positions +2 to +4, no guanines in the positions +1 and −1, and a U-rich stretch located 5′ to the cleavage site) are sufficient for RNase MRP cleavage, we tested a series of oligonucleotides with sequences that were random, except for their middle parts where the sequence features of interest were located (e.g., 5′-N19-UUUU-H*H-CUC-N13-3′, where N is a random nucleotide [G, A, U, C] and H is a random mix of A, U, and C; the random nucleotides were used to eliminate a possible influence of the upstream and downstream sequences or secondary structures on the results). Cleavage assays demonstrate that these oligonucleotides are cleaved by RNase MRP at the position indicated by the asterisk, consistent with the results of the substrate selection (Fig. 3).
FIGURE 3.

RNase MRP cleavage of partially randomized sequences. Random sequences were used to eliminate the potential influence of the upstream and downstream sequences or secondary structures on the results. (A) 5′-N19-UUUU-H*H-AUC-N13 -3′. (B) 5′-N19-UUUU-H*H-CUC-N13 -3′. (C) 5′-N19-UUUU-H*H-CGC-N13-3′. In all panels, N is a random nucleotide (G, A, U, C); H is a random mix of A, U, C; asterisk (*) designates the cleavage site (indicated by an arrow in the figure). (Lane 1) Digest with RNase T1 (a marker); (lane 2) alkaline hydrolysis (a marker); (lane 3) untreated RNA substrate; (lanes 4,5) substrates digested with RNase MRP. Substrates were 5′-end-labeled with 32P and separated on a 6% denaturing polyacrylamide gel.
The most salient feature of the identified substrates is the conserved cytosine in the position +4 (Fig. 2A). To test if the presence of a cytosine in the position +4 is required for RNase MRP cleavage, we replaced this cytosine with other nucleobases (G, A, and U). The results of the cleavage assays show that the presence of a cytosine in the position +4 is required for RNase MRP cleavage (Fig. 4).
FIGURE 4.

Cytosine in the position +4 [C(+4)] is required for RNase MRP cleavage (shown by an arrow); a replacement of C(+4) for any other base eliminates RNase MRP cleavage. (Crossed arrows) Positions of expected cleavage. (Lanes 1,6,11,16) Digest with RNase T1 (markers); (lanes 2,7,12,17) alkaline hydrolysis (markers); (lanes 3,8,13,18) untreated RNA substrates; (lanes 4,5) RNase MRP digest of a substrate containing 5′-UUUUACGC-3′ sequence; (lanes 9,10) RNase MRP digest of a substrate containing 5′-UUUUACGG-3′ sequence; (lanes 14,15) RNase MRP digest of a substrate containing 5′-UUUUACGA-3′ sequence; (lanes 19,20) RNase MRP digest of a substrate containing 5′-UUUUACGU-3′ sequence. Substrates were 5′-end-labeled with 32P and separated on a 6% denaturing polyacrylamide gel.
An analysis of identified substrates indicates that guanines in positions +1 and −1 (i.e., immediately flanking the cleavage site) are selected against (Fig. 2A; Supplemental Table 1). To test if the presence of guanines in the immediate vicinity of the cleavage site affects cleavage, we compared RNase MRP cleavage of a substrate that does not have guanines in positions +1 and −1 with that of substrates that have guanines in positions +1 or −1, or both. The results show that while the presence of a guanine next to the cleavage site does not eliminate RNase MRP cleavage, the efficiency of cleavage is considerably reduced (Fig. 5). The presence of guanines at both the +1 and the −1 positions effectively eliminates cleavage (Fig. 5).
FIGURE 5.

Guanines flanking RNase MRP cleavage site reduce the efficiency of cleavage. (Lanes 1,6,11,16) Digest with RNase T1 (markers); (lanes 2,7,12,17) alkaline hydrolysis (markers); (lanes 3,8,13,18) untreated RNA substrates; (lanes 4,5) RNase MRP digest of the substrate 5′-GGN19-UUUUAACUC-N13-3′; (lanes 9,10) RNase MRP digest of the substrate 5′-GGN19-UUUUAGCUC-N13-3′; (lanes 14,15) RNase MRP digest of the substrate 5′-GGN19-UUUUGACUC-N13-3′; (lanes 19,20) RNase MRP digest of the substrate 5′-GGN19-UUUUGGCUC-N13-3′. Substrates were 5′-end-labeled with 32P and separated on a 6% denaturing polyacrylamide gel.
Most of the identified substrates have a U-rich stretch (typically three to five uridines) localized several (typically two to five) nucleotides 5′ to the cleavage site; the localizations, sizes, and compositions of these stretches vary (Supplemental Table 1). Removal of the U-rich stretch does not eliminate RNase MRP cleavage but does result in a reduction in the efficiency of cleavage (data not shown).
All identified RNase MRP cleavage sites are located in regions of RNA that are expected to be single-stranded. To test if RNase MRP can cleave double-stranded RNA, cleavable RNase MRP substrates were extended using a complementary sequence so the resultant molecules contained cleavage site sequences as part of a double-stranded RNA hairpin. RNase MRP was not able to cleave double-stranded RNA (data not shown).
Cleavage of known RNase MRP substrates
RNase MRP is essential for the survival of the eukaryotic cell (Schmitt and Clayton 1992). Thus far, two natural RNase MRP substrates outside the mitochondria have been identified. First, RNase MRP was shown to cleave the 5′ UTR of the CLB2 (Cyclin B2) mRNA (Gill et al. 2004). (RNase MRP, generally a nucleolar enzyme, was shown to transiently accumulate in a discrete cytoplasmic spot where it co-localizes with the CLB2 mRNA [Gill et al. 2006]). In vitro cleavage assays performed on the 5′ UTR of CLB2 mRNA confirm previously reported (Gill et al. 2004) RNase MRP cleavage at sites that are consistent with the identified consensus (Supplemental Fig. 2).
The other known RNase MRP substrate is the internal transcribed spacer 1 (ITS1), which is cleaved by RNase MRP at the A3 site (Fig. 1C; Lygerou et al. 1996). As expected on the basis of the results of the analysis of RNase MRP specificity, mutations of the cytosine located 4 nt 3′ to the A3 site eliminate RNase MRP cleavage (Fig. 6A,B). A hairpin that may potentially fold in the immediate vicinity of the A3 site (Fig. 1C) is not essential for RNase MRP cleavage (Fig. 7).
FIGURE 6.

Cytosine in the position +4 [C(+4)] is required for RNase MRP cleavage at the A3 site. (A) Cleavage of a 56-nt-long substrate containing the A3 site. (Arrow) The location of the cleavage site. (B) Cleavage of the same substrate as in A, but with C(+4) replaced with a random mix of G, A, and U. (Lanes 1,5) Alkaline hydrolysis (markers); (lanes 2,6) untreated RNA substrates; (lanes 3,4,7,8) RNase MRP digests. All substrates were 5′-end-labeled with 32P and separated on a 6% denaturing polyacrylamide gel.
FIGURE 7.

A putative hairpin located 5′ to the A3 site in the internal transcribed spacer 1 of pre-rRNA (Fig. 1C) is not essential for RNase MRP cleavage. (Lanes 1–6) RNase MRP cleavage of an ITS1 fragment encompassing nucleotides −81 to 66 from the A3 site. (Lanes 7–12) RNase MRP cleavage of a short ITS1 fragment encompassing nucleotides −47 to 7 from the A3 site with the putative hairpin (nucleotides −39 to −7) removed. (Lanes 1,7) Alkaline hydrolysis (markers); (lanes 2,8) RNase T1 digests (markers); (lanes 3,9) untreated RNA substrates; (lanes 4,5,10,11) RNase MRP digests; (lanes 6,12) RNase A digests (markers). The mobilities of the short products of RNase MRP digestion differ slightly from those of the products of alkaline hydrolysis due to the presence of additional terminal phosphates in the latter; RNase A digest markers (lanes 6,12) were treated with T4 polynucleotide kinase to eliminate this effect (Brown and Bevilacqua 2005). All substrates were 5′-end-labeled with 32P and separated on a 6% denaturing polyacrylamide gel.
DISCUSSION
The results of the substrate selection combined with cleavage assays indicate that cleavage by RNase MRP occurs in single-stranded regions of RNA and requires the presence of a cytosine at the position +4 from the cleavage site (Fig. 2A). Positions +2, +3 are typically occupied by CU, UU, CG, AU, or CA (Fig. 2B,C), and a presence of a U-rich region 5′ of the cleavage site improves the efficiency of RNase MRP cleavage, while the presence of a guanine at positions +1 or −1 substantially reduces it.
The specificity demonstrated by RNase MRP in vitro appears to be surprisingly broad. Several explanations can be suggested to reconcile the discrepancy between the abundance of sequences cleavable by RNase MRP in vitro and the apparent rarity of cleavage sites in vivo.
It should be noted that a large fraction of the potential RNase MRP cleavage sites on pre-rRNA is inaccessible to RNase MRP cleavage because of the formation of secondary structures (both in the transcribed spacers and, especially, in the regions corresponding to the mature rRNAs). The binding of ribosomal proteins or factors governing the ribosomal synthesis is likely to further reduce the number of pre-rRNA sites accessible to RNase MRP cleavage in vivo. In general, the relative rarity of “naked” single-stranded RNA in the cell should substantially reduce the number of sites available for RNase MRP cleavage, albeit not necessarily to the extent required to explain the apparent rarity of actually observed sites in vivo.
If RNase MRP has broad substrate specificity in vivo, the cell must employ additional mechanisms to direct RNase MRP cleavage. One of the likely mechanisms might involve the localization of RNase MRP, which could be actively governed by its interactions with other elements of cellular machinery to co-localize the enzyme with its proper substrates such as pre-rRNA. Such co-localization of RNase MRP with proper substrates, when combined with the restrictions imposed by the secondary structure of RNA and the presence of bound proteins, could dramatically reduce the number of in vivo cleavage sites even with the inherently broad specificity of the enzyme itself. The existence of additional “specificity factors” that can interact with RNase MRP in vivo and serve to direct the enzyme to its proper cleavage sites is also a possibility. Indeed, effects of the depletion of Rrp5, an essential nucleolar protein involved in the maturation of rRNA (Venema and Tollervey 1996), are similar to those of the inactivation of RNase MRP (Venema and Tollervey 1996; Lindahl et al. 2009).
While the purified enzyme contained all known essential RNase MRP components (Supplemental Fig. 1), the potential presence of a fraction of the molecules that is missing one or more components could, in principle, result in a broadened specificity. The presence of such a fraction could be the result of a partial component loss during RNase MRP purification (which does not seem to be likely since RNase MRP proved to be very stable even in elevated salt or in the presence of moderate concentrations of denaturants [O Esakova and AS Krasilnikov, unpubl.]), or, more likely, could result from the existence of several subpopulations of RNase MRP in the cell. The latter seems to be a plausible explanation considering that human RNase MRP holoenzymes were suggested to exist in at least two forms that differed in their protein compositions (Welting et al. 2006). The different forms may play a biological role: A change in the composition of RNase MRP could, among other things, result in different specificities in different cellular compartments and/or during different phases of the cell cycle. (RNase MRP was shown to participate in the regulation of the cell cycle [Cai et al. 2002; Gill et al. 2004, 2006].) The potential presence of more than one form of RNase MRP could in principle result in the broadening of the specificity of the purified enzyme; however, the existence of different forms of RNase MRP in yeast and the effects of the potential changes in the composition on RNase MRP specificity need further studies.
The identification of potential “specificity factors” (whether they are novel factors transiently associated with RNase MRP in vivo or bona fide RNase MRP components) would be interesting, especially in light of recent data indicating that the involvement of RNase MRP in the processing of pre-rRNA may go beyond the known cleavage at the A3 site (Lindahl et al. 2009; Schneider et al. 2010).
In addition, it cannot be excluded that RNase MRP interacts with more than one region of its substrate in vivo. It should be noted that pre-tRNA recognition by the related RNase P holoenzyme involves several parts of the substrate, each part contributing to recognition (for review, see Kirsebom 2007; Esakova and Krasilnikov 2010). Given a sufficient sequence or structural complexity of the putative additional parts of the substrate, they could potentially be missed in our in vitro selection.
Domain 1 of RNase MRP and the catalytic domain of the closely related RNase P have multiple common features (Fig. 1A,B) and are likely to have similar structural organizations (Esakova et al. 2008 and references therein). Substrate recognition by RNase MRP, however, appears to be markedly different from pre-tRNA recognition by RNase P. Eukaryotic RNase P does not recognize specific sequences in its pre-tRNA substrates, but rather uses conserved structural features of the substrates for recognition (Esakova and Krasilnikov 2010 and references therein). The key conserved structural features used by RNase P for its recognition of pre-tRNA (T- and D-loops) (Torres-Larios et al. 2005, 2006; Reiter et al. 2010) are located at a considerable distance from the cleavage site. As a result, the RNase P domain responsible for substrate recognition (the specificity domain) (Fig. 1A; for review, see Esakova and Krasilnikov 2010) is positioned away from the catalytic domain (Torres-Larios et al. 2005, 2006; Reiter et al. 2010).
In contrast, RNase MRP proved to be sensitive to sequence features of its substrates that are located in the immediate vicinity of the cleavage site. This suggests that elements of RNase MRP responsible for substrate recognition (most likely, parts of Domain 2) are positioned in the immediate vicinity of the catalytic site, folding back into the catalytic domain and forming an overall more compact (perhaps less flat) structure than RNase P. The only phylogenetically conserved element in Domain 2 of RNase MRP, the GARAR sequence (Fig. 1B), is positioned close enough to the putative catalytic center to fulfill the role of the element folding back to the catalytic domain and interacting with the substrate. This hypothesis is currently under investigation.
MATERIALS AND METHODS
Yeast strains
The S. cerevisiae strain OE1004 (MATa RMP1∷TAPHIS8∷TRP1 sep 1∷URA3 pep4∷LEU2 nuc1∷LEU2 ade2-1 trp1-1 his3-11,15 can1-100 ura3-1 leu2-3,112) used for RNase MRP purification was constructed using strain LSY389-34A (MATa sep 1∷URA3 pep4∷LEU2 nuc1∷LEU2 ade2-1 trp1-1 his3-11,15 can1-100 ura3-1 leu2-3,112; a generous gift from Mark Schmitt) (Salinas et al. 2005) as the starting point. The affinity tag fused to the C-terminus of the Rmp1 gene (Salinas et al. 2005) was generated using PCR-based genomic tagging. To generate the tag, the plasmid pBS1479, which contains the TAP fusion cassette (Rigaut et al. 1999), was amplified using primers Rmp1-HisIg-A (5′-AAAGGAAAAAGAAGAACAAATCAGCCATTGATGGCATATTCGGACATCACCATCATCATCACCATCATGATTATGATATTCCAACTACTG-3′) and Rmp1-HisIg-B (5′-TACTTGGGCAGACAAGGTCATTAATGAATGATTTATTAGCATTTACGACTCACTATAGGG-3′). The resultant tag TAPHIS8 was similar to the standard TAP tag (Rigaut et al. 1999), but with eight histidine residues replacing the calmodulin-binding fragment. The genomic sequence of the Rmp1 gene with the fused tag was confirmed by sequencing. The insertion of the tag did not affect 5.8S rRNA processing in vivo (Fig. 8).
FIGURE 8.
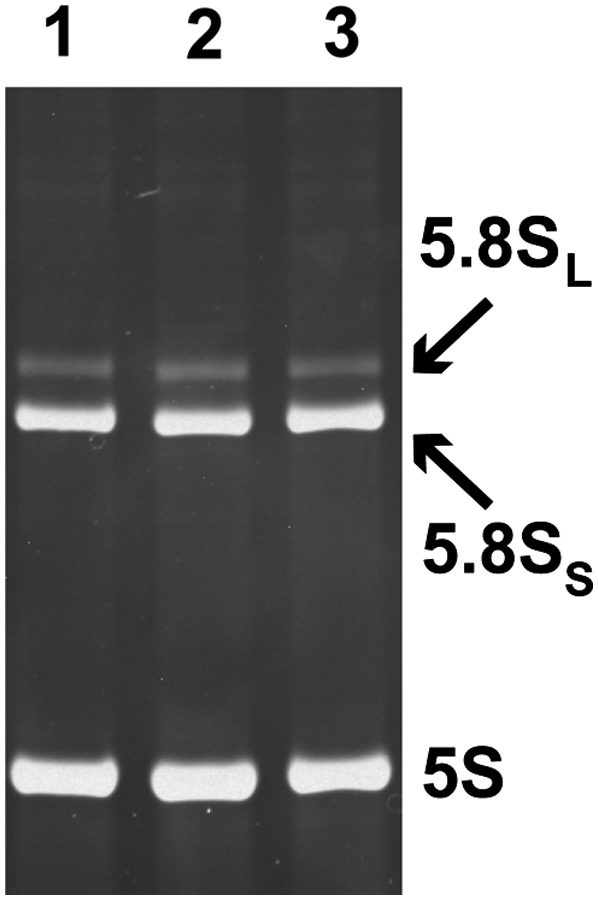
The insertion of the affinity tag does not affect 5.8S rRNA processing by RNase MRP in vivo. Ethidium bromide–stained total RNA separated on an 8% denaturing (8 M urea) polyacrylamide gel. (Lane 1) Wild-type yeast; (lane 2) the affinity tag fused to the C terminus of Pop4; (lane 3) the affinity tag fused to the C terminus of Rmp1.
RNase MRP purification
RNase MRP holoenzyme was purified from S. cerevisiae strain OE1004 (above) using a tandem affinity tag attached to the C terminus of RNase MRP protein component Rmp1. Rmp1 (Salinas et al. 2005) is not found in RNase P, which eliminated the possibility of the contamination of purified RNase MRP with this closely related ribonucleoprotein complex. The tandem affinity tag was based on the commonly used TAP tag (Rigaut et al. 1999), but the original calmodulin-binding fragment was replaced with a His8 tag to avoid depletion of magnesium during RNase MRP purification.
The purification procedure was generally based on one previously described (Gill et al. 2004), with modifications aimed to maintain the presence of magnesium at all purification steps and reflecting the change in the composition of the purification tag.
Sixteen liters of yeast was grown at 30°C with vigorous aeration on YPD media to the late logarithmic phase. The culture was cooled on ice; the cells (∼150 g) were harvested by centrifugation at 4000g (4°C), washed with water, and resuspended in a buffer containing 20 mM Tris-HCl (pH 7.9), 150 mM KCl, 1 mM Mg-Acetate, 10% glycerol, 1 mM PMSF, and 0.1 mM EDTA. The cells were disrupted using a BeadBeater (Biospec) (15 × 15-sec pulses on ice), then Tween 20 was added to 0.1% (v/v), and the extract was clarified by centrifugation at 17,000g for 10 min (4°C) followed by ultracentrifugation at 100,000g for 3 h (4°C). The clarified extract was mixed with 3 mL of rabbit IgG agarose (Sigma) and incubated for 5 h at 4°C with light agitation. The IgG agarose was washed six times with 5 volumes of the buffer containing 20 mM Tris-HCl (pH 7.9), 150 mM KCl, 1 mM Mg-Acetate, 10% glycerol, 1 mM PMSF, 0.1 mM EDTA, and 0.1% (v/v) Tween 20 (Buffer A), and resuspended in 2 mL of the same buffer. Then 300 units of tobacco etch virus protease (TEV) were added and the sample was incubated for 12 h at 4°C with light agitation. The resin was pelleted by centrifugation at 500g for 5 min (4°C), and the supernatant was collected; the resin was additionally washed twice with 5 mL of Buffer A. The three fractions of the supernatant were combined; the buffer was exchanged for Buffer B (20 mM Na-HEPES at pH 7.9, 150 mM KCl, 1 mM Mg-Acetate, 10% glycerol, 1 mM PMSF, 0.1 mM EDTA, 0.1% [v/v] Tween 20) and concentrated to the final volume of 2 mL using an Amicon-Ultra 15 (100 kDa MWCO) concentrator (Millipore). The sample was incubated with 0.5 mL of Ni-NTA Agarose (QIAGEN) for 5 h at 4°C with light agitation. The resin was washed six times with 10 mL of Buffer A supplemented with 10 mM Na-imidazole (pH 7.4). After the final wash, the resin was resuspended in a buffer containing 10 mL of 400 mM Na-imidazole (pH 7.4), 50 mM KCl, 1 mM Mg-Acetate, 10% glycerol, 1 mM PMSF, and 0.1% (v/v) Tween 20, and RNase MRP was eluted for 25 min at 4°C with light agitation. After elution, the elution buffer was exchanged for a buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM KCl, 1 mM Mg-Acetate, 5 mM DTT, and 0.1% (v/v) Tween 20 (Buffer C), and concentrated using an Amicon-Ultra 4 (100 kDa MWCO) concentrator (Millipore). The enzyme was immediately used or transferred into Buffer C supplemented with 50% (v/v) glycerol and stored at −20°C.
Selection of RNase MRP substrates
The selection procedure was based on Pan and Uhlenbeck (1992) and Pan (1995) with modifications. During the first round of substrate selection, to prepare DNA templates for the generation of the initial random RNA library, 30 pmol of synthetic template 5′-GGAAGTCGGCTCTAG-(N68)-CTTCACGATTTCAATAC-3′ (where N was randomized G, A, U, C) was amplified using four cycles of PCR with 0.2 nmol of primers 5′-TAATACGACTCACTATAGGAAGTCGGCTCTAG-3′ (the introduced T7 RNA polymerase promoter sequence is underscored) and 5′-GTATTGAAATCGTGAAG-3′.
The resultant double-stranded DNA was extracted with phenol and used for run-off transcription with T7 RNA polymerase (0.1 μg/mL) in 40 mM Tris-HCl (pH 8.1), 6 mM MgCl2, 5mM DTT, 1 mM spermidine, 50 μg/mL BSA, 1 mM ATP, 1 mM UTP, 1 mM CTP, 1 mM GTP, 5 mM GMP, 100 nM DNA template, and 300 nM of oligonucleotide 5′-TAATACGACTCACTATAGG-3′ for 2.5 h at 37°C. The same procedure was used to generate RNA from double-stranded DNA obtained in all subsequent rounds of substrate selection.
The resulting RNA was purified on a 10% denaturing (8 M urea) polyacrylamide gel; extracted from the gel; dissolved in a buffer containing 50 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 10 mM DTT, 12 μM ATP; incubated for 3 min at 83°C, and cooled on ice. DMSO (to 15% [v/v]) and T4 RNA ligase 1 (1 U/μL) were added to the mixture, and ligation was run for 2 h at 37°C to generate circularized RNA. The resultant circularized RNA was purified using 10% denaturing (8 M urea) polyacrylamide gel. The mobility of circularized RNA on the 10% denaturing gel was significantly (about threefold) lower than that of linear RNA, ensuring a reliable separation of the two bands. Following the extraction from the gel, the RNA was incubated for 2 min at 85°C in 50 mM Tris-HCl (pH 8.1), and for 10 min at room temperature. Following incubation, MgCl2 was added to 10 mM, and the sample was incubated for an additional 5 min at 22°C and for 5 min at 37°C.
RNase MRP cleavage was performed for 30 min at 30°C in 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM DTT, 50 mM KCl, 50 μg/mL BSA, and 70 nM purified RNase MRP in the presence of SUPERase·In RNase inhibitor (Ambion). The reaction was stopped by the addition of an equal volume of 9 M urea.
Linearized RNA was separated from uncut circular RNA on a 10% denaturing (8 M urea) polyacrylamide gel. During the four initial cycles of selection, linearized RNA was not visible and its position was estimated using a linear 100-nt-long RNA marker (of an unrelated sequence) run alongside on the same gel; RNA was extracted from an ∼15-mm slice of the gel centered at the position of the marker. The approximately threefold difference in the mobility of circularized and linear RNA ensured a reliable separation of the two. A faint band corresponding to the linearized RNA appeared in the fifth round of substrate selection. The linearized RNA (enriched in sequences cleavable by RNase MRP) was extracted from the gel, resuspended in the buffer for ligation with T4 RNA ligase 1 (above), incubated for 3 min at 83°C, and cooled on ice. The RNA was self-ligated using T4 RNA ligase 1 in the presence of 15% (v/v) DMSO as described above. Following ligation, the RNA was extracted with phenol.
Extracted RNA was used to generate a library of double-stranded DNA similar to the initial one, but enriched with sequences cleavable by RNase MRP using reverse transcription followed by PCR amplification. RNA was resuspended in the 15 μL of 5 mM Tris-HCl (pH 8.0), 10 mM KCl, and 0.1 mM EDTA, and annealed to a primer (5′-GTATTGAAATCGTGAAG-3′; 1 μM) by heating for 3 min to 83°C followed by incubation for 15 min on ice. A reverse transcription reaction was performed in 20 μL of 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 5 mM MgCl2, 1.5 mM DTT, 1 mM of each dNTPs, and 40 units of SuperScript II reverse transcriptase (Invitrogen) in the presence of the SUPERase·In RNase inhibitor (Ambion) for 15 min at 47°C. PCR amplification was performed with the same primers that were used in the generation of the initial double-stranded DNA template (above). After 27 cycles of amplification, the PCR products were extracted with phenol and used in the next round of substrate selection.
Substrate selection was run as described above for six rounds; most of the RNA substrates generated in the sixth round were cleavable by RNase MRP. Double-stranded DNAs generated at the end of the last selection round were cloned into the Sma I site of the pUC19 plasmid; the resulting plasmids were used to transform E. coli cells. One hundred and twenty individual colonies were randomly picked and used to sequence plasmid DNA.
Analysis of RNase MRP cleavage sites
Plasmid DNA was amplified by PCR using the same primers as described above; RNA was synthesized using standard run-off transcription with T7 RNA polymerase (Milligan and Uhlenbeck 1989) followed by purification on 15% denaturing (8 M urea) polyacrylamide gel. The resulting RNA was dephosphorylated with alkaline phosphatase and 5′-end 32P labeled with T4 polynucleotide kinase. Labeled RNA was gel-purified again and used in RNase MRP cleavage assays performed in 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, and 50 μg/mL BSA at 37°C in the presence of SUPERase·In RNase inhibitor (Ambion).
To analyze RNase MRP cleavage of the CLB2 mRNA substrate, the substrate was synthesized using standard run-off transcription with SP6 RNA polymerase. Plasmid pJA108 (a generous gift from Mark Schmitt) (Gill et al. 2004) linearized with EcoRI was used as the template for in vitro transcription. RNase MRP cleavage assays were performed as described above. Cleavage products were analyzed by primer extension using primers RT270-1 (5′-ACAATGATTAAAATTTCTCC-3′) and RT270-2 (5′-CAAAAGGAAAACAGATGCTC-3′) following the protocol that was previously described (Esakova et al. 2008); the same primers annealed to plasmid pJA108 were used to obtain sequencing ladders.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank Phil Bevilacqua, Joe Reese, and Mark Schmitt for valuable comments and suggestions. We thank the staff of the Proteomics Core Facility at the Huck Institute of Life Sciences (PSU) for their help with mass spectrometry analysis. This work was supported by NIH Grant GM085149 to A.S.K.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2393711.
REFERENCES
- Altman S, Kirsebom L 1999. Ribonuclease P. In The RNA world, 2nd ed (ed. Gesteland RF, et al. ), pp. 351–380 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Brown TS, Bevilacqua PC 2005. Method for assigning double-stranded RNA structures. Biotechniques 38: 368–372 [DOI] [PubMed] [Google Scholar]
- Cai T, Aulds J, Gill T, Cerio M, Schmitt ME 2002. The Saccharomyces cerevisiae RNase mitochondrial RNA processing is critical for cell cycle progression at the end of mitosis. Genetics 161: 1029–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JR, Lee Y, Lane WS, Engelke DR 1998. Purification and characterization of the nuclear RNase P holoenzyme complex reveals extensive subunit overlap with RNase MRP. Genes Dev 12: 1678–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA 1987a. A novel endoribonuclease cleaves at a priming site of mouse mitochondrial DNA replication. EMBO J 6: 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DD, Clayton DA 1987b. A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science 235: 1178–1184 [DOI] [PubMed] [Google Scholar]
- Chu S, Archer RH, Zengel JM, Lindahl L 1994. The RNA of RNase MRP is required for normal processing of ribosomal RNA. Proc Natl Acad Sci 91: 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Krasilnikov AS 2010. Of proteins and RNA: The RNase P/MRP family. RNA 16: 1725–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O, Perederina A, Quan C, Schmitt ME, Krasilnikov AS 2008. Footprinting analysis demonstrates extensive similarity between eukaryotic RNase P and RNase MRP holoenzymes. RNA 14: 1558–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T, Cai T, Aulds J, Wierzbicki S, Schmitt ME 2004. RNase MRP cleaves the CLB2 mRNA to promote cell cycle progression: Novel method of mRNA degradation. Mol Cell Biol 24: 945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill T, Aulds J, Schmitt ME 2006. A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae. J Cell Biol 173: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Karwan R, Bennett JL, Clayton DA 1991. Nuclear RNase MRP processes RNA at multiple discrete sites: interaction with an upstream G box is required for subsequent downstream cleavages. Genes Dev 5: 1264–1276 [DOI] [PubMed] [Google Scholar]
- Kikovska E, Svard SG, Kirsebom LA 2007. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc Natl Acad Sci 104: 2062–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsebom LA 2007. RNase P RNA-mediated cleavage: Substrate recognition and catalysis. Biochimie 89: 1183–1194 [DOI] [PubMed] [Google Scholar]
- Kiss T, Filipowicz W 1992. Evidence against a mitochondrial location of the 7-2/MRP RNA in mammalian cells. Cell 70: 11–16 [DOI] [PubMed] [Google Scholar]
- Krasilnikov AS, Yang X, Pan T, Mondragon A 2003. Crystal structure of the specificity domain of ribonuclease P. Nature 421: 760–764 [DOI] [PubMed] [Google Scholar]
- Krasilnikov AS, Xiao Y, Pan T, Mondragon A 2004. Basis for structural diversity in homologous RNAs. Science 306: 104–107 [DOI] [PubMed] [Google Scholar]
- Lindahl L, Fretz S, Epps N, Zengel JM 2000. Functional equivalence of hairpins in the RNA subunits of RNase MRP and RNase P in Saccharomyces cerevisiae. RNA 6: 653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl L, Bommankanti A, Li X, Hayden L, Jones A, Khan M, Oni T, Zengel JM 2009. RNase MRP is required for entry of 35S precursor rRNA into the canonical processing pathway. RNA 15: 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MD, Rosenblad MA, Samuelsson T 2009. Conserved and variable domains of RNase MRP RNA. RNA Biol 6: 208–220 [DOI] [PubMed] [Google Scholar]
- Lu Q, Wierzbicki S, Krasilnikov AS, Schmitt ME 2010. Comparison of mitochondrial and nucleolar RNase MRP reveals identical RNA components with distinct enzymatic activities and protein components. RNA 16: 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z, Mitchell P, Petfalski E, Seraphin B, Tollervey D 1994. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev 8: 1423–1433 [DOI] [PubMed] [Google Scholar]
- Lygerou Z, Allmang C, Tollervey D, Seraphin B 1996. Accurate processing of a eukaryotic precursor ribosomal RNA by Ribonuclease MRP in vitro. Science 272: 268–270 [DOI] [PubMed] [Google Scholar]
- Martin AN, Li Y 2007. RNase MRP RNA and human genetic diseases. Cell Res 17: 219–226 [DOI] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci 101: 7287–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62 [DOI] [PubMed] [Google Scholar]
- Pan T 1995. Novel RNA substrates for the ribozyme from Bacillus subtilis Ribonuclease P identified by in vitro selection. Biochemistry 34: 8458–8464 [DOI] [PubMed] [Google Scholar]
- Pan T, Uhlenbeck OC 1992. In vitro selection of RNAs that undergo autolytic cleavage with Pb2+. Biochemistry 31: 3887–3895 [DOI] [PubMed] [Google Scholar]
- Pannucci JA, Haas ES, Hall TA, Harris JK, Brown JW 1999. RNase P RNAs from some Archaea are catalytically active. Proc Natl Acad Sci 96: 7803–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perederina A, Esakova O, Quan C, Khanova E, Krasilnikov AS 2010. Eukaryotic ribonucleases P/MRP: the crystal structure of the P3 domain. EMBO J 29: 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccinelli P, Rosenblad MA, Samuelsson T 2005. Identification and analysis of Ribonuclease P and MRP RNA in a broad range of eukaryotes. Nucleic Acids Res 33: 4485–4495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragon A 2010. Structure of a bacterial ribonuclease P in complex with tRNA. Nature 468: 784–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridanpaa M, Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, vanVenrooij W, Pruijn G, Salmela R, Rockas S, et al. 2001. Mutations in the RNA component of RNase MRP cause a pleiotropic human disease, Cartilage-Hair Hypoplasia. Cell 104: 195–203 [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Rosenblad MA, Lopez MD, Piccinelli P, Samuelsson T 2006. Inventory and analysis of the protein subunits of the ribonucleases P and MRP provides further evidence of homology between the yeast and human enzymes. Nucleic Acids Res 34: 5145–5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas K, Wierzbicki S, Zhou L, Schmitt ME 2005. Characterization and purification of Saccharomyces cerevisiae RNase MRP reveals a new unique protein component. J Biol Chem 280: 11352–11360 [DOI] [PubMed] [Google Scholar]
- Schmitt ME, Clayton DA 1992. Yeast site-specific ribonucleoprotein endoribonuclease MRP contains an RNA component homologous to mammalian RNase MRP RNA and essential for cell viability. Genes Dev 6: 1975–1985 [DOI] [PubMed] [Google Scholar]
- Schmitt ME, Clayton DA 1993. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol Cell Biol 13: 7935–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MD, Bains AK, Rajendra TK, Dominski Z, Matera AG, Simmons AJ 2010. Functional characterization of the Drosophila MRP (mitochondrial RNA processing) RNA gene. RNA 16: 2120–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Chamberlain J, Engelke DR, Gegenheimer P 2000. Evidence for an RNA-based catalytic mechanism in eukaryotic nuclear ribonuclease P. RNA 6: 554–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Larios A, Swinger KK, Krasilnikov AS, Pan T, Mondragon A 2005. Crystal structure of the RNA component of bacterial ribonuclease P. Nature 437: 584–587 [DOI] [PubMed] [Google Scholar]
- Torres-Larios A, Swinger KK, Pan T, Mondragon A 2006. Structure of ribonuclease P—a universal ribozyme. Curr Opin Struct Biol 16: 327–335 [DOI] [PubMed] [Google Scholar]
- Venema J, Tollervey D 1996. RRP5 is required for formation of both 18S and 5.8S rRNA in yeast. EMBO J 15: 5701–5714 [PMC free article] [PubMed] [Google Scholar]
- Welting TJM, Kikkert BJ, van Venrooij WJ, Pruijn GJM 2006. Differential association of protein subunits with the human RNase MRP and RNase P complexes. RNA 12: 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]