

Abstract
Like other forms of engineering, metabolic engineering requires knowledge of the components (the ‘parts list’) of the target system. Lack of such knowledge impairs both rational engineering design and diagnosis of the reasons for failures; it also poses problems for the related field of metabolic reconstruction, which uses a cell’s parts list to recreate its metabolic activities in silico. Despite spectacular progress in genome sequencing, the parts lists for most organisms that we seek to manipulate remain highly incomplete, due to the dual problem of ‘unknown’ proteins and ‘orphan’ enzymes. The former are all the proteins deduced from genome sequence that have no known function, and the latter are all the enzymes described in the literature (and often catalogued in the EC database) for which no corresponding gene has been reported. Unknown proteins constitute up to about half of the proteins in prokaryotic genomes, and much more than this in higher plants and animals. Orphan enzymes make up more than a third of the EC database. Attacking the ‘missing parts list’ problem is accordingly one of the great challenges for post-genomic biology, and a tremendous opportunity to discover new facets of life’s machinery. Success will require a co-ordinated community-wide attack, sustained over years. In this attack, comparative genomics is probably the single most effective strategy, for it can reliably predict functions for unknown proteins and genes for orphan enzymes. Furthermore, it is cost-efficient and increasingly straightforward to deploy owing to a proliferation of databases and associated tools.
Keywords: comparative genomics, metabolic reconstruction, orphan enzyme, pathway hole, unknown protein
INTRODUCTION
Metabolic engineering, the targeted manipulation of pathways and transporters using recombinant DNA, is a fairly mature technology for micro-organisms [1] and a maturing one for plants [2-4]. However, its potential is often limited by ignorance of the components of the target metabolic network, i.e. the system’s ‘parts list’. In many cases, this ignorance extends to the core components of target pathways, such as metabolite transporters in plants [5,6], but even more often it involves not knowing what else besides the core components is ‘out there’ in the system. An illustration of this is that engineering the lysine biosynthesis pathway in plants uncovered a previously unknown lysine catabolism enzyme as a key component of the lysine network [7].
Ignorance of parts lists also limits the effectiveness of metabolic reconstructions [8-10] (using the genome sequence to reproduce an organism’s complete metabolic network in silico) by giving rise to metabolic gaps [11] or ‘missing network content’ [12]. Besides its many applications in metabolic engineering [13], metabolic reconstruction is being increasingly used to explore the interaction of microbes with their environments [14,15] and to understand pathogen function inside and outside the host [16-18]. But if the parts list of proteins in the genome is highly incomplete, as it often is, an organism’s capabilities will inevitably be underestimated. Similarly, in biomedicine, proteomic and transcriptomic screens for disease states have uncovered many unknowns (biomarkers) that correlate with these states [19,20]. But going from there to mechanistic understanding and rational drug therapies demands knowledge of function [21,22].
It is therefore clearly crucial to know organisms’ metabolic parts lists, i.e. to assign functions to all of the proteins associated with metabolism. But we are still far from this goal, and the gap between the richness of genomic information and our knowledge of protein function is, in a certain sense, actually growing. Because this ‘unknown’ protein problem has not had coverage commensurate with its importance, the first section of this review documents its scope. Because one of the most powerful ways of attacking the problem, i.e. comparative genomics (taken to mean the integrated analysis of genomes and post-genomic data), is still underutilized by biochemists, the second section outlines the principles whereby comparative genomics can predict functions for unknown proteins. The last section illustrates application of these principles using as examples enzymes that bacteria and eukaryotes have in common.
‘UNKNOWN’ PROTEINS: THE ELEPHANT IN THE ROOM
The scope of the unknown protein problem
The large-scale sequencing of genomes has revealed that 30–40% of the proteins encoded by typical bacterial genomes have no clearly known function [8,23]. Moreover, many of the ‘known’ functions may be uncertain inasmuch as they are unsupported by experimental evidence; even in an organism as well studied as Escherichia coli, there is experimental information for only 54% of the gene products [24]. The prevalence of unknowns is even greater in archaeal and eukaryotic genomes, and is well over 50% in higher plants and animals [8,24-26] (Figure 1A).
Figure 1. Scale and relentless growth of the unknown protein and orphan enzyme problems.
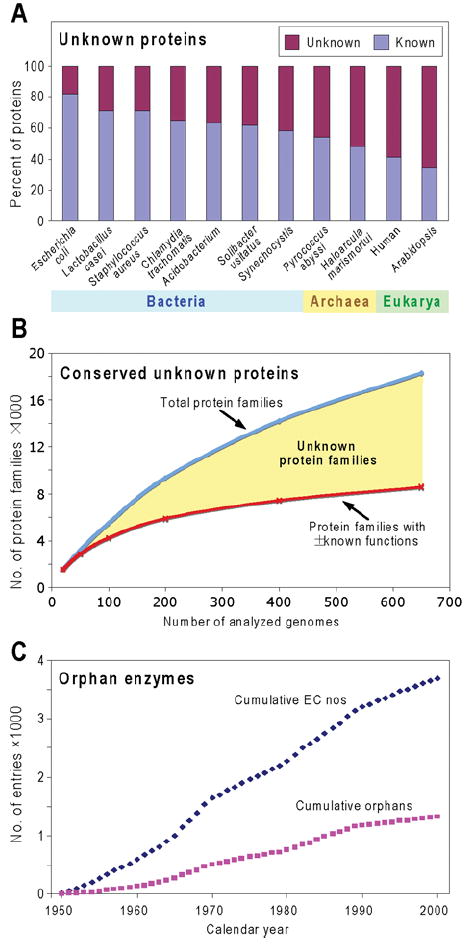
(A) The percentages of known and unknown proteins encoded by representative genomes. The numbers of known proteins were estimated from the SEED database by summing protein-encoding genes included in subsystems and those with non-hypothetical functions not in subsystems. Since this assumes that all proteins in subsystems have known functions, and some such functions are merely reasonable hypotheses, this gives a generous estimate of known proteins. (B) A qualitative sketch of the relationship between the number of conserved unknown proteins and the number of genomes sequenced, from an exploratory analysis by R. Overbeek and A. L. Osterman (personal communication). The SEED database was used to estimate the number of protein families (corresponding roughly to orthologues) comprising at least five members from genomes representing two or more genera (thereby excluding very local families). A jackknife approach was then used to compute an average number of families (blue curve) in bundles (‘runs’) of progressively increased size from 20 genomes up to 650 genomes per run. The lower curve in red shows the number of families having at least some elements of function assigned (i.e. at least a general function such as ‘sugar kinase’ deduced from homology). Note again the generosity of this estimate of the number of proteins that have a known function. The yellow area between the curves represents the number of unknown families. (C) Cumulative total numbers of biochemical activities (EC numbers) characterized between 1950 and 2000, and those that are still orphans. Data are derived from Figure 1 of [33].
Thus with ~ 1000 genomes now completed, if a conservative average of 3000 genes per genome is assumed, it follows that today’s databases contain ~ 106 unknown proteins. Some of these are organism-specific (so-called ‘ORFans’ [8,27]), but the vast majority belong to unknown orthologue families, of which there are thousands [8,28]. Furthermore, as more genomes are sequenced, more protein families are found (Figure 1B, blue line) and only a minority of them have known or partially known functions (red line). Of course, only a fraction of unknown protein families are associated with metabolism (as enzymes, transporters or regulators). But there is reason to think that it is a significant fraction given (i) the prevalence of gaps in known metabolic networks [12], (ii) the fact that new metabolic functions continue to be discovered even in well-characterized organisms such as E. coli [29], and (iii) the many cases where the same pathway step turns out to be mediated by totally different proteins in different organisms (‘non-orthologous displacement’) [30].
The reverse side of the unknown protein problem is that some 36% of the 3736 enzymes with an EC number have no matching protein or gene sequences; these have been termed ‘orphan enzymes’ [9,31-33] and are listed in the ORENZA and ADOMETA databases (Table 1). Since only 60–80%of enzymes have EC numbers [9], this implies that there are ~ 1900 orphan enzymes in total. Like unknown protein families, the number of orphan enzymes is growing (Figure 1C).
Table 1.
Publicly available databases and analysis platforms for comparative genomics research
The dual problem of proteins with no matching function and biochemical functions with no matching protein is thus a huge one. Making these matches presents one of the most urgent challenges of the post-genomic era; it can only be met by community-wide mobilization [8,34,35].
Ubiquitous unknowns: the top targets
As noted above, most unknown proteins belong to orthologue families that occur in a range of genomes. In some cases, this range is extremely broad, and includes most or even all forms of life from bacteria and archaea to higher plants and mammals [8,28]. There are many such widely distributed (henceforth ‘ubiquitous’) proteins, as shown by the OrthoMCL database of orthologous protein families [36]. For instance, most bacteria share >400 orthologue families with Arabidopsis and humans; of these, about half lack known functions. Ubiquitous proteins are plainly ancient in origin [37] and must have crucial functions in metabolism, transport or core cellular processes such as translation that are shared by all organisms [8,10]. Thus, among all the families of unknown proteins, the ubiquitous ones merit the highest priority for functional characterization because they have the greatest potential payoff in new biological knowledge [8,10]. Fortunately, they are also the best targets for comparative genomics approaches, as we now discuss.
THE PREDICTIVE POWER OF COMPARATIVE GENOMICS
Beyond homology-based predictions
Homology-based approaches to predicting function, from pairwise sequence comparisons [38] to fold-recognition algorithms [39], obviously only work when at least one of the orthologues in a family has an experimentally verified function. Although long-range homology can sometimes correctly place unknown proteins in a general class (e.g. ‘esterase’), assigning a precise function calls for approaches that go beyond homology. Enter comparative genomics. Broadly defined, comparative genomics is the integration of different types of genomic and post-genomic evidence to link protein with function. It began in the late 1990s just after the first set of genomes was sequenced. Ten years later, the success stories are now plentiful and several reviews have covered both techniques and specific examples (e.g. [40-43]).
The ‘guilt by association’ principle
The basic principle by which comparative genomics predicts functions is ‘guilt by association’: it finds associations between known and unknown genes in sequenced genomes, and deduces probable functions from these associations [44]. A familiar example is the grouping of bacterial genes into operons, in which the genes encode related functions such as steps in a metabolic pathway. In this case, the function of an unknown gene can be inferred from those of known genes in the operon. Many other types of associations besides operonic arrangements can be derived from whole-genome datasets and their attendant post-genomic resources [41,45-47]. These are summarized in Figure 2 and briefly described below, along with relevant databases and tools (which are listed with their URLs in Table 1). The databases listed are primarily for bacteria and plants, reflecting the authors’ expertise.
Figure 2. Types of associations in comparative genomics.
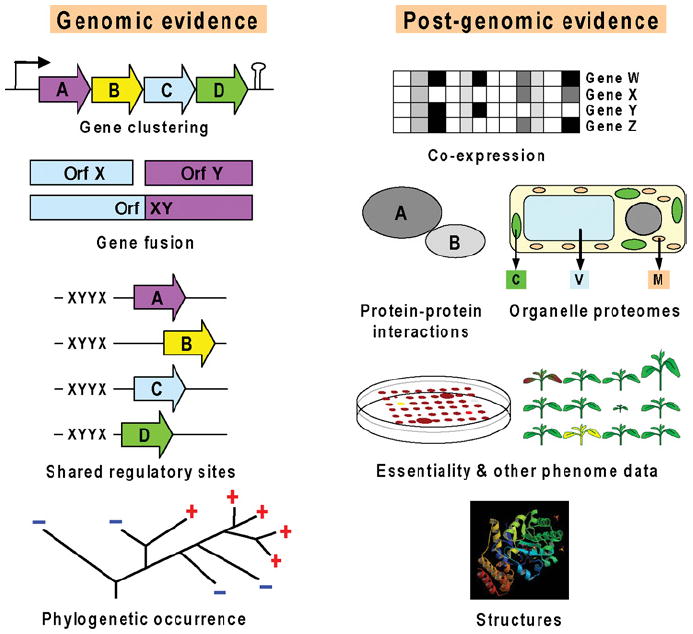
Multiple types of evidence gathered from genomic and post-genomic resources are integrated in order to make predictions on gene function. The more lines of evidence converge, the more robust predictions become.
Associations based on genomes
Gene clustering
Of the ways in which genes can be associated, gene clustering, i.e. proximity in the genome, is the most generally useful. Although not absent from eukaryotes [48,49], clustering is far more marked in prokaryotes, where functionally related genes not only are arranged in operons, but also can be divergently transcribed from the same promoter region [45] or may simply be neighbours or near-neighbours, even though not co-transcribed [45,50]. On average, ~ 35% of bacterial metabolic genes are in conserved clusters [45]. Clusters that are conserved across diverse genomes are the most informative [45,50], which is one reason ubiquity is so helpful. Gene clustering can be analysed using the STRING, SEED and MicrobesOnline databases, among others.
Phylogenetic occurrence profiles
Another very useful type of association is phylogenetic co-occurrence, whose underlying principles are that enzymes of the same pathway will be either all present in or all absent from a given organism [23,41] and that genes that functionally replace each other will have reciprocal (anticorrelated) distributions [51]. The presence/absence patterns of genes among genomes can often identify candidates for ‘missing’ genes [52] such as those encoding orphan enzymes, or link unknown genes to known pathways. Phylogenetic profiles can be analysed using STRING, PHYDBAC, MBGD, the Signature Genes tool at NMPDR and the Phylogenetic Profiler at JGI. The two latter tools are designed to detect genes whose occurrence is correlated or anticorrelated among user-specified sets of organisms.
Gene fusions
In a gene-fusion event, separate parent gene products are encoded in a single multifunctional polypeptide. Such fusions, which have been called ‘Rosetta stone’ proteins, suggest a high probability of functional interaction between the two proteins, e.g. as enzymes in the same pathway or as components of a protein complex [53,54]. Just as with gene clustering, if the function of one of the fused genes is known and the other is not, the fusion allows strong functional predictions. Prokaryotic gene-fusion events are catalogued in the FusionDB database.
Shared regulatory sites
Genes participating in the same pathway or process are often regulated by a common protein recognizing a specific DNA sequence, or by common riboswitches [55,56]. Finding shared regulatory sites is thus a powerful way to find genes that are functionally linked. Gene regulation databases include SwissRegulon and PRODORIC.
Metabolic reconstruction
Metabolic reconstruction is both a goal and a method; the quest to reconstruct an organism’s full metabolic repertoire in silico itself helps discover and rationalize that repertoire. Thus reconstructing a complete functional pathway from the set of genes in a genome using reference biochemical knowledge, as pioneered by E. Selkov, is of great value in inferring function from various kinds of genomic data because it imposes consistency [57,58]. The completeness of the reconstructed pathway indicates the correctness of initial gene function assignments and establishes which pathway steps are not yet connected to a gene. Metabolic reconstruction is most effective when applied iteratively; problems of wrong functional assignments and missing genes become apparent, and are resolved, in successive cycles [59]. One way to implement metabolic reconstruction is via a ‘subsystems’ approach, in which a metabolic pathway (a ‘subsystem’) is analysed by experts across a large collection of genomes in parallel [60,61]. This approach is particularly helpful in identifying and making sense of pathway variants (e.g. truncated pathways or non-orthologous displacements). Another, more widespread, approach is genome-wide metabolic reconstruction and modelling, which has a wider scope of metabolism coverage but is essentially focused on a single organism. It can nonetheless reveal pathway gaps or inconsistencies that may otherwise be missed [12,58].
Associations based on post-genomic resources
As well as genomes themselves, various kinds of functional genomic data can yield functional associations between proteins. Although such post-genomic data are often still too noisy to be used as primary sources, they can be very effectively combined with genomics-based data.
Gene expression profiles
Associations can be derived from co-expression datasets (from microarrays), which are now well developed for model bacteria as well as for plants and animals (e.g. [62-64]). Moreover, the sets of conditions and (for plants and animals) the site or developmental stage in which a gene is expressed can provide vital clues about function [43]. Microarray databases and tools include MicrobesOnline and GenExpDB for bacteria, and ATTED and the Golm Transcriptome Database for Arabidopsis.
Proteomics data
At the protein level, protein–protein interaction datasets (e.g. [65]) [from two-hybrid or TAP (tandem affinity purification) tag experiments] have analogous value to those from microarrays. Also, for plants or other eukaryotes, organellar proteome data can sometimes rule in or rule out a possible function, for instance in the case of an enzyme of a pathway whose organellar location is known [43]. Protein–protein interaction databases include DIP, APID and (for E. coli) eNet. Plant proteome databases are PPDB and SUBA II.
Essentiality and other phenotype data
The availability of large-scale bacterial and plant knockout collections, along with databases on knockout phenotypes, can quickly show whether a gene is essential or is associated with a particular phenotype [66-68]. Besides revealing associations directly (e.g. when auxotrophy connects a gene with a biosynthetic pathway) phenotype data, especially essentiality data, pinpoint important genes. Essentiality data for bacteria are integrated into the SEED database; plant phenome databases include RAPID, SeedGenes and Chloroplast2010.
Three-dimensional structures
Structural genomics projects have determined the structures of hundreds of proteins of unknown function, many of which are ubiquitous [69]. Although structural genomics is usually unable to assign a specific function to a target protein, three-dimensional structures help, via fold recognition, to establish long-range homology when this is obscured at the sequence level, and thus contribute to general class functional assignments. Furthermore, a structure can be very helpful for comparative genomics because the ligands that the protein is computationally predicted to bind (e.g. [70]) can be compared with possible substrates inferred from, e.g., gene clustering evidence. Protein structures are compiled in the Protein Data Bank. If no structure is available, structure prediction algorithms such as PHYRE and PSIPRED GenTHREADER can be useful substitutes.
The genome deluge
A total of over 1000 prokaryotic and eukaryotic genomes have now been completely sequenced, approx. 4000 more are in the pipeline (Figure 3A), and the pace continues to quicken [71]. This progress is highly favourable for comparative genomics, because a crucial feature of comparative genomics associations is that the number that can be found grows roughly at the square of the number of genomes [45], as shown schematically in the inset of Figure 3(A). The power of comparative genomics to identify functional associations between genes will thus keep growing rapidly. Moreover, since post-genomic datasets are also expanding rapidly, and analysing multiple types of associations improves predictions [41,44,45], the specificity and robustness of predictions will also keep growing. This means that many functions that are elusive today will become predictable in the foreseeable future.
Figure 3. The genome deluge and its implications.
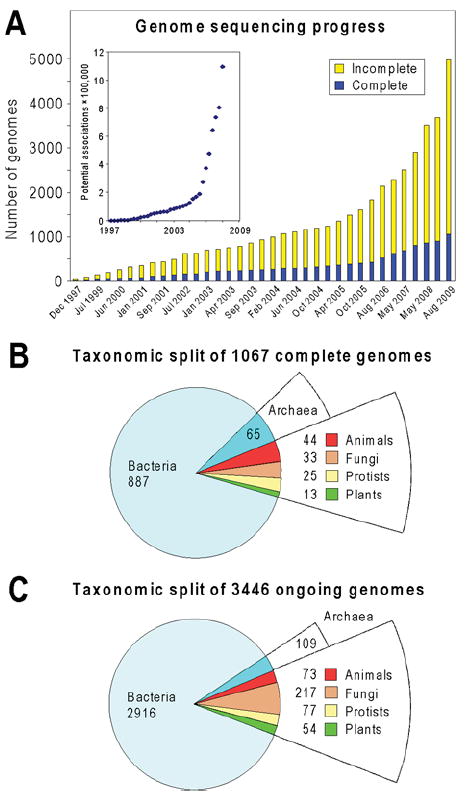
Statistics are from the Genomes OnLine Database (http://genomesonline.org/index2.htm). (A) Progress in genome sequencing since 1997. The inset plots the square of the number of completely sequenced genomes against time; this value is roughly proportional to the potential for recognizing functional associations from genome data. Note its explosive growth since about 2006. The incomplete genomes include several hundred comprehensive EST (expressed sequence tag) projects. (B) Taxonomic breakdown of the 1067 genomes completed by August 2009. (C) Taxonomic breakdown of 3446 full genome sequencing projects that were ongoing (incomplete) as of August 2009. EST and genome survey projects are excluded from the total.
SYNERGY OF PROKARYOTE–EUKARYOTE INTEGRATIONS
Of the genomes completed so far, approx. 10% come from a diverse set of eukaryotes in which all major groups are represented (Figure 3B); the percentage of eukaryotes among ongoing genomes is similar and their absolute number is almost 4-fold higher (Figure 3C) [71]. These eukaryotic genomes, which already collectively encode some 1.8×106 ORFs (open reading frames), can now or soon will be included in comparative genomics analyses. Such inclusion is very valuable because analysing prokaryotic and eukaryotic genomes together yields information that cannot be obtained by looking at either group alone, and many discoveries have now been made this way. This section illustrates the synergy using three historical examples involving metabolic pathways of engineering interest, i.e. folate synthesis, NAD synthesis and leucine degradation, plus a case study showing how much faster an engineering target enzyme can be found with comparative genomics than without it.
Example 1: a missing folate biosynthesis enzyme
The folate biosynthesis pathway is an attractive engineering target in bacteria [72,73] and plants [74]. Although the other pathway genes had been identified, until recently the gene for one enzyme (dihydroneopterin triphosphate pyrophosphatase) was missing in both groups (Figure 4A). This enzyme can be viewed as mediating the committing step in folate biosynthesis since its substrate, dihydroneopterin triphosphate, has three other known fates in various organisms (Figure 4A). Partial purification and characterization of dihydroneopterin triphosphate pyrophosphatase from E. coli had shown that it is a small (17 kDa) protein that requires Mg2+ for activity and is optimally active at pH 8.5 [75]. Comparative genomics analysis (Figure 4B) revealed a gene (ylgG) encoding a small protein belonging to the Nudix family embedded in a folate synthesis operon in Lactococcus lactis and other bacteria. This made YlgG a prime candidate for the missing enzyme as Nudix family members include nucleoside triphosphate pyrophosphatases (dihydroneopterin triphosphate is structurally analogous to a nucleoside triphosphate) and Nudix enzymes characteristically require a bivalent cation and have an alkaline pH optimum. Experimental tests showed that inactivating ylgG in L. lactis resulted in dihydroneopterin triphosphate accumulation and folate depletion, and that recombinant YlgG had high dihydroneopterin triphosphate pyrophosphatase activity; ylgG was consequently renamed folQ [76]. The equivalent E. coli gene (nudB) was identified 2 years later via a classical strategy involving cloning and characterizing all 13 E. coli Nudix proteins [77], which demanded notably more effort than the comparative genomics approach. Lastly, having identified the L. lactis enzyme, it was possible to show that its closest homologue in Arabidopsis also had high dihydroneopterin triphosphate pyrophosphatase activity (Figure 4B) [76].
Figure 4. FolQ, a missing folate synthesis enzyme in bacteria and plants.
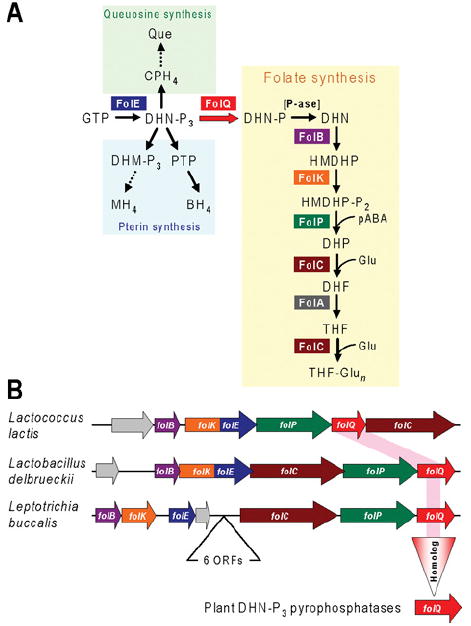
(A) The tetrahydrofolate biosynthesis pathway and its branches leading to queuosine and pterins. Note that the previously missing enzyme FolQ (YlgG in Lactococcus lactis) is the first step unique to the folate pathway. Abbreviations: BH4, 5,6,7,8-tetrahydrobiopterin; CPH4, 6-carboxy-5,6,7,8-tetrahydropterin; DHF, 7,8-dihydrofolate; DHM, 7,8-dihydromonapterin; DHN, 7,8-dihydroneopterin; DHP, 7,8-dihydropteroate; Glu, glutamate; HMDHP, 6-hydroxymethyl-7,8-dihydropterin; MH4, 5,6,7,8-tetrahydromonapterin; -P, phosphate; -P2, pyrophosphate; -P3, triphosphate; pABA, p-aminobenzoate; P-ase, non-specific phosphatase; PTP, 6-pyruvoyl-5,6,7,8-tetrahydropterin; Que, queuosine; THF, 5,6,7,8-tetrahydrofolate; THF-Glun, tetrahydrofolate polyglutamates. (B) Clustering in operonic arrangements of folQ with genes encoding other folate synthesis enzymes in two lactobacteria (phylum Firmicutes) and Leptotrichia buccalis (phylum Fusobacteria). Arrows indicate transcriptional direction; overlapping arrows indicate translational coupling. Genes are colour-coded in agreement with (A); non-conserved genes are coloured grey. A short intervening block of six genes separates the two clusters of folate synthesis genes in L. buccalis. The rose highlight linking the bacterial folQ genes and the vertical triangle represent the projection of the bacterial gene function to plants.
Example 2: the tryptophan to quinolinate route in NAD synthesis
Manipulating levels of NAD and related cofactors, i.e. NAD(P)(H), is a useful tool for metabolic engineering [78,79]. Such engineering requires knowledge of the NAD biosynthesis pathway genes, to which comparative genomics has contributed significantly for the early pathway steps leading to quinolinate, the universal de novo precursor of the pyridine ring of NAD [80,81]. Before the advent of comparative genomics, two different pathways to quinolinate were known: the two-enzyme ‘prokaryotic’ pathway from aspartate and the five-enzyme ‘eukaryotic’ route from tryptophan (Figure 5A). However, in certain bacteria, classical radiotracer studies had demonstrated 14C incorporation from tryptophan into NAD and some of the ‘eukaryotic’ pathway enzyme activities had been detected, pointing to the existence of an alternative pathway in these organisms. Comparative genomics analysis identified candidates for all five bacterial genes of this pathway, all of which were then validated by complementation and biochemical assays [80]. The most crucial observations leading to identification of the genes for the alternative pathway were the absence from some genomes of genes encoding both enzymes (NadA and NadB) of the ‘prokaryotic’ pathway (Figure 5B) and the presence of various operon-like gene clusters containing homologues of four out of the five ‘eukaryotic’ pathway enzymes (Figure 5C). The one missing enzyme, KFA (N-formylkynurenine formamidase), of the bacterial pathway (which is non-orthologous to eukaryotic KFA) was correctly predicted from its tendency to cluster with the other four (Figure 5C).
Figure 5. A tryptophan to quinolinate pathway in bacteria.
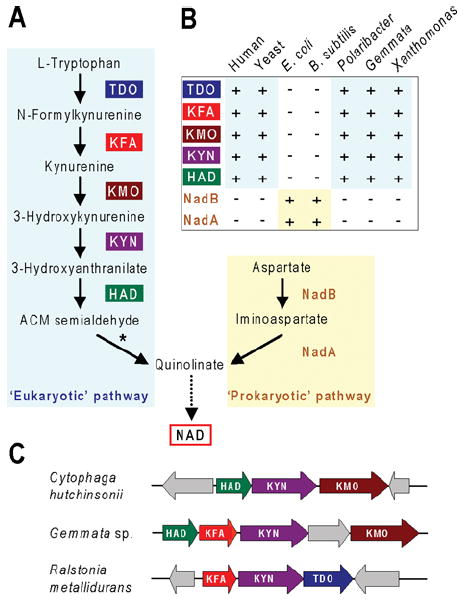
(A) The two biosynthetic routes to quinolinate: the five-step ‘eukaryotic’ route and the two-step ‘prokaryotic’ one. ACM semialdehyde, 2-amino-3-carboxymuconate semialdehyde. Conversion of ACM semialdehyde into quinolinate (asterisked) is non-enzymatic. (B) Schematic profile of the presence and absence of the seven genes of the ‘eukaryotic’ and ‘prokaryotic’ pathways among two representative eukaryotes, two representative bacteria with the ‘prokaryotic’ pathway (Escherichia coli and Bacillus subtilis), and three bacteria with the ‘eukaryotic’ pathway (Polaribacter filamentus, Gemmata sp., and Xanthomonas axonopodis). +, gene present; -, gene absent. (C) Clustering in operonic arrangements of various ‘eukaryotic’ pathway genes in representative bacteria. Arrows indicate transcriptional direction. Non-conserved genes are coloured grey.
Example 3: the leucine-degradation pathway
Leucine degradation yields acetyl-CoA and acetoacetate, which are important intermediates in primary and secondary metabolism [82], including the synthesis of hydroxymethylglutaryl-CoA and thence isoprenoids and sterols (Figure 6A). The leucine degradation pathway has been well studied in humans and all of the human genes are elucidated and characterized (Figure 6B). In contrast, before comparative genomics work, relatively little was known about this pathway in bacteria, and no bacterial genes had been connected directly to steps after isovaleryl-CoA.
Figure 6. Leucine catabolism in bacteria and humans.
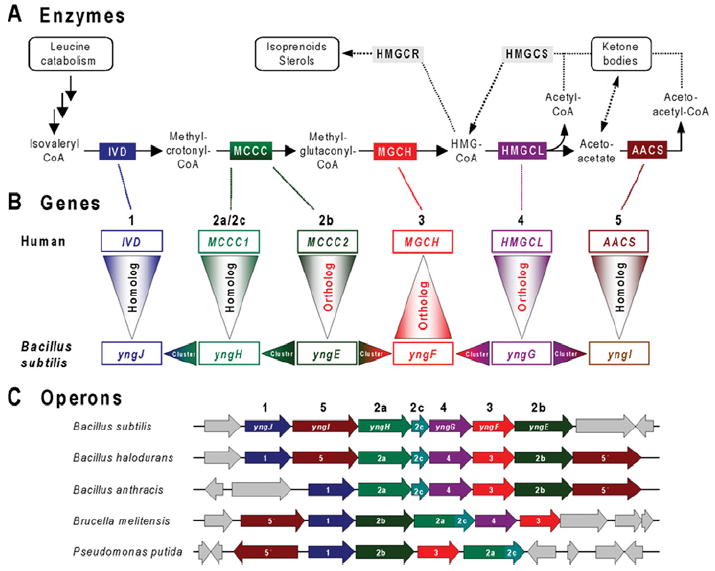
(A) Enzymatic steps involved in the later steps of leucine catabolism and the metabolism of hydroxymethylglutaryl-CoA (HMG-CoA). Intermediates are shown by chemical names. Enzymes conserved in humans, Bacillus subtilis and certain other bacteria are as follows: IVD, isovaleryl-CoA dehydrogenase (EC 1.3.99.10); MCCC, methylcrotonoyl-CoA carboxylase (EC 6.4.1.4) [2a/2c, biotin-carboxylase subunit/biotin carboxyl carrier domain/subunit; 2b, carboxyl transferase subunit]; MGCH, methylglutaconyl-CoA hydratase (EC 4.2.1.18); HMGCL, hydroxymethylglutaryl-CoA lyase (EC 4.1.3.4); and AACS, acetoacetate-CoA synthetase (EC 6.2.1.16). These five enzymes are colour-coded. Two enzymes related to the mevalonate pathway of isoprenoid/sterol biosynthesis (present in humans, but not B. subtilis) are shown in grey: HMGCS, HMG-CoA synthase (EC 2.3.3.10); and HMGCR, HMG-CoA reductase (EC 1.1.1.34). Enzymes catalysing early steps of leucine catabolism (from leucine to isovaleryl-CoA) are similar in humans and bacteria (not shown). (B) Projection of functional assignments between human and bacterial genes. Gene names (human and B. subtilis) corresponding to pathway enzymes are colour-coded and numbered in agreement with (A). The reasoning used in analysing leucine catabolism in bacteria is illustrated by arrowheads pointing in the direction of functional projections. Vertical triangles with red lettering correspond to unambiguous projections based on orthology (same specific function). The triangles point in the direction of the projection. Vertical triangles with black lettering indicate homologues that belong to large families that contain multiple paralogues that share a ‘general class’ function, but differ in substrate specificity. Horizontal triangles indicate conjectures based on gene clustering (refinement of ‘general class’ functions and genuine functional predictions). (C) Large operon-like clusters of genes related to leucine catabolism detected in a number of Gram-positive and Gram-negative bacteria. Conserved homologous genes are colour-coded and numbered in agreement with (A) and (B). Genes without homologues in a given chromosomal neighbourhood are coloured grey.
Attempts to identify bacterial genes solely by homology of their products with those of eukaryotic genes produced ambiguous results since most leucine-degradation enzymes belong to large families of paralogues. Such paralogues usually retain a ‘general class’ function (e.g. ‘dehydrogenase’), but differ widely in substrate specificity. However, a comparative genomics approach (outlined in Figure 6B) provided convincing evidence for the presence of the entire pathway of leucine catabolism in a number of diverse bacteria [59]. The first step was identification of a conserved gene cluster containing the bacterial orthologues (genes 2b and 4) of two of the human genes (Figure 6C). This observation enabled upgrading functional predictions for two additional bacterial genes in the same cluster (genes 1 and 2a) from a general class to a specific function. At the time that this analysis was performed, no methylglutaconyl-CoA hydratase gene had been identified in any organism. Another conserved bacterial gene in the cluster (gene 3), a member of the enoyl-CoA hydratase family, was predicted to fulfil this functional role, and this prediction was projected to the orthologous gene in the human genome. The prediction for the human gene has since been verified experimentally [83,84], nicely illustrating ‘two-way comparative genomics traffic’ between prokaryotes and eukaryotes.
Another functional inference concerns the last conserved member of the same cluster (gene 5) (Figure 6C). Its assignment as acetoacetyl-CoA synthetase is supported by homology with other acyl-CoA synthetases and by clustering with the leucine-catabolism pathway where acetoacetate is a final product. The gene cluster in Bacillus halodurans contains two paralogous forms (genes 5 and 5′), whereas each of the very similar clusters in Bacillus anthracis and Bacillus subtilis has either one or the other, suggesting that they are isofunctional. Traditionally, acetoacetyl-CoA synthetase has not been considered to be closely tied to leucine catabolism, but the gene clustering evidence strongly suggests that this is so, at least in some bacteria.
Case study: identifying the plant choline-oxidizing enzyme
The two-step pathway from choline to glycine betaine (Figure 7A) has long been a target for metabolic engineering of resistance to salinity and water deficit in bacteria and plants because glycine betaine is a potent osmoprotectant [85,86]. The genes for the E. coli pathway had been cloned and sequenced by 1991 [87]: betA, encoding a membrane-bound FAD-containing choline dehydrogenase, and betB, encoding a soluble NAD-linked betaine aldehyde dehydrogenase. In E. coli, these genes are clustered with betT, specifying a choline transporter and betI, coding for a transcriptional repressor (Figure 7B) [87]. Identical or similar choline oxidation pathways and gene clusters occur in many other bacteria [88]; these clusters can also include betC, coding for choline sulfatase (Figure 7B) or choline transporter genes other than betT (e.g. opuAC).
Figure 7. A hypothetical shortcut to the plant choline-oxidizing enzyme.
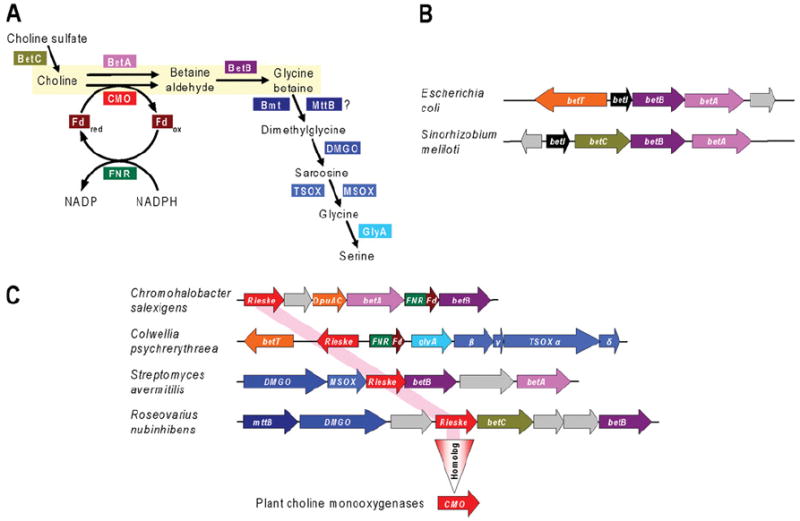
(A) The choline to glycine betaine pathway in bacteria and plants, and related reactions of choline metabolism in bacteria. BetA, choline dehydrogenase (EC 1.1.99.1); BetB, betaine aldehyde dehydrogenase (EC 1.2.1.8); BetC, choline sulfatase (EC 3.1.6.6); Bmt, betaine–homocysteine S-methyltransferase (EC 2.1.1.5); CMO, choline mono-oxygenase (EC 1.14.15.7), a Rieske-type [2Fe–2S] protein; DMGO, dimethylglycine oxidase (EC 1.5.3.10); Fd, ferredoxin; FNR, ferredoxin–NADP+ reductase (EC 1.18.1.2); GlyA, serine hydroxymethyltransferase (EC 2.1.2.1); MSOX, monomeric sarcosine oxidase; MttB, homologue of trimethylamine methyltransferase often clustered with dimethylglycine oxidase; TSOX, heterotetrameric sarcosine oxidase. Other bacterial enzymes (not shown) that mediate oxidation of choline to betaine aldehyde are choline oxidase (EC 1.1.3.17) and GbsB, a soluble, NAD-linked type III alcohol dehydrogenase. (B) Typical clustering arrangements of the choline–glycine betaine pathway genes betA and betB with betI (encoding a transcriptional repressor) and betT (encoding a choline transporter) or betC. Genes are colour-coded in agreement with (A). (C) Clustering in diverse bacteria of genes for Rieske-type proteins homologous with choline mono-oxygenase with up to 13 different genes of choline metabolism. Genes are colour-coded in agreement with (A) and (B). The rose highlight linking the bacterial Rieske-type genes and the vertical triangle represent the projection of the hypothetical bacterial gene function (choline oxidation) to plant choline mono-oxygenases. The gene labelled opuAC encodes a homologue of the periplasmic choline-binding component of an ABC (ATP-binding cassette) transporter. The genes labelled α, β, γ, and δ encode the four subunits of heterotetrameric sarcosine oxidase. Non-conserved genes are coloured grey.
Investigation of the plant pathway showed that it is plastid-localized, that the second enzyme is a betaine aldehyde dehydrogenase as in bacteria [89], and that the first is not a dehydrogenase, but a ferredoxin-dependent choline mono-oxygenase [90,91]. The ferredoxin electron donor can be reduced photosynthetically or by ferredoxin–NADP reductase plus NADPH in darkness [91]. The plant choline-oxidizing system thus comprises three proteins: choline mono-oxygenase, ferredoxin and ferredoxin-NADP reductase (Figure 7A). Cloning and characterization of choline mono-oxygenase showed it to be a Rieske-type [2Fe–2S] enzyme [92,93]. Rieske-type oxygenases with reductase and ferredoxin components were already well known in bacteria [94], but choline mono-oxygenase was the first such case from plants.
After discovery of choline mono-oxygenase activity in 1989 [91], it took 8 years to identify the gene: 6 years to purify the protein [92] and 2 more to clone the cDNA from peptide sequence data [93]. But had it been possible to apply comparative genomics to the search for the plant choline mono-oxygenase gene, it could, as we now explain, have been identified in approx. 2 h using the SEED database and its tools.
The starting point for our retrospective analysis is the sequence of plant betaine aldehyde dehydrogenase, which appeared in 1990 [89]. This protein has many strong homologues in bacteria, first among them being betB proteins, whose genes cluster with betA and other bet genes (Figure 7B). However, certain of these homologues are encoded by genes in a different sort of cluster. This sort contains a gene for a Rieske-type protein as well as various other genes of choline and glycine betaine metabolism, including betC and dimethylglycine and sarcosine oxidases (Figure 7C). A gene specifying a reductase–ferredoxin fusion protein to service the Rieske-type protein is sometimes present, as is a betA gene (Figure 7C). These clusters strongly implicate the Rieske-type protein in choline metabolism, most probably as a choline oxygenase. (The co-occurrence of the Rieske-type gene with betA is not inconsistent with this inference because these enzymes could be alternatives. They have opposite cofactor requirements: an electron donor for choline mono-oxygenase compared with an electron acceptor for BetA, and choline mono-oxygenase has an oxygen requirement which BetA does not.) When the Rieske-type protein from clusters such as those in Figure 7C is used to search plant genomes, the only BlastP hits are choline mono-oxygenases.
The inference that the bacterial Rieske-type proteins are choline mono-oxygenases awaits experimental validation, but this makes no difference to the chain of reasoning. This chain would be quite strong enough to warrant experimental tests of the plant homologues were their function unknown.
CONCLUSION
In the present review, we have sought to convince biochemists that the unknown protein problem is vast, and that comparative genomics can help to solve it, especially when prokaryote and eukaryote genomes are analysed together. Although comparative genomics approaches are being adopted by more and more researchers, they remain underutilized. Given how fast the power of these approaches is increasing, and will continue to increase (Figure 3A), this underutilization means the loss of many opportunities and even, as the choline mono-oxygenase case suggests, significant waste of time and effort (“8 years in the lab can save 2 hours at the computer”).
Barriers to the adoption of comparative genomics have been noted briefly elsewhere [43], but they bear additional comment here. First, there is a perception that the necessary bioinformatic skills are specialist ones. This is not the case; powerful but fairly intuitive websites such as STRING and SEED (Table 1) now bring comparative genomics tools within the reach of any experimentalist after a few hours of instruction. Another perception is that a prerequisite for comparative genomics is high-level literacy in the metabolism, physiology, ecology and systematics of a wide range of prokaryotes and eukaryotes. This barrier is minimal; online databases now make all the necessary background knowledge just a few mouse-clicks away, so it can easily be acquired ‘on the fly’. Lastly, solving the unknown protein problem can seem daunting. But, as noted at the outset, it can be achieved by a sustained community effort. In practice, such an effort will require sharing of unpublished ideas, predictions and observations in a co-ordinated fashion [35]. Although this requires some change of mindset away from the classical ‘single-PI specialist’ model, this is not a utopian dream, because adopting the new mindset requires only enlightened self-interest: researchers rapidly realize that much more progress can be made with it than without it.
Acknowledgments
We thank Dr Elizabeth Vierling for discussions that prompted this review, Dr Alex Toker for the invitation to write it, and Dr Ross Overbeek and Dr Andrei Osterman for providing the analyses summarized in Figure 1(B) and for insightful criticism of the manuscript. We also thank Dr Tim Helentjaris for the “8 years in the lab … ” quote.
FUNDING Supported by the National Science Foundation [grant number MCB-0839926 to A. D. H.], the National Institutes of Health [grant number AI066244-01 to V.deC.-L.], and by an endowment from the C. V. Griffin Sr. Foundation.
Abbreviations used
- KFA
N-formylkynurenine formamidase
References
- 1.Stephanopoulos GN, Aristidou AA, Nielsen J. Metabolic Engineering: Principles and Methodologies. Academic Press; San Diego: 1998. [Google Scholar]
- 2.Hanson AD, Shanks JV. Plant metabolic engineering: entering the S curve. Metab Eng. 2002;4:1–2. [Google Scholar]
- 3.Capell T, Christou P. Progress in plant metabolic engineering. Curr Opin Biotechnol. 2004;15:148–154. doi: 10.1016/j.copbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 4.Wu S, Chappell J. Metabolic engineering of natural products in plants; tools of the trade and challenges for the future. Curr Opin Biotechnol. 2008;19:145–152. doi: 10.1016/j.copbio.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Kunze R, Frommer WB, Flügge UI. Metabolic engineering of plants: the role of membrane transport. Metab Eng. 2002;4:57–66. doi: 10.1006/mben.2001.0207. [DOI] [PubMed] [Google Scholar]
- 6.Yazaki K. Transporters of secondary metabolites. Curr Opin Plant Biol. 2005;8:301–307. doi: 10.1016/j.pbi.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Stepansky A, Less H, Angelovici R, Aharon R, Zhu X, Galili G. Lysine catabolism, an effective versatile regulator of lysine level in plants. Amino Acids. 2006;30:121–125. doi: 10.1007/s00726-005-0246-1. [DOI] [PubMed] [Google Scholar]
- 8.Galperin MY, Koonin EV. ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004;32:5452–5463. doi: 10.1093/nar/gkh885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karp PD. Call for an enzyme genomics initiative. Genome Biol. 2004;5:401. doi: 10.1186/gb-2004-5-8-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koonin EV, Galperin MY. Sequence – Evolution – Function: Computational Approaches in Comparative Genomics. Kluwer; Dordrecht: 2002. [PubMed] [Google Scholar]
- 11.Durot M, Bourguignon PY, Schachter V. Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol Rev. 2009;33:164–190. doi: 10.1111/j.1574-6976.2008.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feist AM, Herrgård MJ, Thiele I, Reed JL, Palsson BØ. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smid EJ, Molenaar D, Hugenholtz J, de Vos WM, Teusink B. Functional ingredient production: application of global metabolic models. Curr Opin Biotechnol. 2005;16:190–197. doi: 10.1016/j.copbio.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Pérez-Pantoja D, De la Iglesia R, Pieper DH, González B. Metabolic reconstruction of aromatic compounds degradation from the genome of the amazing pollutant-degrading bacterium Cupriavidus necator JMP134. FEMS Microbiol Rev. 2008;32:736–794. doi: 10.1111/j.1574-6976.2008.00122.x. [DOI] [PubMed] [Google Scholar]
- 15.Borenstein E, Kupiec M, Feldman MW, Ruppin E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc Natl Acad Sci U S A. 2008;105:14482–14487. doi: 10.1073/pnas.0806162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osterman AL, Begley TP. A subsystems-based approach to the identification of drug targets in bacterial pathogens. Prog Drug Res. 2007;64:132–170. doi: 10.1007/978-3-7643-7567-6_6. [DOI] [PubMed] [Google Scholar]
- 17.Pinney JW, Papp B, Hyland C, Wambua L, Westhead DR, McConkey GA. Metabolic reconstruction and analysis for parasite genomes. Trends Parasitol. 2007;23:548–554. doi: 10.1016/j.pt.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Thiele I, Vo TD, Price ND, Palsson BØ. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. J Bacteriol. 2005;187:5818–5830. doi: 10.1128/JB.187.16.5818-5830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh D, Poisson LM. “Omics” data and levels of evidence for biomarker discovery. Genomics. 2009;93:13–16. doi: 10.1016/j.ygeno.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Dhamoon AS, Kohn EC, Azad NS. The ongoing evolution of proteomics in malignancy. Drug Discov Today. 2007;12:700–708. doi: 10.1016/j.drudis.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Weinglass AB, Whitelegge JP, Kaback HR. Integrating mass spectrometry into membrane protein drug discovery. Curr Opin Drug Discov Dev. 2004;7:589–599. [PubMed] [Google Scholar]
- 22.Walgren JL, Thompson DC. Application of proteomic technologies in the drug development process. Toxicol Lett. 2004;149:377–385. doi: 10.1016/j.toxlet.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 23.Osterman A, Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Curr Opin Chem Biol. 2003;7:238–251. doi: 10.1016/s1367-5931(03)00027-9. [DOI] [PubMed] [Google Scholar]
- 24.Frishman D. Protein annotation at genomic scale: the current status. Chem Rev. 2007;107:3448–3466. doi: 10.1021/cr068303k. [DOI] [PubMed] [Google Scholar]
- 25.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 26.Horan K, Jang C, Bailey-Serres J, Mittler R, Shelton C, Harper JF, Zhu JK, Cushman JC, Gollery M, Girke T. Annotating genes of known and unknown function by large-scale coexpression analysis. Plant Physiol. 2008;147:41–57. doi: 10.1104/pp.108.117366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siew N, Azaria Y, Fischer D. The ORFanage: an ORFan database. Nucleic Acids Res. 2004;32:D281–D283. doi: 10.1093/nar/gkh116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voit EO, Riley M. Extending knowledge of Escherichia coli metabolism by modeling and experiment. Genome Biol. 2003;4:235. doi: 10.1186/gb-2003-4-11-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galperin MY, Koonin EV. Functional genomics and enzyme evolution: homologous and analogous enzymes encoded in microbial genomes. Genetica. 1999;106:159–170. doi: 10.1023/a:1003705601428. [DOI] [PubMed] [Google Scholar]
- 31.Pouliot Y, Karp PD. A survey of orphan enzyme activities. BMC Bioinformatics. 2007;8:244. doi: 10.1186/1471-2105-8-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lespinet O, Labedan B. ORENZA: a web resource for studying ORphan ENZyme activities. BMC Bioinformatics. 2006;7:436. doi: 10.1186/1471-2105-7-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Vitkup D. Distribution of orphan metabolic activities. Trends Biotechnol. 2007;25:343–348. doi: 10.1016/j.tibtech.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Janitz M. Assigning functions to genes: the main challenge of the post-genomics era. Rev Physiol Biochem Pharmacol. 2007;159:115–129. doi: 10.1007/112_2007_0703. [DOI] [PubMed] [Google Scholar]
- 35.Roberts RJ. Identifying protein function: a call for community action. PLoS Biol. 2004;2:E42. doi: 10.1371/journal.pbio.0020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen F, Mackey AJ, Stoeckert CJ, Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hedges SB, Blair JE, Venturi ML, Shoe JL. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol Biol. 2004;4:2. doi: 10.1186/1471-2148-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333:863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 39.Bhaduri A, Ravishankar R, Sowdhamini R. Conserved spatially interacting motifs of protein superfamilies: application to fold recognition and function annotation of genome data. Proteins. 2004;54:657–670. doi: 10.1002/prot.10638. [DOI] [PubMed] [Google Scholar]
- 40.Galperin MY, Koonin EV. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- 41.Kharchenko P, Chen L, Freund Y, Vitkup D, Church GM. Identifying metabolic enzymes with multiple types of association evidence. BMC Bioinformatics. 2006;7:177. doi: 10.1186/1471-2105-7-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Crécy-Lagard V. Identification of genes encoding tRNA modification enzymes by comparative genomics. Methods Enzymol. 2007;425:153–183. doi: 10.1016/S0076-6879(07)25007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Crécy-Lagard V, Hanson AD. Finding novel metabolic genes through plant–prokaryote phylogenomics. Trends Microbiol. 2007;15:563–570. doi: 10.1016/j.tim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Aravind L. Guilt by association: contextual information in genome analysis. Genome Res. 2000;10:1074–1077. doi: 10.1101/gr.10.8.1074. [DOI] [PubMed] [Google Scholar]
- 45.Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci U S A. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Date SV, Marcotte EM. Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat Biotechnol. 2003;21:1055–1062. doi: 10.1038/nbt861. [DOI] [PubMed] [Google Scholar]
- 47.von Mering C, Zdobnov EM, Tsoka S, Ciccarelli FD, Pereira-Leal JB, Ouzounis CA, Bork P. Genome evolution reveals biochemical networks and functional modules. Proc Natl Acad Sci U S A. 2003;100:15428–15433. doi: 10.1073/pnas.2136809100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JM, Sonnhammer EL. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003;13:875–882. doi: 10.1101/gr.737703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Field B, Osbourn AE. Metabolic diversification: independent assembly of operon-like gene clusters in different plants. Science. 2008;320:543–547. doi: 10.1126/science.1154990. [DOI] [PubMed] [Google Scholar]
- 50.Yanai I, Mellor JC, DeLisi C. Identifying functional links between genes using conserved chromosomal proximity. Trends Genet. 2002;18:176–179. doi: 10.1016/s0168-9525(01)02621-x. [DOI] [PubMed] [Google Scholar]
- 51.Makarova KS, Koonin EV. Filling a gap in the central metabolism of archaea: prediction of a novel aconitase by comparative-genomic analysis. FEMS Microbiol Lett. 2003;227:17–23. doi: 10.1016/S0378-1097(03)00596-2. [DOI] [PubMed] [Google Scholar]
- 52.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suhre K. Inference of gene function based on gene fusion events: the Rosetta-stone method. Methods Mol Biol. 2007;396:31–41. doi: 10.1007/978-1-59745-515-2_3. [DOI] [PubMed] [Google Scholar]
- 54.Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 55.Gelfand MS, Novichkov PS, Novichkova ES, Mironov AA. Comparative analysis of regulatory patterns in bacterial genomes. Brief Bioinform. 2000;1:357–371. doi: 10.1093/bib/1.4.357. [DOI] [PubMed] [Google Scholar]
- 56.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 57.Selkov E, Maltsev N, Olsen GJ, Overbeek R, Whitman WB. A reconstruction of the metabolism of Methanococcus jannaschii from sequence data. Gene. 1997;197:GC11–GC26. doi: 10.1016/s0378-1119(97)00307-7. [DOI] [PubMed] [Google Scholar]
- 58.Bono H, Ogata H, Goto S, Kanehisa M. Reconstruction of amino acid biosynthesis pathways from the complete genome sequence. Genome Res. 1998;8:203–210. doi: 10.1101/gr.8.3.203. [DOI] [PubMed] [Google Scholar]
- 59.Overbeek R, Devine D, Vonstein V. Curation is forever: comparative genomics approaches to functional annotation. Targets. 2003;2:138–146. [Google Scholar]
- 60.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crécy-Lagard V, Diaz N, Disz T, Edwards R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ye Y, Osterman A, Overbeek R, Godzik A. Automatic detection of subsystem/pathway variants in genome analysis. Bioinformatics. 2005;21:i478–i486. doi: 10.1093/bioinformatics/bti1052. [DOI] [PubMed] [Google Scholar]
- 62.Gollub J, Ball CA, Sherlock G. The Stanford Microarray Database: a user’s guide. Methods Mol Biol. 2006;338:191–208. doi: 10.1385/1-59745-097-9:191. [DOI] [PubMed] [Google Scholar]
- 63.Obayashi T, Kinoshita K, Nakai K, Shibaoka M, Hayashi S, Saeki M, Shibata D, Saito K, Ohta H. ATTED-II: a database of co-expressed genes and cis elements for identifying co-regulated gene groups in Arabidopsis. Nucleic Acids Res. 2007;35:D863–D869. doi: 10.1093/nar/gkl783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laule O, Hirsch-Hoffmann M, Hruz T, Gruissem W, Zimmermann P. Web-based analysis of the mouse transcriptome using Genevestigator. BMC Bioinformatics. 2006;7:311. doi: 10.1186/1471-2105-7-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, Osterman A. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17:448–456. doi: 10.1016/j.copbio.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez-Ricaud L, Warringer J, Ericson E, Glaab K, Davidsson P, Nilsson F, Kemp GJ, Nerman O, Blomberg A. PROPHECY: a yeast phenome database, update 2006. Nucleic Acids Res. 2006;35:D463–D467. doi: 10.1093/nar/gkl1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tzafrir I, Dickerman A, Brazhnik O, Nguyen Q, McElver J, Frye C, Patton D, Meinke D. The Arabidopsis SeedGenes Project. Nucleic Acids Res. 2003;31:90–93. doi: 10.1093/nar/gkg028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Todd AE, Marsden RL, Thornton JM, Orengo CA. Progress of structural genomics initiatives: an analysis of solved target structures. J Mol Biol. 2005;348:1235–1260. doi: 10.1016/j.jmb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 70.Hermann JC, Marti-Arbona R, Fedorov AA, Fedorov E, Almo SC, Shoichet BK, Raushel FM. Structure-based activity prediction for an enzyme of unknown function. Nature. 2007;448:775–779. doi: 10.1038/nature05981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu T, Pan Z, Domagalski N, Koepsel R, Ataai MM, Domach MM. Engineering of Bacillus subtilis for enhanced total synthesis of folic acid. Appl Environ Microbiol. 2005;71:7122–7129. doi: 10.1128/AEM.71.11.7122-7129.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wegkamp A, Starrenburg M, de Vos WM, Hugenholtz J, Sybesma W. Transformation of folate-consuming Lactobacillus gasseri into a folate producer. Appl Environ Microbiol. 2004;70:3146–3148. doi: 10.1128/AEM.70.5.3146-3148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bekaert S, Storozhenko S, Mehrshahi P, Bennett MJ, Lambert W, Gregory JF, 3rd, Schubert K, Hugenholtz J, Van Der Straeten D, Hanson AD. Folate biofortification in food plants. Trends Plant Sci. 2008;13:28–35. doi: 10.1016/j.tplants.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 75.Suzuki Y, Brown GM. The biosynthesis of folic acid. XII Purification and properties of dihydroneopterin triphosphate pyrophosphohydrolase. J Biol Chem. 1974;249:2405–2410. [PubMed] [Google Scholar]
- 76.Klaus SM, Wegkamp A, Sybesma W, Hugenholtz J, Gregory JF, 3rd, Hanson AD. A Nudix enzyme removes pyrophosphate from dihydroneopterin triphosphate in the folate synthesis pathway of bacteria and plants. J Biol Chem. 2005;280:5274–5280. doi: 10.1074/jbc.M413759200. [DOI] [PubMed] [Google Scholar]
- 77.Gabelli SB, Bianchet MA, Xu W, Dunn CA, Niu ZD, Amzel LM, Bessman MJ. Structure and function of the E. coli dihydroneopterin triphosphate pyrophosphatase: a Nudix enzyme involved in folate biosynthesis. Structure. 2007;15:1014–1022. doi: 10.1016/j.str.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 78.Berríos-Rivera SJ, San KY, Bennett GN. The effect of NAPRTase overexpression on the total levels of NAD, the NADH/NAD+ ratio, and the distribution of metabolites in Escherichia coli. Metab Eng. 2002;4:238–247. doi: 10.1006/mben.2002.0229. [DOI] [PubMed] [Google Scholar]
- 79.Heuser F, Schroer K, Lütz S, Bringer-Meyer S, Sahm H. Enhancement of the NAD(P)(H) pool in Escherichia coli for biotransformation. Eng Life Sci. 2007;7:343–353. [Google Scholar]
- 80.Kurnasov O, Goral V, Colabroy K, Gerdes S, Anantha S, Osterman A, Begley TP. NAD biosynthesis: identification of the tryptophan to quinolinate pathway in bacteria. Chem Biol. 2003;10:1195–1204. doi: 10.1016/j.chembiol.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 81.Lima WC, Varani AM, Menck CF. NAD biosynthesis evolution in bacteria: lateral gene transfer of kynurenine pathway in Xanthomonadales and Flavobacteriales. Mol Biol Evol. 2009;26:399–406. doi: 10.1093/molbev/msn261. [DOI] [PubMed] [Google Scholar]
- 82.Khannapho C, Zhao H, Bonde BK, Kierzek AM, Avignone-Rossa CA, Bushell ME. Selection of objective function in genome scale flux balance analysis for process feed development in antibiotic production. Metab Eng. 2008;10:227–233. doi: 10.1016/j.ymben.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 83.IJlst L, Loupatty FJ, Ruiter JP, Duran M, Lehnert W, Wanders RJ. 3-Methylglutaconic aciduria type I is caused by mutations in AUH. Am J Hum Genet. 2002;71:1463–1466. doi: 10.1086/344712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ly TB, Peters V, Gibson KM, Liesert M, Buckel W, Wilcken B, Carpenter K, Ensenauer R, Hoffmann GF, Mack M, Zschocke J. Mutations in the AUH gene cause 3-methylglutaconic aciduria type I. Hum Mutat. 2003;21:401–407. doi: 10.1002/humu.10202. [DOI] [PubMed] [Google Scholar]
- 85.Le Rudulier D, Strom AR, Dandekar AM, Smith LT, Valentine RC. Molecular biology of osmoregulation. Science. 1984;224:1064–1068. doi: 10.1126/science.224.4653.1064. [DOI] [PubMed] [Google Scholar]
- 86.McCue KF, Hanson AD. Drought and salt tolerance: towards understanding and application. Trends Biotechnol. 1990;8:358–362. [Google Scholar]
- 87.Lamark T, Kaasen I, Eshoo MW, Falkenberg P, McDougall J, Strøm AR. DNA sequence and analysis of the bet genes encoding the osmoregulatory choline–glycine betaine pathway of Escherichia coli. Mol Microbiol. 1991;5:1049–1064. doi: 10.1111/j.1365-2958.1991.tb01877.x. [DOI] [PubMed] [Google Scholar]
- 88.Kempf B, Bremer E. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch Microbiol. 1998;170:319–330. doi: 10.1007/s002030050649. [DOI] [PubMed] [Google Scholar]
- 89.Weretilnyk EA, Hanson AD. Molecular cloning of a plant betaine–aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought. Proc Natl Acad Sci U S A. 1990;87:2745–2749. doi: 10.1073/pnas.87.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lerma C, Hanson AD, Rhodes D. Oxygen-18 and deuterium labeling studies of choline oxidation by spinach and sugar beet. Plant Physiol. 1988;88:695–702. doi: 10.1104/pp.88.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brouquisse R, Weigel P, Rhodes D, Yocum CF, Hanson AD. Evidence for a ferredoxin-dependent choline monooxygenase from spinach chloroplast stroma. Plant Physiol. 1989;90:322–329. doi: 10.1104/pp.90.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Burnet M, Lafontaine PJ, Hanson AD. Assay, purification, and partial characterization of choline monooxygenase from spinach. Plant Physiol. 1995;108:581–588. doi: 10.1104/pp.108.2.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rathinasabapathi B, Burnet M, Russell BL, Gage DA, Liao PC, Nye GJ, Scott P, Golbeck JH, Hanson AD. Choline monooxygenase, an unusual iron–sulfur enzyme catalyzing the first step of glycine betaine synthesis in plants: prosthetic group characterization and cDNA cloning. Proc Natl Acad Sci U S A. 1997;94:3454–3458. doi: 10.1073/pnas.94.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mason JR, Cammack R. The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu Rev Microbiol. 1992;46:277–305. doi: 10.1146/annurev.mi.46.100192.001425. [DOI] [PubMed] [Google Scholar]