

Abstract
Elevated Src tyrosine kinase activity is commonly observed in breast cancer and likely contributes to neoplasia and malignancy. p130Cas (“Crk-associated substrate”) is a major Src substrate found at the sites where integrins mediate cell adhesion to the extracellular matrix. Src phosphorylates multiple tyrosines in the p130Cas “substrate domain” (SD) and this signaling event has been implicated in the promotion of cell motility, primarily from studies on fibroblasts. In breast cancer, studies on p130Cas have focused on its role in conferring antiestrogen resistance to cells that express the estrogen receptor (ER+). However, little is known regarding the role of p130Cas in the more aggressive estrogen receptor negative (ER-) breast cancers for which there is a need for development of effective targeted therapies. We found high levels of p130Cas SD tyrosine phosphorylation to be a common characteristic of ER− breast cancer cell lines, with particularly high levels observed for the BT-549 cell line. Using RNA interference to knock down p130Cas expression in BT-549 cells, combined with rescue by WT p130Cas versus a signaling-deficient control, we provide evidence that p130Cas SD tyrosine phosphorylation is an important signaling event in the migration, invasion, proliferation, and survival of this ER-breast cancer cell line.
Keywords: adhesion, BCAR1, integrins, Src, FAK, tyrosine phosphorylation
Introduction
Elevated activity of the oncogenic tyrosine kinase, Src, is commonly seen in breast cancer.1,2 Major sites of Src signaling in adherent cells are focal adhesions, where integrins mediate anchorage to the extracellular matrix (ECM). Phosphorylation of Src substrates in focal adhesions may contribute to cancer progression by activating pathways leading to increased tumor growth, invasion, and metastasis.3 One of the major Src substrates in focal adhesions is p130Cas (“Crk-associated substrate”).
p130Cas was first recognized as a major tyrosine-phosphorylated protein in cells transformed by viral oncogenes v-crk and v-src.4,5 Molecular cloning revealed p130Cas to be a nonenzymatic “docking” protein consisting of an N-terminal SH3 domain, a Src-binding domain near the C-terminus, and a central “substrate domain” (SD) containing 15 YxxP motifs.6 p130Cas localizes prominently to focal adhesions, where SD YxxP tyrosines are subject to phosphorylation.7 Src is recruited to phosphorylate the SD sites through both direct binding and by interaction with focal adhesion kinase (FAK) that binds to the p130Cas SH3 domain.8 At least 10 of the 15 SD YxxP tyrosines can be phosphorylated9,10 to generate sites for recruitment of SH2-containing effectors that trigger downstream signaling events. Recruitment of adaptor proteins, Crk and Nck, to the phosphorylated p130Cas SD have been implicated in plasma membrane protrusion associated with cell motility.10–13 p130Cas SD tyrosine phosphorylation has further been implicated in the invasive and metastatic properties of Src-transformed cells.14,15
Apart from its recognition as a Src substrate in focal adhesions, p130Cas is distinguished as the product of the human gene BCAR1 identified in a screen for antiestrogen (tamoxifen) resistance of estrogen receptor positive (ER+) breast cancer cells.16 In breast cancer patients, high p130Cas levels are associated with a poor response to tamoxifen therapy, early disease recurrence, and lower long-term survival.17,18 Tamoxifen resistance conferred by p130Cas does not appear to result from alternative activation of ER target genes,19 but has been linked to Src-driven cell proliferation and survival pathways mediated either in complex with the ER to promote ERK signaling and cyclin D1 induction,20,21 or to an ER–independent manner involving EGFR and Stat5b.22 These studies have also revealed a role for adhesion-dependent p130Cas signaling in promoting protein kinase B (AKT) activation and resistance to apoptosis in response to ER antagonism by antiestrogens.23,24
While previous investigations on the role of p130Cas in breast cancer have focused on its involvement in antiestrogen resistance, little is known regarding its role in the malignant behavior of ER− breast cancer cells. About one-third of all breast cancers are ER–, so are not treatable by targeted antiestrogen therapies.25,26 ER– breast cancers tend to be more aggressive than ER+ breast cancers, which is reflected in the properties of breast cancer cell lines.27–30 ER-breast cancer cell lines characteristically express the mesenchymal marker vimentin, exhibit a fibroblast-like appearance in monolayer, and grow on Matrigel as loose colonies with large stellate projections indicative of their invasive behavior. In contrast, ER+ breast cancer cell lines express luminal epithelial cell markers including E-cadherin, grow as epithelial sheets in monolayer, and form tightly-adherent cysts or fused colonies on Matrigel indicative of poor invasive capacity.
In this study, we investigated the role of p130Cas signaling in the neoplastic properties of mesenchymal-like ER− breast cancer cells. p130Cas SD tyrosine phosphorylation was found to be commonly elevated in ER− breast cancer cell lines as compared to ER+ cell lines. The p130Cas SD is phosphorylated to particularly high levels in the BT-549 ER− cell line, which was thus chosen for further study of the impact of p130Cas signaling on ER− breast cancer cell behavior. Using RNA interference to knock down p130Cas expression, combined with rescue by WT p130Cas versus a signaling-deficient control, we present evidence that p130Cas SD tyrosine phosphorylation is an important signaling event in the migration, invasion, proliferation, and survival of ER− breast cancer cells.
Materials and methods
Antibodies
Mouse monoclonal antibodies against p130Cas (clone 21, designated here as CAS-TL) and FAK (clone 77) were from BD Transduction Laboratories (San Jose, CA). Rabbit polyclonal antibodies against FAK pTyr397, pTyr576/577 and pTyr861 were from Biosource International, Inc. (Camarillo, CA). The rabbit monoclonal antibodies against AKT pThr308, AKT pSer473, and AKT (pan), and the rabbit polyclonal antibodies against Src pTyr419, p130Cas pTyr165, p130Cas pTyr249, and p130Cas pTyr 410 were from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibody clone 327 ascites against Src was a gift from Dr. Christine Cartwright (Stanford University). Mouse monoclonal anti-pan-actin was from Thermo Fischer Scientific (Fremont, CA). The mouse monoclonal antibody against green fluorescent protein (GFP) was from Roche Applied Science (Mannheim, Germany). Horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgGs were obtained from BD Transduction Laboratories. Alexa 594-conjugated phalloidin was from Molecular Probes (Eugene, OR), and FITC-conjugated anti-rabbit IgG was from Jackson Immunoresearch Laboratories (Westgrove, PA).
Cells and cell culture
MCF-10A, MDA-MB-231, Hs578T, BT-549, MDA-MB-435s, MCF-7, T-47D, BT-474 and MDA-MB-468 cells were obtained from American Type Culture Collection. Cells were cultured at 37°C with 5% CO2 in a humidified incubator. MCF-10A cells were grown in complete mammary epithelial cell growth media (MEGM; Cambrex Bio Science Walkersville, Inc., Walkersville, MD) supplemented with growth factors and antibiotics (MEGM singlequots; Cambrex Bio Science Walkersville, Inc.), 100 ng/mL cholera toxin, and 5% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA). Human breast cancer cells were cultured in Dubecco’s modified Eagle’s medium (DMEM) containing L-glutamine, glucose, and sodium pyruvate (Mediatech, Herndon, VA) supplemented with 10% FBS, 1% nonessential amino acids (Invitrogen, Carlsbad, CA), 1% antimycotic/antibiotic (Mediatech), and 5 μg/mL plasmocin (InvivoGen, San Diego, CA).
Stable expression of Venus-tagged mouse p130Cas variants
Using standard molecular biology methods, plasmids expressing mouse p130Cas-Venus variants, WT31 or the 15F signaling-deficient mutant,10 were first constructed by subcloning the cDNAs into the vector pCS2-Venus.32 Then, for the purpose of stable expression, the p130Cas-Venus cDNAs from the pCS2 constructs were subcloned into the LZRS-MS-zeo plasmid.14 The resulting LZRS-MS-CASWT-Venus and LZRS-MS-CAS15F-Venus plasmids along with the LZRS-Venus “empty” vector were verified by sequencing then transfected into the Phoenix A packaging cell line for production of viral supernatants that were used to infect BT-549. Cells stably expressing similar levels of the Venus fluorophore were obtained through sorting by flow cytometry, and expression was verified by immunoblotting using antibodies against total p130Cas protein or GFP (which also detects Venus).
siRNA
BT-549 cells stably expressing p130Cas-Venus WT, p130Cas-Venus 15F, or Venus only, were grown to 60%–80% confluence on 60 mm diameter dishes. Cells were transfected with 500 pmol of the siGenome BCAR1 duplex 2 siRNA or the siGenome nontargeting siRNA #1 purchased from Dharmacon (Lafayette, CO) using 15 μL of Lipofectamine 2000 (Invitrogen) in Opti-Mem (Invitrogen) according to the manufacturer’s instructions. After six hours, the media was replaced with antibiotic-free growth media, and cells were allowed to grow overnight at 37°C. Complete growth media was added the following day. Cells were lysed 48 hours after transfection and subjected to immunoblotting.
Immunoblotting
For immunoblot analysis of total cell lysates, subconfluent cells were washed in phosphate-buffered saline (PBS), lysed in modified RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 50 mM NaF, 1% aprotinin, and 0.1 mM Na3VO4), and protein concentrations of cleared lysates were determined using the BCA Protein assay (Pierce Biotechnology, Rockford, IL). In some experiments, to enhance the ability to detect tyrosine phosphorylation of the p130Cas SD, cells were treated with 500 μM sodium vanadate (Na3VO4) for 3 hours prior to lysis. Lysates were diluted in 2x SDS-PAGE sample buffer for immunoblot analysis of whole cell extracts. Samples (20–30 μg total protein) were separated by SDS-polyacrylamide (7%) gel electrophoresis and transferred to nitrocellulose membranes. Nonspecific activity was blocked by incubating membranes in either 3% nonfat dried milk in Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBS-T) for one hour at room temperature or Blocking Buffer for Near Infra Red Fluorescent Western Blotting (Rockland Immunochemicals, Gilbertsville, PA) for 30 minutes at room temperature. Membranes were incubated in primary antibody overnight at 4°C, washed extensively with TBS-T, then incubated with HRP-conjugated secondary antibody for one hour at room temperature. After final extensive washes, blots were developed using enhanced chemiluminescence (ECL, Amersham Biosciences, Chicago, IL) and exposure to autoradiographic film.
Immunofluorescence microscopy
BT-549 cells were plated on glass coverslips coated with 10 μg/mL human fibronectin (Sigma, St. Louis, MO) and grown for 48 hours in Dubecco’s modified Eagle’s medium (DMEM) containing 10% FBS. Cells were fixed for five minutes in 50% methanol/50% acetone at −20°C. After washing in PBS, nonspecific binding sites were blocked with PBS containing 1% bovine serum albumin for one hour at room temperature. Cells were incubated overnight with the polyclonal antibody against p130Cas Tyr165 (1:100 dilution) at 4°C. After extensive washing with PBS, cells were incubated with FITC-conjugated anti-rabbit secondary antibody (1:300) for two hours at room temperature. Cells were washed again in PBS then incubated with 3.3 nmol/L Alexa 594-conjugated phalloidin for one hour at room temperature in the dark. Cells were rinsed again in PBS and mounted on glass slides with ProLong Antifade media (Molecular Probes). Cellular fluorescence was visualized using a Nikon Eclipse 80i microscope equipped with the X-Cite 120 fluorescence illumination system (EXFO Life Sciences Division, Mississauga, Ontario). Images were acquired using a CoolSNAP ES camera (Photometrics, Tuscon, AZ) and QED Capture software (Media Cybernetics, Inc., Bethesda, MD).
TIRF microscopy
To visualize localization of p 130Cas-Venus variants to focal adhesions, total internal reflection fluorescence cell (TIRF) microscopy was performed after transient transfection of a plasmid that expresses mCherry-paxillin as a red fluorescent marker protein. Cells were grown on coverslips coated with 1 μg/ml fibronectin, and visualized using a Nikon ECLIPSE TE2000-E inverted microscope (Melville, NY) equipped with a Perfect Focus System and a TIRF 100 1.49 NA oil-immersion lens. Fluorophores were excited with an 18 mW argon laser (Melles Griot, Albuquerque, NM) and 10 mW DPSS laser 85YCA010 (Melles Griot). A custom-made double-dichroic TIRF mirror and emission filters (Chroma, Rockingham, VT) in a Ludl filter wheel were used in conjunction with a neutral density (2) filter. Images were captured using a back-illuminated EM-CCD camera Cascade 512B (Photometrics), driven by IPLab software. Captured images were imported into ImageJ software33 for processing.
Wound healing migration assay
Cells were grown to confluence by overnight culture on coverslips coated with 1 μg/mL fibronectin, and wounds were made in the culture by scratching the confluent monolayer using a plastic pipette tip. Cells were allowed to migrate into the denuded wound area over twelve hours in DMEM containing 10% FBS and buffered with 10 mM HEPES. Images of migrating cells at the wound edge were taken every five minutes using a Nikon TE2000E microscope equipped with a Perfect Focus System and using a 20x 1.49 NA lens and CoolSNAP HQ CCD camera (Photometrics) driven by IPLab software (Scanalytics BD Biosciences Bioimaging, Rockville, MA). For each cell population, the rate of migration (μm/hour) of individual cells at the wound edge was determined using ImageJ software. Beginning two hours after the initial wounding, the nuclear centers of 20 cells per wound were tracked over a 10-hour period. The average rate of cell migration from three independent experiments analyzing a total of 120 cells was plotted in the Results. The standard error of the mean was determined, and statistical differences were established using the nonpaired one-tailed Student’s t-test (P < 0.001).
Modified Boyden chamber migration and Matrigel invasion assay
Modified Boyden chamber migration and Matrigel invasion assays were performed using Biocoat Matrigel invasion chamber inserts and control uncoated inserts containing 8 μm pores (BD Transduction Laboratories) according to the manufacturer’s instructions. Matrigel membranes were rehydrated for two hours in DMEM, and all inserts were placed in wells containing 10% FBS in DMEM. For each experiment, a total of 100,000 cells suspended in 0.5 mL of serum-free DMEM were added to the top of each of three chambers then incubated at 37°C in 5% CO2. After 24 hours, the chambers were washed with PBS, and cells were removed from the top side of the membranes with a cotton swab. Invading cells were fixed in 4% paraformaldehyde for 30 minutes and stained with 2% crystal violet. From each chamber, five representative fields were documented by photomicroscopy, and the numbers of migrating or invading cells were counted. The average number of migrating or invading cells per chamber in the triplicate samples is presented. Statistical significance for migration and invasion was determined using the nonpaired one-tailed Student’s t-test (P < 0.05).
Cell proliferation assay
Growth curves were determined for analysis of adherent growth. Twenty thousand cells in 0.5 ml of DMEM with 10% FBS were plated in triplicate on 24-well plates. At daily intervals, cells were harvested by trypsinization and counted using a Z1 Coulter Particle Counter (Beckman Coulter, Inc. Fullerton, CA). The mean cell number at each day was plotted. Statistical significance was determined using the nonpaired one-tailed Student’s t-test (P < 0.002).
TUNEL assay
Apoptosis was evaluated by the TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay. siRNA transfected cells were grown on four-chamber slides for 24 hours. Adherent cultured cells were fixed in 1 % paraformaldehyde and made permeable in a pre-cooled ethanol:acetic acid (2:1) mixture. Endogenous peroxidase was quenched with 3.0% hydrogen peroxide. The ApopTag® Peroxidase In situ Apoptosis Detection Kit (Chemicon, Billerica, MA) was then used according to the manufacturer’s protocol. Coverslips were mounted on the specimens using Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Cells were analyzed using a Nikon Eclipse 80i microscope with the X-Cite 120 Fluorescence illumination system (EXFO Life Sciences Division), a 40x oil immersion lens, and CoolSNAP ES camera (Photometrics). Images were acquired using QED Capture software (Media Cybernetics, Inc., Bethesda, MD). The number of TUNEL-positive cells was counted from at least twenty-five microscopic fields per experiment (> 1200 cells) for each cell type. This number was divided by the total number of cells from the same field to determine the percentage of TUNEL-positive cells. The percentage of TUNEL-positive cells from three to four separate experiments was combined to give the average percentage of TUNEL-positive cells for each cell type. Significance was determined using the nonpaired one-tailed Student’s t-test (P < 0.05).
Results
p130Cas SD phosphorylation is commonly elevated in mesenchymal-like ER− breast cancer cell lines
p130Cas expression and tyrosine phosphorylation were examined in eight breast cancer cell lines representative of either the epithelial-like class (MCF-7, T-47D, BT-474, and MDA-MB-468) or the mesenchymal-like class (MDA-MB-231, Hs578T, BT-549, and MDA-MB-435s) (Table 1). All four cell lines of the mesenchymal-like class are ER−, while three of the four cell lines of the epithelial-like class are ER+. Also included in the analysis was MCF-10A, an immortalized “normal” breast epithelial cell line derived from a patient with fibrocystic disease. Total cell lysates prepared from subconfluent monolayer cultures were analyzed by immunoblotting. The relative extent of p130Cas SD tyrosine phosphorylation was determined using a mixture of pCas165, pCas249, and pCas410 antibodies that specifically detect phosphorylated SD YxxP tyrosines.7
Table 1.
Features of breast cancer cell linesa
| Class | Cell line | Originb | ERc | Vimc | E-cadc | ^HER2c | ^EGFRc |
|---|---|---|---|---|---|---|---|
| Mesenchymal-like, highly invasive | |||||||
| MDA-MB-231 | adenocarcinoma, pleural effusion | − | + | − | − | − | |
| Hs578T | carcinosarcoma, primary | − | + | − | − | − | |
| BT-549 | papillary IDC, primary | − | + | − | − | − | |
| MDA-MB-435s | IDC, pleural effusion | − | + | − | − | − | |
| Epithelial-like, weakly invasive | |||||||
| MCF-7 | IDC, pleural effusion | + | − | + | − | − | |
| T-47D | IDC, pleural effusion | + | − | + | − | + | |
| BT-474 | IDC, primary | + | − | + | + | − | |
| MDA-MB-468 | adenocarcinoma, pleural effusion | − | − | + | − | + | |
IDC, invasive ductal carcinoma; the MDA-MB-435s line is noted for a gene expression profile characteristic of melanoma cells, suggesting a possible non breast cancer origin.
ER, estrogen receptor; Vim, vimentin;
HER2, elevated expression of HER2 receptor tyrosine kinase;
EGFR, elevated expression of EGF receptor tyrosine kinase.
Phosphorylation of the p 130Cas SD was readily detected in two of the ER− cell lines, Hs578T and BT-549, belonging to the mesenchymal-like class (Figure 1A). SD phosphorylation was remarkably high in the BT-549 cells. After pretreatment with sodium vanadate to inhibit tyrosine phosphatases, p130Cas SD phosphorylation could also be detected in the other two mesenchymal-like ER− lines, MDA-MB-231 and MDA-MB-435s (Figure 1B). Of the mesenchymal-like ER− cell lines, SD phosphorylation was considerably lower in the MDA-MB-435s cells, which also express relatively less total p130Cas protein (Figure 1C). Even with vanadate treatment, p130Cas SD phosphorylation was not detectable in the epithelial-like ER+ cell lines, while a low level of SD phosphorylation was detected in the ER− MDA-MB-468 cells (Figure 1B). In the normal MCF-10A breast epithelial cells, p130Cas SD phosphorylation was also detected weakly, and only after vanadate treatment.
Figure 1.
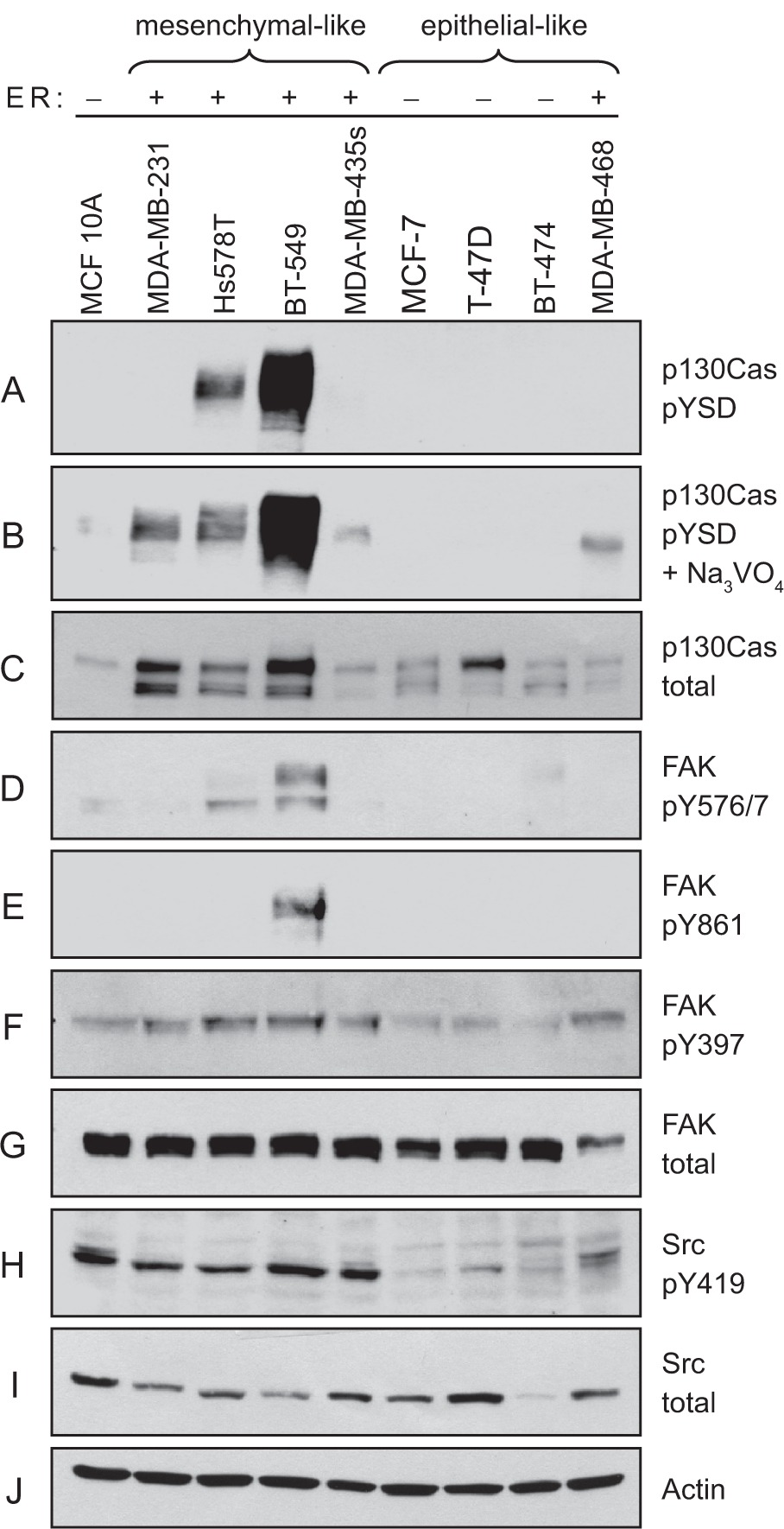
Elevated p130Cas SD tyrosine phosphorylation and Src activity in ER-breast cancer cell lines. Total cell lysates prepared from four representative mesenchymal-like breast cancer cell lines (MDA-MB-231, Hs578T, BT-549, and MDA-MB-435s) and four representative epithelial-like breast cancer cell lines (MCF-7, T-47D, BT-474, and MDA-MB-468) were analyzed by immunoblotting. MCF-10A was also included as a control representative of noncancerous breast epithelial cells. A and B) P130Cas SD tyrosine phosphorylation (pYSD) was detected using equimolar amounts of phosphospecific antibodies pCAS-165, pCAS-249, pCAS-410. In B) the cells were pretreated with sodium vanadate (Na3VO4) to enhance detection by the pCas antibodies. C) Total p130Cas protein was detected using the CAS-TL antibody. D–G) FAK tyrosine phosphorylation levels were evaluated using phosphospecific antibodies against known Src targets (Tyr-576/Tyr577 and Tyr-861), and the FAK autophosphorylation site Tyr-397. H and I) Activated Src was detected using a phosphospecific antibody against the autophosphorylation site Tyr-419 (H). J) Actin was detected as a loading control.
To gain evidence for the subcellular localization of p130Cas SD tyrosine phosphorylation in BT-549 cells, immunostaining was done using the pCas-165 antibody, reported to have the best specificity for detecting p130Cas by this method.7 The pCas-165 antibody prominently stained focal adhesions located at the cell periphery and under the cell body (Figure 2A), indicating that the p130Cas SD tyrosine phosphorylation observed by immunoblotting reflects integrin-mediated signaling.
Figure 2.
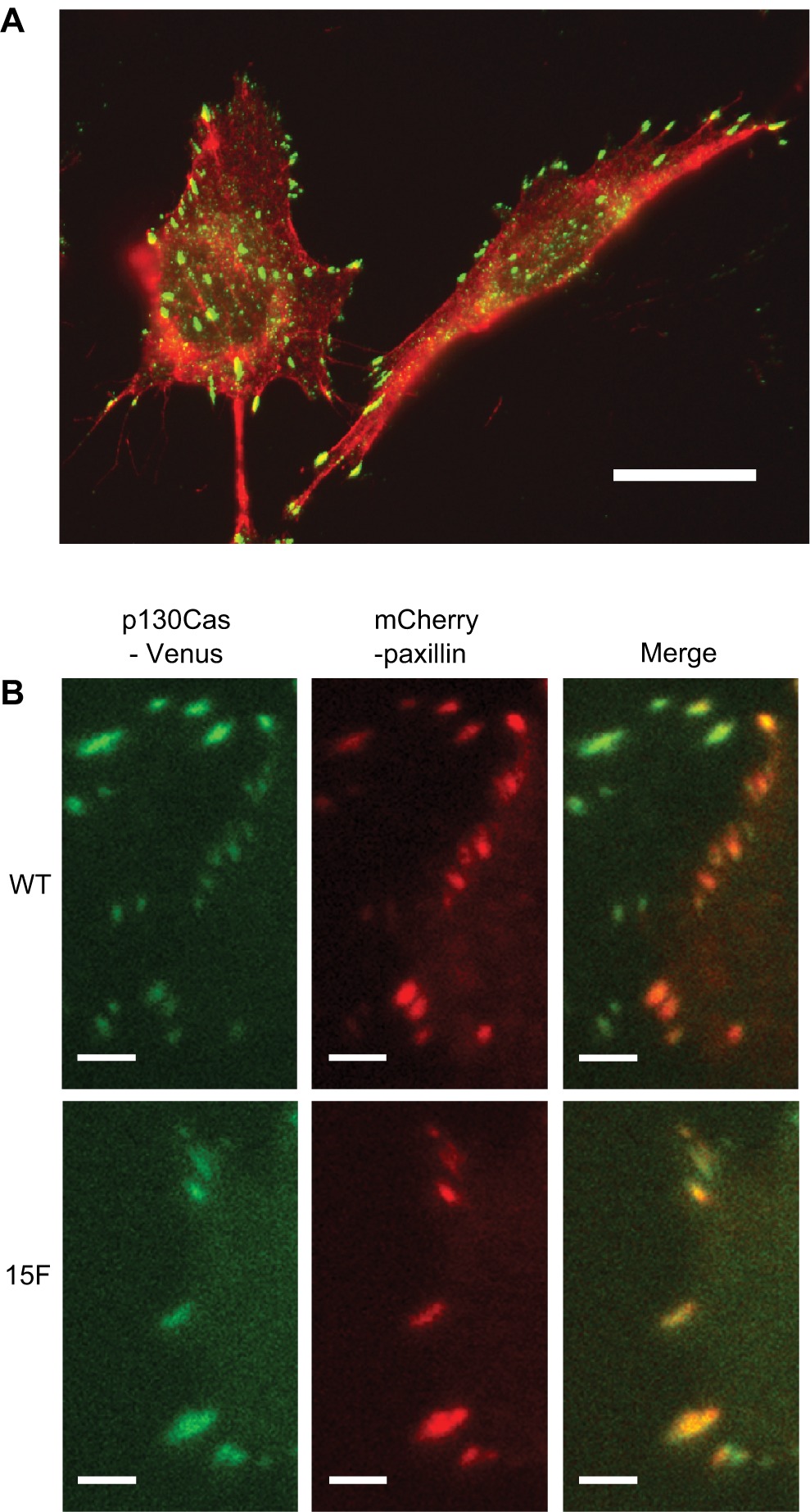
Localization of p130Cas SD tyrosine phosphorylation and p130Cas-Venus variants to focal adhesions. A) Cell immunostaining was performed on BT-549 cells grown on fibronectin. Phosphorylated p130Cas SD was detected using pCAS-165 antibody followed by the FITC-conjugated anti-rabbit IgG (green). F-actin, including stress fibers associated with focal adhesions, was visualized using Alexa 594-phalloidin (red). Bar indicates 20 μm. B) TIRF microscopy was used to show the localization of WT and 15F p130Cas-Venus variants to focal adhesions within BT-549 cells. Cells expressing p130Cas-Venus variants (green) were transiently transfected with plasmid mCherry-C1-paxillin (red), an established focal adhesion marker. The images show focal adhesions located near the cell peripheries. Bars indicate 5 μm.
The expression and tyrosine phosphorylation of FAK, another prominent Src substrate in focal adhesions, were also examined. Similar to the results obtained for p130Cas, known sites of Src-mediated FAK phosphorylation, including Tyr-576/577 (Figure 1D) and Tyr-861 (Figure 1E), were detected at relatively high levels in the BT-549 cell line. The FAK autophosphorylation site, Tyr-397 (Figure 1F), showed much less variation in its phosphorylation level among the breast cancer cell lines, although this site also tended to be less phosphorylated in the epithelial-like ER+ lines. These findings further indicate that Src-mediated signaling through cell/ECM adhesion substrates is a common feature of mesenchymal-like ER− breast cancer cells, and is particularly elevated in the BT-549 cell line.
To examine Src activity in the breast cancer cell lines, an antibody that recognizes the Tyr-419 autophosphorylation site in the kinase domain activation loop was used for immunoblotting. This analysis indicated that Src activity was uniformly higher in the mesenchymal-like ER− breast cancer cell lines as compared to the epithelial-like ER+ lines (Figure 1H). Notably, the BT-549 cell line did not exhibit an exceptionally high degree of Src activation, indicating that additional factors contribute to the exceptionally high levels of p130Cas and FAK phosphorylation in these cells.
Taken together, these immunoblotting data indicate that adhesion-associated Src signaling, including p130Cas SD tyrosine phosphorylation, is commonly associated with the mesenchymal-like, invasive phenotype of ER− breast cancer cells.
Knockdown and rescue of p130Cas expression in BT-549 breast cancer cells
To evaluate the role of p130Cas in conferring the neoplastic and/or malignant properties to mesenchymal-like ER-breast cancer cells, we used RNA interference to reduce p130Cas expression in the BT-549 cell line. BT-459 cells were chosen because they exhibit the greatest extent of p130Cas SD tyrosine phosphorylation. For these studies, BT-549 cells were transiently transfected with a duplex siRNA that targets human p130Cas (designated “CAS”), or a nontargeting (NT) control. The CAS siRNA consistently reduced p130Cas protein levels by ~70% (maximum at 48 hours post-transfection), compared to the NT control which had no effect on p130Cas expression (Figure 3, top panel, and data not shown). Because “off-target” effects of siRNAs are a concern when interpreting results from this loss-of-function approach,34 a rescue control was included in our p130Cas knockdown studies. For the rescue, mouse p130Cas carrying a C-terminal Venus fluorophore tag was stably expressed in BT-549 cells. The mouse p130Cas transcript is refractory to the human CAS siRNA, and the Venus tag reduces SDS-PAGE mobility such that the rescue protein can be readily distinguished from endogenous p130Cas.
Figure 3.
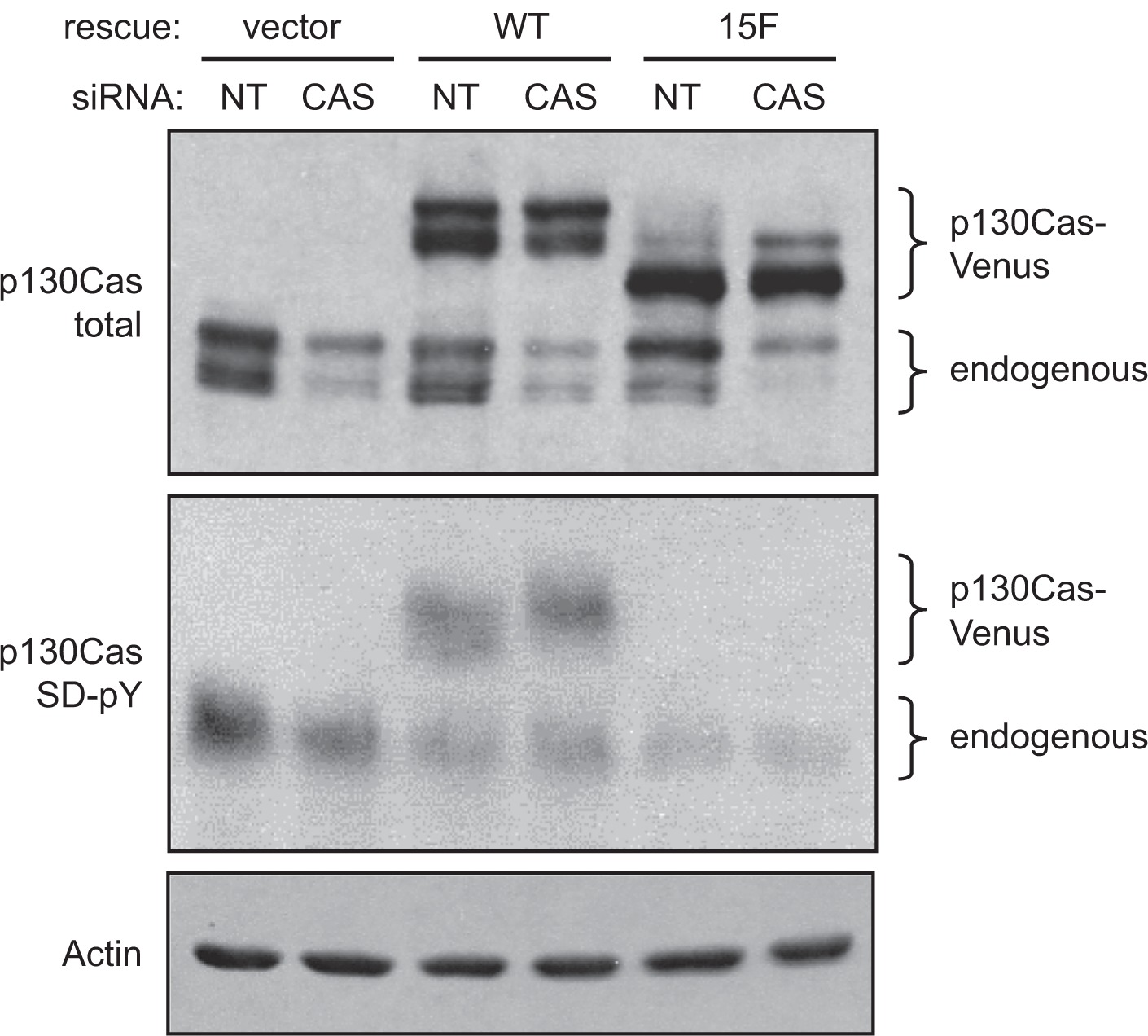
Knockdown of p130Cas in BT-549 cells and expression of siRNA-resistant mouse p130Cas-Venus variants. BT-549 cells transfected with either the CAS siRNA or NT control siRNA were lysed 48 hours post-transfection, and total cell lysates were assessed by immunoblot analysis of replicate blots. The p130Cas knockdown was performed on BT-549 cells that stably express either WT mouse p130Cas (resistant to the siRNA) or the signaling-deficient 15F mouse p130Cas variant, both carrying the Venus fluorophore tag. Cells stably expressing Venus only (empty rescue vector) were also included in the analysis. (Top panel) Both endogenous p130Cas and the p130Cas-Venus variants were detected by immunoblotting with the CAS-TL antibody. (Middle panel) Tyrosine phosphorylation of the p130Cas SD was assessed using a mixture of phosphospecific antibodies (pCAS-165, pCAS-249, pCAS-410). Prior to lysis, cells were treated with vanadate to enhance the detection of p130Cas SD tyrosine phosphorylation. (Bottom panel) Actin was detected as a loading control.
BT-549 cell populations that stably express mouse wild type (WT) p130Cas-Venus were prepared using a retroviral vector and sorting to select cells expressing the Venus fluorophore. As a negative control for the rescue, cells expressing a signaling-deficient p130Cas-Venus variant (15F, in which all 15 YxxP tyrosines in the SD were mutated to phenylalanine) were similarly prepared. As an additional control, cells expressing Venus alone (rescue vector only) were also prepared. Expression of the p130Cas-Venus variants was confirmed by immunoblotting (Figure 3, top panel). Transfection of the human CAS siRNA effectively reduced endogenous p130Cas expression but had no effect on the expression of the mouse p130Cas-Venus variants (Figure 3, top panel). The signaling capacity of WT p130Cas-Venus was demonstrated by immunoblotting using the pCas antibodies specific for phosphorylated SD YxxP tyrosines (Figure 3, middle panel; note that tyrosine-phosphorylated p130Cas migrates more slowly as a much broader band as compared to the unphosphorylated protein). As expected, the pCas antibodies did not recognize 15F p130Cas-Venus. The efficient focal adhesion targeting of both WT and 15F p130Cas-Venus variants was also documented, using TIRF microscopy (Figure 2B).
p130Cas SD tyrosine phosphorylation promotes BT-549 cell migration and invasion
Utilizing our system for knockdown and rescue of p130Cas expression, we initially investigated the role of p130Cas signaling in BT-549 cell migration using monolayer scratch wound assays. Cells at the wound edge were monitored by time-lapse microscopy, and the rate of migration was determined for 120 individual cells per experimental condition. In BT-549 cells expressing the empty rescue vector, transfection with the CAS siRNA significantly reduced the cell migration rate by ~40% as compared to cells transfected with the NT siRNA (Figure 4). This migration defect was largely rescued by expression of WT mouse p130Cas-Venus, but not by the signaling-deficient 15F p130Cas-Venus variant (Figure 4), indicating that p130Cas signaling via SD tyrosine phosphorylation promotes BT-549 cell migration. The effect of p130Cas knockdown and rescue on BT-549 cell migration was also examined using a modified Boyden chamber assay, in which cells were allowed to migrate through porous filters over a 24-hour period. In this assay, the CAS siRNA significantly reduced the number of migrating cells, by ~3-fold, compared to the NT siRNA (Figure 5A). Again, WT p130Cas-Venus, but not the 15F variant, was able to rescue the Boyden chamber migration defect caused by the CAS siRNA (Figure 5A).
Figure 4.
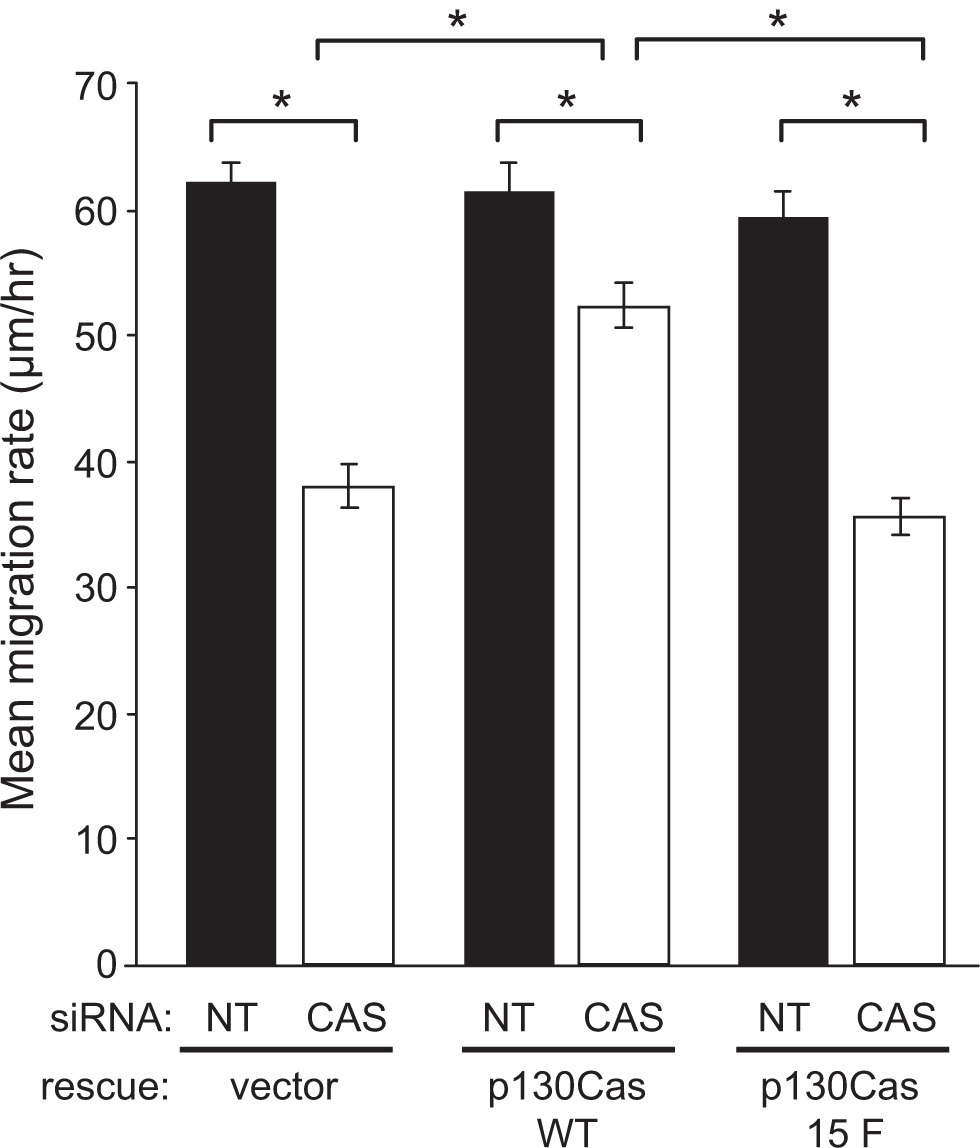
p130Cas SD tyrosine phosphorylation is required for efficient BT-549 cell migration. BT-549 cells stably expressing mouse p130Cas-Venus variants (WT vs 15F) or Venus only (rescue vector) were transfected with either the CAS or NT siRNA and analyzed for migration by scratch wound assay. After wounding, cell movement into the denuded area was recorded over a 10-hour period and mean migration rates (distance in μm/hour) were determined from measurements of the distance moved by 120 individual cells from each experimental condition. Error bars represent standard error of the mean. Statistical significance was determined by Student’s t-test (*P < 0.001).
Figure 5.
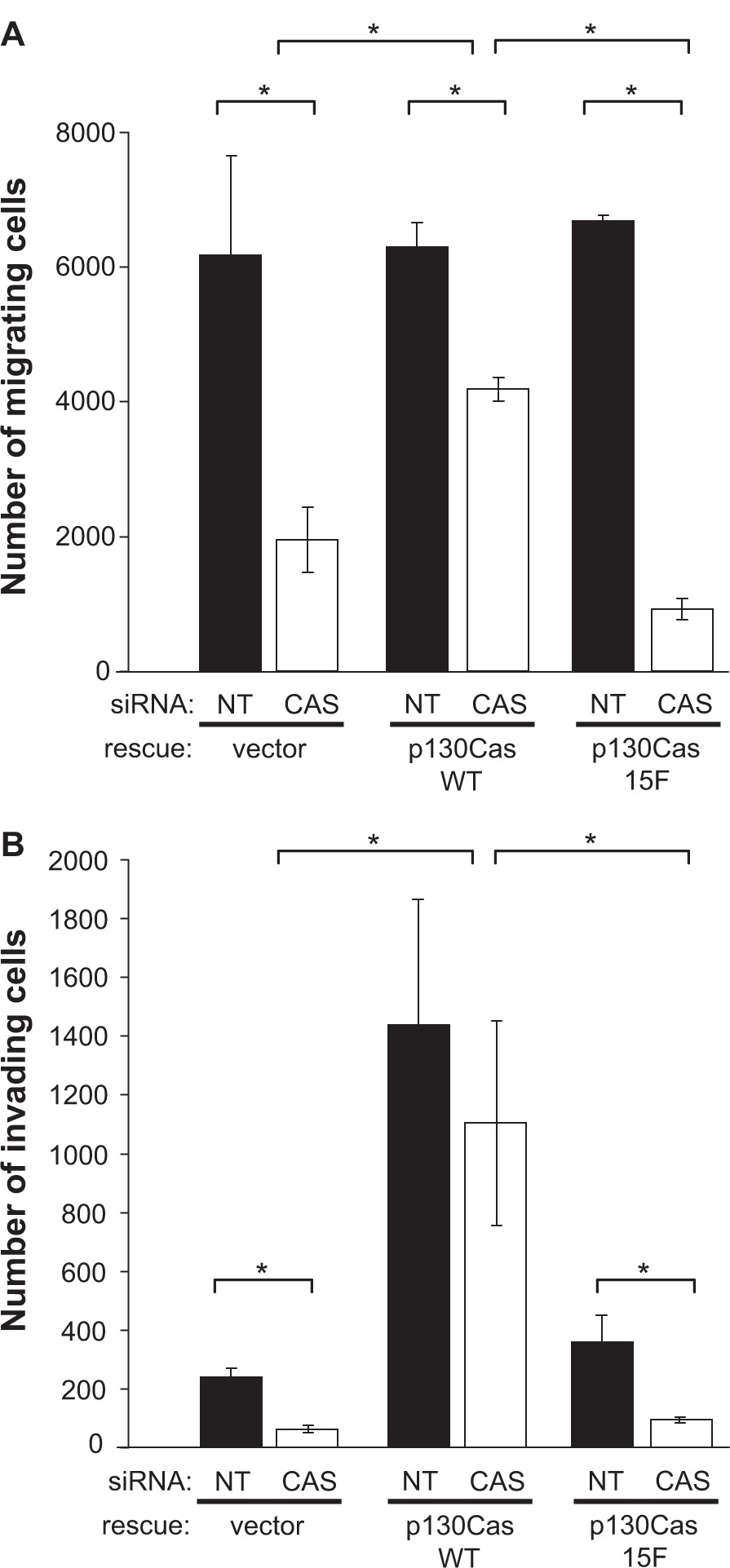
p130Cas SD tyrosine phosphorylation promotes BT549 cell invasion. BT549 cells stably expressing mouse p130Cas-Venus variants (WT vs 15F) or Venus only (rescue vector) were transfected with either the CAS or NT siRNA and analyzed for migration and invasion by modified Boyden chamber assays. For each experimental condition, triplicate assays were performed wherein 100,000 cells suspended in serum-free media were placed in the top of the chamber and allowed to migrate, over 24 hours, through porous filters towards media containing 10% serum. A) Migration through uncoated filters. B) Invasion through Matrigel-coated filters. Mean numbers of migrating or invading cells per chamber are shown for a representative experiment. Error bars represent the standard error of the mean. Statistical significance was determined by Student’s t-test (*P < 0.05).
The role of p130Cas in the ability of BT-549 cells to degrade and invade through the ECM was investigated using Matrigel invasion chambers. Transfection of the CAS siRNA resulted in a significant reduction in cell invasion through the Matrigel chamber over a 24-hour period, in comparison to the NT control (Figure 5B). Expression of WT p130Cas-Venus, but not 15F CAS-Venus, restored the invasive capacity of BT-549 cells (Figure 5B). Indeed, expression of WT p130Cas-Venus, but not 15F p130Cas-Venus, greatly enhanced the invasive capacity of BT-549 cells as compared to cells expressing the empty rescue vector (Figure 5B). Thus p130Cas SD tyrosine phosphorylation promotes the invasive capacity of BT-549 cells, in a manner that exceeds the capacity for cell migration.
p130Cas SD tyrosine phosphorylation promotes BT-549 cell proliferation and survival
In addition to having a role in cell motility and migration, integrin-mediated cell/ECM adhesions can transmit proliferation and survival signals into the cell. To determine if p130Cas signaling contributes to BT-549 cell proliferation, growth curves were generated for cell populations under the various experimental conditions of p130Cas knockdown and rescue. For cells expressing the empty rescue vector, transfection with the CAS siRNA resulted in a significant decrease in the growth rate, as compared to transfection with the NT siRNA (Figure 6, compare CAS:vector only to NT:vector only). Expression of WT p130Cas-Venus effectively rescued this proliferation deficiency caused by the CAS siRNA (Figure 6, compare CAS:p130Cas(WT) to CAS:vector only). However 15F p130Cas-Venus was unable to achieve the rescue (Figure 6, compare CAS:p130Cas(15F) to CAS:p130Cas(WT)), indicating that p130Cas signaling has a positive impact on BT-549 cell proliferation.
Figure 6.
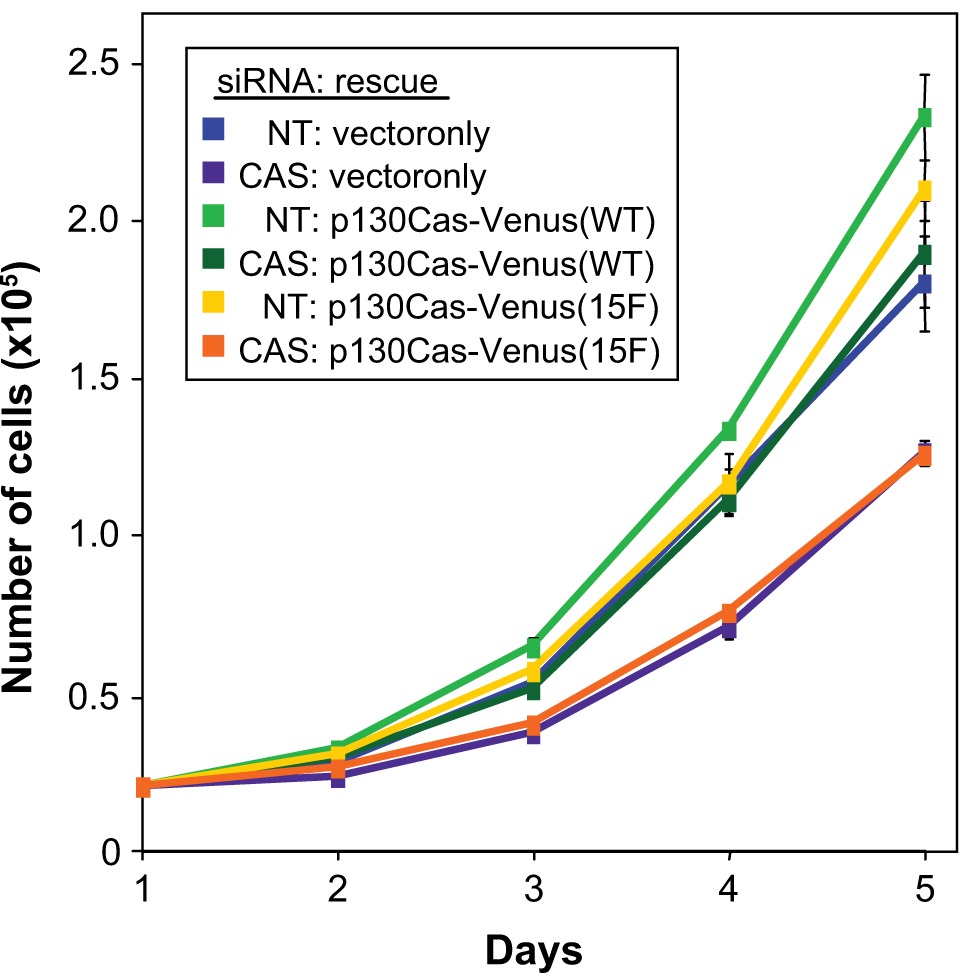
p130Cas SD tyrosine phosphorylation is required for efficient BT-549 cell proliferation. BT-549 cells stably expressing mouse p130Cas-Venus variants (WT vs 15F) or Venus only (rescue vector) were transfected with either the CAS or NT siRNA and analyzed for proliferation rate by determining growth curves. For each experimental condition, 20,000 cells were plated in replicate dishes in media containing 10% FBS and cells were counted, in triplicate, at daily intervals over the subsequent five-day period. Shown are resulting adherent growth curves from a representative experiment, obtained by plotting the mean number of cells counted at each time point Error bars represent standard deviations. Statistical significance was assessed on days 3–5 by Student’s t-test. Proliferation rates of two populations, CAS siRNA:vector only (purple line) and CAS siRNA:p130Cas-Venus (15F) (orange line), were significantly less than the others (P < 0.002).
To determine whether p130Cas has a role in BT-549 cell survival signaling, we performed TUNEL assays to monitor apoptosis within the cells, 48 hours after siRNA transfection. The human CAS siRNA caused a significant ~2.5-fold increase in the percentage of TUNEL-positive cells (Figure 7), indicative of apoptosis. Expression of WT p130Cas-Venus rescued the apoptosis triggered by the CAS siRNA, while 15F p130Cas-Venus did not (Figure 7). Thus a survival signaling pathway in BT-549 breast cancer cells appears to be activated in response to p130Cas SD tyrosine phosphorylation.
Figure 7.
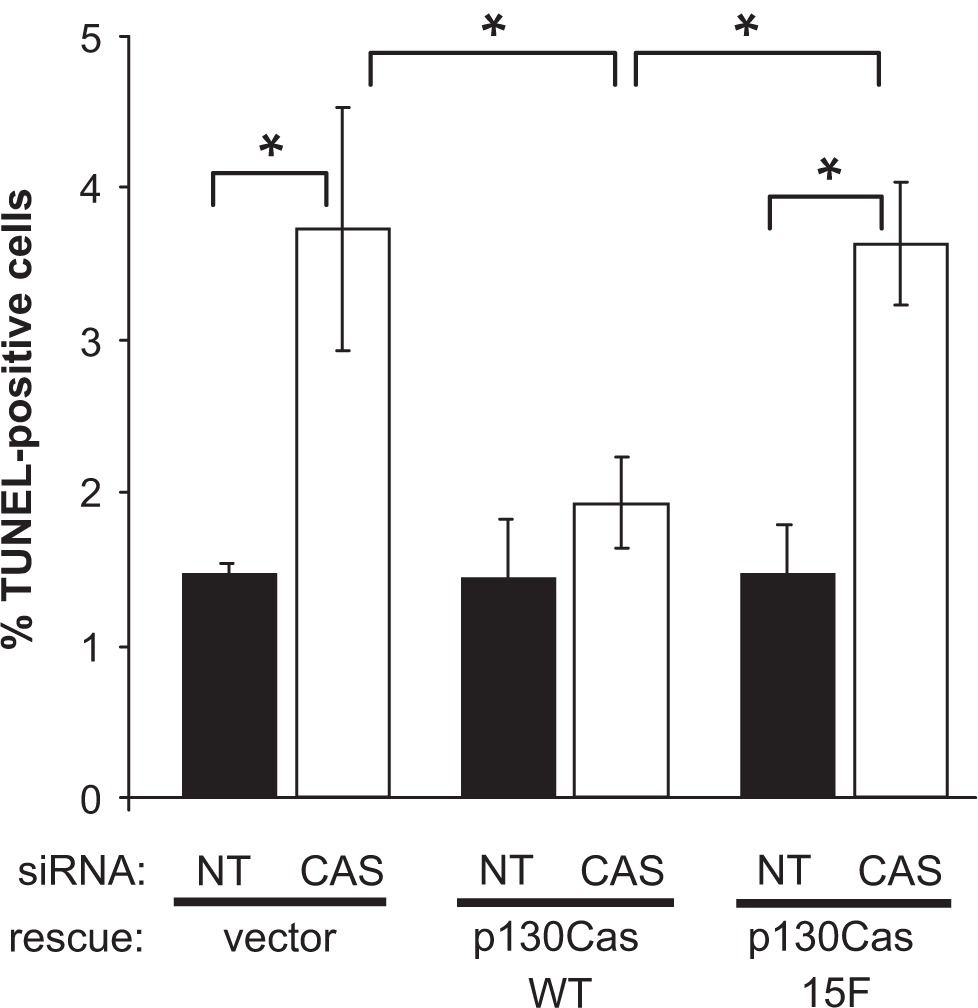
p130Cas SD tyrosine phosphorylation enhances BT-549 cell survival. BT-549 cells stably expressing mouse p130Cas-Venus variants (WT vs 15F) or Venus only (rescue vector) were transfected with either the CAS or NT siRNA and analyzed for apoptosis using the TUNEL assay. For each experimental condition, the percentage of TUNEL-positive cells was determined by scoring at least 1200 cells. The plots show average values obtained from three independent experiments. Error bars represent standard error of the mean. Significant differences were determined by Student’s t-test (*P < 0.05).
p130Cas SD tyrosine phosphorylation promotes AKT activation in BT-549 cells
The protein kinase AKT has been recognized as a mediator of pro-survival, anti-apoptotic, signaling pathways downstream of integrin-mediated adhesion in epithelial cells.35,36 Therefore, we examined AKT levels and activation state within the BT-549 cells under the various experimental conditions of p130Cas knockdown and rescue. AKT activation was assessed by immunoblotting with two different phosphospecific antibodies: pThr-308 in the kinase domain activation loop and pSer-473 in the C-terminal region. Phosphorylation of both sites is required for full activation of AKT.37 In BT-549 cells carrying the empty rescue vector, transfection of the human CAS siRNA resulted in a significant reduction in the phosphorylation of AKT Thr-308 and Ser-473, compared to the NT siRNA, even though the total AKT protein level became elevated (Figure 8, compare lanes 1 and 2). Expression of WT p130Cas-Venus, but not 15F p130Cas-Venus, rescued the defect in AKT activation caused by the CAS siRNA (Figure 8). Thus p130Cas signaling via SD tyrosine phosphorylation is part of a pathway leading to activation of AKT.
Figure 8.
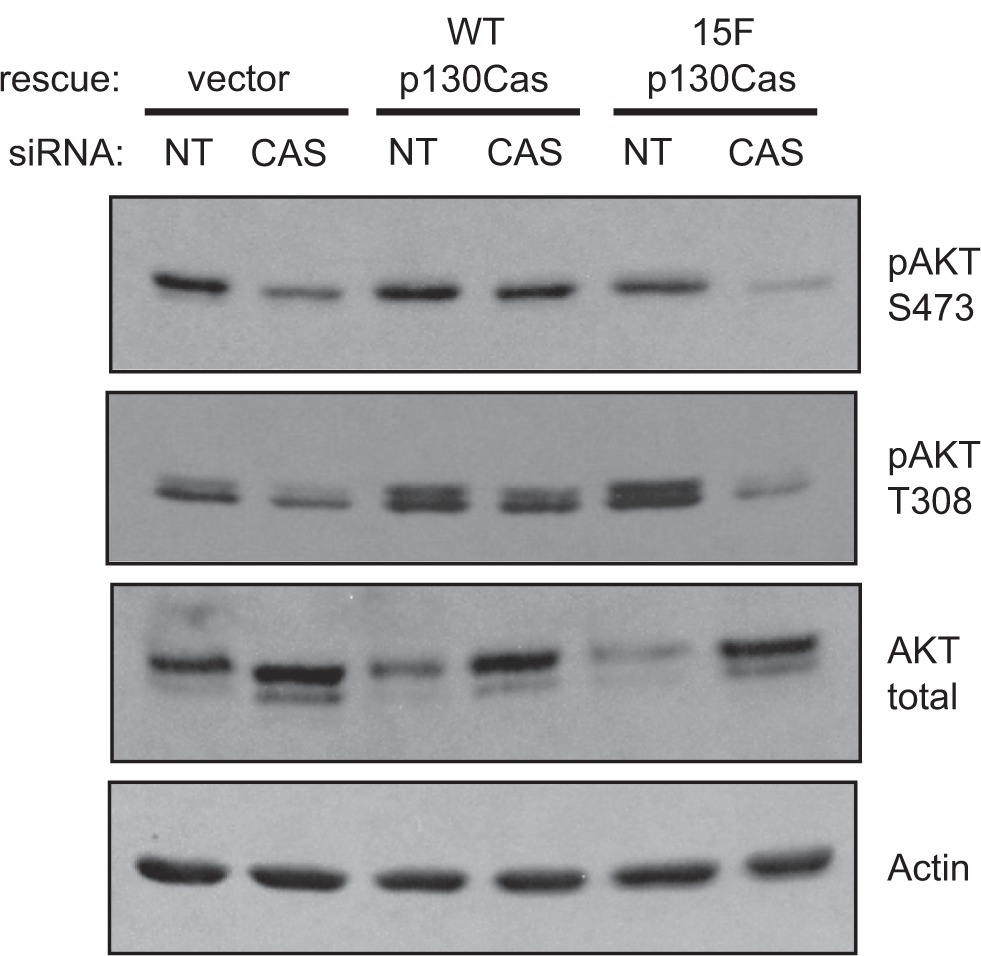
p130Cas SD tyrosine phosphorylation promotes AKT activation in BT-549 cells. BT-549 cells stably expressing mouse p130Cas-Venus variants (WT vs 15F) or Venus only (rescue vector) were transfected with either the CAS or NT siRNA. Forty-eight hours post-transfection, total cell lysates were prepared and analyzed by immunoblotting. (Top three panels) The activation state of AKT was assessed using phosphospecific antibodies against either the Ser-473 or Thr-308 sites, relative to the total AKT protein levels. (Bottom panel) Actin was detected as a loading control.
Note: This analysis was performed on replicate blots of the same samples used in Figure 3 to assess p130Cas.
Discussion
While past studies on p130Cas in breast cancer have focused on its role in conferring antiestrogen resistance to ER+ cells, little has been learned regarding the role of p130Cas in ER− breast cancer. Compared to ER+ breast cancer cells, ER− cells generally have a less differentiated mesenchymal phenotype and are highly invasive. This more aggressive nature, together with the desire for targeted approaches for therapy, make understanding the signaling mechanisms that drive the neoplastic and malignant behavior of ER− breast cancer cells an imperative. Previous studies implicating p130Cas SD tyrosine phosphorylation in the invasive behavior of Src-transformed mouse fibroblasts14,15 led us to hypothesize that p130Cas similarly contributed to malignant behavior of the mesenchymal-like ER− breast cancer cells.
We found elevated p130Cas signaling, as assessed by SD tyrosine phosphorylation, to be a distinguishing feature of ER− breast cancer cell lines as compared to ER+ lines. Two of the ER− lines included in our analysis, Hs578T and particularly BT-549, exhibited markedly higher levels of p130Cas SD phosphorylation that could be detected even in the absence of vanadate treatment to block tyrosine phosphatases. Of the ER− lines we analyzed, MDA-MB-435s cells exhibited a much lower level of p130Cas SD phosphorylation, which is notable in that these cells are reported to be the least invasive.27,28 Also it should be noted that the breast cancer origin of MDA-MB-435s parental cells has been questioned on the basis of gene expression profile analysis.38 Of the epithelial-like breast cancer cell lines, p130Cas SD phosphorylation was detected only in the ER− MDA-MB-468 cells, and at a relatively low level. MDA-MB-468 cells are noted for expressing high levels of the EGF receptor, which may impact on the observed p130Cas SD phosphorylation, possibly through synergistic effects with Src.39,40 Indeed MDA-MB-468 cells also exhibited higher Src activity, compared to the other epithelial-like cell lines, as assessed by the kinase domain activation loop autophosphorylation site.
The mesenchymal-like ER− breast cancer cell lines may be expected to more actively establish integrin-mediated cell/ECM adhesions, as opposed to the cadherin-mediated cell-cell adhesions characteristic of the epithelial phenotype. That ER-breast cancer cells generally exhibit elevated signaling associated with integrin-mediated adhesion is borne out by our observation that FAK Tyr-397, a site of autophosphorylation associated with focal adhesion assembly,41 is phosphorylated in the ER− cell lines to a uniform level that is elevated in comparison to the ER+ lines. When phosphorylated, the FAK Tyr-397 site acts to recruit and activate Src,41 and this mechanism likely contributes to the increased Src activity apparent in the ER− lines as compared to the ER+ lines. That p130Cas SD tyrosine phosphorylation is also generally elevated in the ER− breast cancer cell lines can be taken as a further indication of the active integrin-mediated signaling in these cells.
Yet it is also evident that the increased p130Cas SD tyrosine phosphorylation observed in the ER− breast cancer cell lines is not strictly due to elevated Src activity. Most notably, the high level of p130Cas SD tyrosine phosphorylation observed in the BT-549 cell line is not associated with equally elevated Src activity or FAK autophosphorylation. The finding that FAK sites of phosphorylation by Src (Tyr-576/577 and Tyr-861) are highest in BT-549 cells, suggests that integrin-mediated Src signaling is generally elevated in these cells. A likely explanation as to why BT-549 cells have remarkably elevated levels of p130Cas and FAK phosphorylation is that these cells do not express the tumor suppressor phosphatase PTEN,42 which has been implicated in the negative regulation of integrin signaling including tyrosine phosphorylation of p130Cas and FAK.43,44 With their high level of p130Cas SD tyrosine phosphorylation, BT-549 cells appealed to us as an ideal model for investigating the role of p130Cas signaling in the neoplastic behavior of ER− breast cancer cells.
A role for p130Cas signaling in the motile behavior of fibroblasts has been well documented. For example, impaired migration capacity of p130Cas-null mouse embryo fibroblasts45 can be restored by ectopic expression of WT p130Cas but not by mutants lacking the sites of tyrosine phosphorylation in the SD.10 Here, we showed that siRNA-mediated knockdown of p130Cas significantly reduced BT-549 cell motility rate in a scratch wound assay and also in the number of cells migrating toward a serum attractant in the modified Boyden chamber assay. Moreover, these migratory deficiencies could be rescued by expression of WT p130Cas but not by the signaling-deficient 15F mutant, where the SD YxxP tyrosine residues were substituted with nonphosphorylatable phenylalanine residues. These findings are significant in that they document the importance of p130Cas SD tyrosine phosphorylation in the migratory behavior of epithelium-derived cancer cells that have undergone the mesenchymal transition. Using a similar experimental approach, p130Cas SD signaling has been shown to have an important role in the migration of the well-differentiated CaCo-2 human intestinal cells which maintain strong cell-cell adhesion and migrate as an epithelial sheet.46
High Src kinase activity has been associated with breast cancer progression,47–49 and p130Cas SD tyrosine phosphorylation has been implicated in the invasive behavior of Src-transformed fibroblasts.14,15 Thus a major objective of our study was to determine if this Src-mediated signaling event is also involved in the invasive capacity of ER− mesenchymal-like breast cancer cells. Indeed, we found that the p130Cas knockdown significantly reduced the capacity of BT-549 cells to invade through a Matrigel barrier, and the p130Cas 15F mutant was unable to rescue this invasion deficiency. Not only did expression of WT p130Cas-Venus fully rescue the invasion deficiency associated with the knockdown, it also significantly increased invasion when expressed in the control cells treated with the nontargeting siRNA (by ~6-fold as compared to the rescue vector only). Such a striking effect of WT p130Cas overexpression was not evident in our analyses of cell migration, proliferation, and survival. A possible explanation for the unique sensitivity of BT-549 cell invasive behavior to the level of WT p130Cas is that cellular process(es) specific to the invasion process become activated only when a high threshold level of SD tyrosine phosphorylation is achieved. This is consistent with past studies on Src-transformed mouse fibroblasts where p130Cas SD tyrosine phosphorylation was strongly implicated in the formation of invasive podosome structures and activation of a matrix-degrading metalloproteinase.14,15 We also note the lack of a dominant-negative effect of 15F p130Cas-Venus on BT-549 cell invasiveness, even though expression of this signaling-deficient variant resulted in a lower level of endogenous p130Cas SD tyrosine phosphorylation. This observation is consistent with the notion that the significant increase in invasive behavior occurs only when SD tyrosine phosphorylation is elevated above the level achieved by endogenous p130Cas.
Our p130Cas knockdown and rescue studies also implicated p130Cas in the proliferation of adherent BT-549 cells, and further associated this property with enhanced survival. Another study has similarly linked p130Cas knockdown to an apoptotic response in a breast carcinoma cell line that expresses high levels of the HER2 oncogene.21 Since BT-549 cells do not overexpress HER2, our findings indicate that the effect of p130Cas on cell survival can occur independently of concomitant HER2 oncogenic signaling. We further demonstrated that the survival deficiency associated with the knockdown could be rescued by WT p130Cas but not by the 15F mutant. Thus p130Cas SD tyrosine phosphorylation, which occurs prominently at sites of cell/ECM adhesion, appears to be an important signaling event in a breast cancer cell survival pathway.
The protein kinase AKT has been linked to cell survival resulting from integrin engagement.35 Thus we examined the consequences of p130Cas knockdown and rescue on AKT activation. We found AKT phosphorylation at sites (Thr-308 and at Ser-473) indicative of its activation to be much reduced as a consequence of p130Cas knockdown, and this deficiency of survival signaling could also be rescued by WT p130Cas but not by the 15F mutant. Using a dominantnegative approach, p130Cas SD tyrosine phosphorylation was also recently linked to AKT activation and cell survival in a tamoxifen-resistant ER+ breast cancer cell line.24 Thus AKT activation downstream of p130Cas SD tyrosine phosphorylation has been implicated in tumorigenic growth of both ER− breast cancer and antiestrogen-resistant ER+ breast cancer. How phosphorylation of p130Cas SD tyrosine sites promote AKT activation is not fully understood. Both the AKT Thr-308 and Ser-473 sites are known to be phosphorylated downstream of PI3K,50 so how SD phosphorylation might lead to PI3K activation is relevant. It has been reported that p130Cas interacts directly with the PI3K p85 subunit,51,52 but this interaction does not involve SD phosphorylation. A C-terminal region of 130Cas interacts with AND-34/BCAR3,53 which has been reported to activate PI3K,54 but this interaction is also independent of SD phosphorylation. The connection between p130Cas SD tyrosine phosphorylation and AKT activation may involve Rac activation mediated by p130Cas/Crk coupling.55 While Rac activation can occur downstream of PI3K, Rac can also promote activation of PI3K indicative of a positive feedback loop.56 The lack of PTEN in BT-549 cells, in addition to contributing to higher levels of p130Cas SD tyrosine phosphorylation, would further impact on AKT activation by virtue of loss of the well established PTEN function as a lipid phosphatase to inhibit PI3K.57 Thus elevated p130Cas SD signaling coupled to a PTEN deficiency could act in a cooperative or synergistic fashion to promote survival and tumorigenic growth in ER− or antiestrogen-resistant ER+ breast cancer.
Conclusion
We have found that elevated tyrosine phosphorylation of the p130Cas SD is a distinguishing feature of mesenchymal-like ER− breast cancer cell lines, and provided evidence from p130Cas knockdown and rescue studies that this signaling event contributes to migration, invasion, proliferation, and survival of one such cell line, BT-549. While our results support a role for p130Cas SD tyrosine phosphorylation in the malignant progression of ER− breast cancer, additional studies are needed to extend these observations to other ER− breast cancer cells. In addition to the interest in p130Cas as a target in the treatment of antiestrogen-resistant ER+ breast cancer, Src-mediated signaling though p130Cas may also be relevant in the development of new therapeutic strategies in the treatment of aggressive ER− breast cancer.
Acknowledgments
We thank our colleagues Irina Kaverina, Larisa Ryzhova, Leslie Meenderink, Dominique Donato, and Jeremy Goettel for advice and expert technical assistance. Supported by NIH grants R01-GM49882 and T32-CA119925, and by a postdoctoral fellowship from the Vanderbilt Scientist-Educator Program.
Footnotes
Disclosures
The authors report no conflicts of interest in this work.
References
- 1.Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602(2):114–130. doi: 10.1016/s0304-419x(02)00040-9. [DOI] [PubMed] [Google Scholar]
- 2.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22(4):337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 3.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23(48):7928–7946. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 4.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332(6161):272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds AB, Kanner SB, Wang HC, Parsons JT. Stable association of activated pp60src with two tyrosine-phosphorylated cellular proteins. Mol Cell Biol. 1989;9(9):3951–3958. doi: 10.1128/mcb.9.9.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakai R, Iwamatsu A, Hirano N, et al. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994;13(16):3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fonseca PM, Shin NY, Brábek J, Ryzhova L, Wu J, Hanks SK. Regulation and localization of CAS substrate domain tyrosine phosphorylation. Cell Signal. 2004;16(5):621–629. doi: 10.1016/j.cellsig.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of CAS substrate domain tyrosine phosphorylation by FAK and Src. Mol Cell Biol. 2001;21(22):7641–7652. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg GS, Alexander DB, Pellicena P, Zhang ZY, Tsuda H, Miller WT. Src phosphorylates Cas on tyrosine 253 to promote migration of transformed cells. J Biol Chem. 2003;278(47):46533–46540. doi: 10.1074/jbc.M307526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin NY, Dise RS, Schneider-Mergener J, Ritchie MD, Kilkenny DM, Hanks SK. Subsets of the major tyrosine phosphorylation sites in Crk-associated substrate (CAS) are sufficient to promote cell migration. J Biol Chem. 2004;279(37):38331–38337. doi: 10.1074/jbc.M404675200. [DOI] [PubMed] [Google Scholar]
- 11.Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140(4):961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Hamasaki H, Nakamoto T, et al. Differential regulation of cell migration, actin stress fiber organization, and cell transformation by functional domains of Crk-associated substrate. J Biol Chem. 2002;277(30):27265–27272. doi: 10.1074/jbc.M203063200. [DOI] [PubMed] [Google Scholar]
- 13.Rivera GM, Antoku S, Gelkop S, et al. Requirement of Nck adaptors for actin dynamics and cell migration stimulated by platelet-derived growth factor B. Proc Natl Acad Sci U S A. 2006;103(25):9536–9541. doi: 10.1073/pnas.0603786103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brábek J, Constancio SS, Shin NY, Pozzi A, Weaver AM, Hanks SK. CAS promotes invasiveness of Src-transformed cells. Oncogene. 2004;23(24):7406–7415. doi: 10.1038/sj.onc.1207965. [DOI] [PubMed] [Google Scholar]
- 15.Brábek J, Constancio SS, Siesser PF, Shin NY, Pozzi A, Hanks SK. Crk-associated substrate tyrosine phosphorylation sites are critical for invasion and metastasis of Src-transformed cells. Mol Cancer Res. 2005;3(6):307–315. doi: 10.1158/1541-7786.MCR-05-0015. [DOI] [PubMed] [Google Scholar]
- 16.Brinkman A, van der Flier S, Kok EM, Dorssers LC. BCAR1, a human homologue of the adapter protein p130Cas, and antiestrogen resistance in breast cancer cells. J Natl Cancer Inst. 2000;92(2):112–120. doi: 10.1093/jnci/92.2.112. [DOI] [PubMed] [Google Scholar]
- 17.van der Flier S, Brinkman A, Look MP, et al. Bcar1/p130Cas protein and primary breast cancer: prognosis and response to tamoxifen treatment. J Natl Cancer Inst. 2000;92(2):120–127. doi: 10.1093/jnci/92.2.120. [DOI] [PubMed] [Google Scholar]
- 18.Dorssers LC, Grebenchtchikov N, Brinkman A, et al. The prognostic value of BCAR1 in patients with primary breast cancer. Clin Cancer Res. 2004;10(18):6194–6202. doi: 10.1158/1078-0432.CCR-04-0444. [DOI] [PubMed] [Google Scholar]
- 19.Dorssers LC, van Agthoven T, Brinkman A, Veldscholte J, Smid M, Dechering KJ. Breast cancer oestrogen independence mediated by BCAR1 or BCAR3 genes is transmitted through mechanisms distinct from the oestrogen receptor signalling pathway or the epidermal growth factor receptor signaling pathway. Breast Cancer Res. 2005;7(1):R82–R92. doi: 10.1186/bcr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabodi S, Moro L, Baj G, et al. p130Cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. J Cell Sci. 2004;117(8):1603–1611. doi: 10.1242/jcs.01025. [DOI] [PubMed] [Google Scholar]
- 21.Cabodi S, Tinnirello A, Di Stefano P, et al. p130Cas as a new regulator of mammary epithelial cell proliferation, survival, and HER2-neu oncogene-dependent breast tumorigenesis. Cancer Res. 2006;66(9):4672–4680. doi: 10.1158/0008-5472.CAN-05-2909. [DOI] [PubMed] [Google Scholar]
- 22.Riggins RB, Thomas KS, Ta HQ, et al. Physical and functional interactions between Cas and c-Src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res. 2006;66(14):7007–7015. doi: 10.1158/0008-5472.CAN-05-3952. [DOI] [PubMed] [Google Scholar]
- 23.Cowell LN, Graham JD, Bouton AH, Clarke CL, O’Neill GM. Tamoxifen treatment promotes phosphorylation of the adhesion molecules, p130Cas/BCAR1, FAK and Src, via an adhesion-dependent pathway. Oncogene. 2006;25(58):7597–7607. doi: 10.1038/sj.onc.1209747. [DOI] [PubMed] [Google Scholar]
- 24.Soni S, Lin BT, August A, Nicholson RI, Kirsch KH. Expression of a phosphorylated p130(Cas) substrate domain attenuates the phosphatidylinositol 3-kinase/Akt survival pathway in tamoxifen resistant breast cancer cells. J Cell Biochem. 2009;107(2):364–375. doi: 10.1002/jcb.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheikh MS, Garcia M, Pujol P, Fontana JA, Rochefort H. Why are estrogen-receptor-negative breast cancers more aggressive than the estrogen-receptor-positive breast cancers? Invasion Metastasis. 1994–1995;14(1–6):329–336. [PubMed] [Google Scholar]
- 26.Rochefort H, Glondu M, Sahla ME, Platet N, Garcia M. How to target estrogen receptor-negative breast cancer? Endocr Relat Cancer. 2003;10(2):261–266. doi: 10.1677/erc.0.0100261. [DOI] [PubMed] [Google Scholar]
- 27.Thompson EW, Paik S, Brünner N, et al. Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J Cell Physiol. 1992;150(3):534–544. doi: 10.1002/jcp.1041500314. [DOI] [PubMed] [Google Scholar]
- 28.Sommers CL, Byers SW, Thompson EW, Torri JA, Gelmann EP. Differentiation state and invasiveness of human breast cancer cell lines. Breast Cancer Res Treat. 1994;31(2–3):325–335. doi: 10.1007/BF00666165. [DOI] [PubMed] [Google Scholar]
- 29.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83(3):249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 30.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci U S A. 1995;92(23):10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20(1):87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 33.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with Image. J Biophotonics International. 2004;11(7):36–42. [Google Scholar]
- 34.Whither RNAi? Nat Cell Biol. 2003;5(6):489–490. doi: 10.1038/ncb0603-490. Editorial. [DOI] [PubMed] [Google Scholar]
- 35.Khwaja A, Rodriguez-Viciana P, Wennström S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16(10):2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng TL, Symons M, Jou TS. Regulation of anoikis by Cdc42 and Rac1. Exp Cell Res. 2004;295(2):497–511. doi: 10.1016/j.yexcr.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15(23):6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 38.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24(3):227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 39.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274(12):8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 40.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96(4):1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116(8):1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 42.Weng LP, Smith WM, Dahia PL, et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59(22):5808–5814. [PubMed] [Google Scholar]
- 43.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 44.Tamura M, Gu J, Takino T, Yamada KM. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999;59(2):442–449. [PubMed] [Google Scholar]
- 45.Honda H, Nakamoto T, Sakai R, Hirai H. p130(Cas), an assembling molecule of actin filaments, promotes cell movement, cell migration, and cell spreading in fibroblasts. Biochem Biophys Res Commun. 1999;262(1):25–30. doi: 10.1006/bbrc.1999.1162. [DOI] [PubMed] [Google Scholar]
- 46.Sanders MA, Basson MD. p130Cas but not paxillin is essential for Caco-2 intestinal epithelial cell spreading and migration on collagen IV. J Biol Chem. 2005;280(25):23516–23522. doi: 10.1074/jbc.M413165200. [DOI] [PubMed] [Google Scholar]
- 47.Lotz M, Wang HH, Cance W, Matthews J, Pories S. Epidermal growth factor stimulation can substitute for c-Src overexpression in promoting breast carcinoma invasion. J Surg Res. 2003;109(2):123–129. doi: 10.1016/s0022-4804(02)00100-2. [DOI] [PubMed] [Google Scholar]
- 48.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res. 2002;8(7):2430–2436. [PubMed] [Google Scholar]
- 49.Myoui A, Nishimura R, Williams PJ, et al. C-Src tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003;63(16):5028–5033. [PubMed] [Google Scholar]
- 50.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88(4):435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 51.Li E, Stupack DG, Brown SL, Klemke R, Schlaepfer DD, Nemerow GR. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275(19):14729–14735. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- 52.Riggins RB, DeBerry RM, Toosarvandani MD, Bouton AH. Src-dependent association of Cas and p85 phosphatidylinositol 3’-kinase in v-crk-transformed cells. Mol Cancer Res. 2003;1(6):428–437. [PubMed] [Google Scholar]
- 53.Gotoh T, Cai D, Tian X, Feig LA, Lerner A. p130Cas regulates the activity of AND-34, a novel Ral, Rap1, and R-Ras guanine nucleotide exchange factor. J Biol Chem. 2000;275(39):30118–30123. doi: 10.1074/jbc.M003074200. [DOI] [PubMed] [Google Scholar]
- 54.Felekkis KN, Narsimhan RP, Near R, et al. AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. Mol Cancer Res. 2005;3(1):32–41. [PubMed] [Google Scholar]
- 55.Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim Biophys Acta. 2004;1692(2–3):63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Barber MA, Welch HC. PI3K and RAC signaling in leukocyte and cancer cell migration. Bull Cancer. 2006;93(5):E44–E52. [PubMed] [Google Scholar]
- 57.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]