

Abstract
We report the long-term follow-up results of a phase II trial of thalidomide for early stage multiple myeloma (MM). Patients were eligible if they had smoldering multiple myeloma (SMM) or indolent MM without need for immediate therapy. Thalidomide was initiated at a dose of 200 mg/day and adjusted as tolerated. Disease progression was defined using modified American Society of Hematology/Food and Drug Administration consensus panel criteria for SMM. Thirty-one patients were enrolled; 29 (19 SMM, 10 indolent MM) were eligible. The median age was 61 years. Median follow up of living patients was 10.2 years (range, 7.5–11.0 years). Ten patients (34%) had a partial response (PR) and nine had minimal response (MR) for an MR plus PR rate of 66%. The median time to progression (TTP) to symptomatic myeloma was 35 months. Median TTP was 61 months in those achieving PR, 39 months with MR, and 9 months among those failing to achieve either MR or PR, p=0.005. Median overall survival from diagnosis was 86 months; median survival from onset of symptomatic myeloma was 49 months. Grade 3–4 non-hematologic adverse events were noted in 55% of patients. Randomized trials are needed to determine the role of early therapy in SMM.
Keywords: myeloma, Thalidomide, therapy
Introduction
Smoldering multiple myeloma (SMM) is an asymptomatic plasma cell dyscrasia characterized by a higher tumor burden than MGUS, and without any evidence of end-organ damage.[1] Patients with SMM have a high risk of progression to symptomatic multiple myeloma. Overall, different series have shown that the diagnosis of SMM makes up approximately 10%–15% of all patients with MM.[2] The risk for progression to active disease in the Mayo Clinic study was 10% per year for the first 5 years, 3% for the next five years, and 1% per year after that time for an over-all progression rate of 73% over 15 years. [1] Despite advances in the treatment of MM, the median survival remains 4 to 5 years.[3] The high risk of progression of SMM to active disease and the relatively short median survival of MM makes it important to investigate preventive strategies aimed at decreasing the time to progression (TTP) to active disease.
In 2003, we reported on the single-agent activity of thalidomide in patients with SMM or indolent MM (minimal end-organ damage without need for immediate anti-MM therapy).[4] With a median follow-up of just over 2 years, the median TTP was 22.5 months, and the 2-year progression free survival (PFS) was 47%. We now present the long-term results of this study with a median of follow up of over 10 years in surviving patients, including an update on adverse effects (AEs).
Materials and Methods
Eligibility
Patients were deemed eligible for enrollment in the study if they met the diagnostic criteria for SMM or indolent MM.[5] SMM was defined as presence of a serum IgG or IgA M protein ≥ 3gm/dL and/or bone marrow plasma cells ≥ 10%, plus absence of anemia, hypercalcemia, lytic bone lesions, or renal failure that can be attributed to the plasma cell proliferative disorder.[5–7] Indolent MM was defined as bone marrow plasma cells ≥ 10%, mild anemia or few small lytic bone lesions, absence of symptoms, and no immediate need for treatment.[5] All patients were required to have ≥10% bone marrow plasma cells and measurable disease defined as a serum monoclonal spike (M spike) of ≥ 2 gm/dL and/or urine M spike ≥ 400 mg/24 hours. Patients had to be also willing to adhere to the reproductive requirements of the System of Thalidomide Education and Prescribing Safety program.[8] All patients gave written informed consent prior to enrollment in the study. Approval for the study and written informed consent form was obtained from the Mayo Institutional Review Board.
Treatment schedule
Thalidomide was initiated at 200 mg/day for 2 weeks, and thereafter increased as tolerated by 200 mg/day every two weeks to the maximum dose of 800 mg/day. Patients were evaluated every 4 weeks for response and potential drug toxicities. The thalidomide could be decreased to 50 mg/day if needed due to serious toxicities at a higher dose. Patients were continued on treatment until disease progression or serious toxicity for 12 months. Patients could continue therapy beyond 12 months at physicians’ discretion; those with less than minor response to therapy were only treated for one year.
Response and toxicity criteria
Response categories (partial and complete response) were assessed using the International Myeloma Working Group uniform response criteria.[9] Partial response (PR) was defined as ≥50% reduction in the level of the serum monoclonal (M) protein and a reduction in 24-hour urinary M protein ≥90% or to <200 mg, plus no increase in the number or size of lytic bone lesions or any other evidence of progressive disease. Complete response (CR) required confirmed disappearance of the monoclonal protein in the serum and urine by immunofixation and <5% plasma cells on bone marrow examination. In addition, MR was defined as a 25% reduction in serum and/or urine M protein. All response categories required confirmation by two consecutive measurements. In addition, minimal response was assessed using the standard European Group for Blood and Bone Marrow Transplant (ie, Blade criteria).[10] Progressive disease was defined according to modified American Society of Hematology/Food and Drug Administration consensus panel on endpoints in myeloma as development of symptomatic myeloma or initiation of conventional myeloma therapy for progressive disease.[11]
Overall survival was measured from the date of study entry until the date of death or last follow-up. PFS was measured from the date of study entry until death or progression to active disease, whichever was earlier. TTP was measured from the date of study entry until progression to active disease. Toxicities were graded using the National Cancer Institute Common toxicity criteria (version 2).
Statistical analysis
Ninety-five percent confidence intervals for the confirmed response probability was calculated using exact binomial 95% confidence intervals. Toxicity incidence was estimated and summarized using frequency and descriptive techniques to assess any patterns. The Fisher exact test was used to compare differences in nominal variables. For continuous variables, the rank-sum test was used for unpaired comparisons and the Wilcoxon signed rank test was used for paired comparisons. Differences between survival curves were tested for statistical significance using two-tailed log-rank and Breslow-Gehan-Wilcoxon tests. Prognostic factors for survival, TTP and PFS were assessed using Cox’s proportional hazards model.
Results
Patient Characteristics
A total of 31 patients were enrolled between April 1999 and March 2002. Two were later found to be ineligible due to having received prior therapies and were excluded from the study. Patients were followed through June 2010, and median follow up of living patients from study entry was 10.2 years (range, 7.5–11.0 years). Nineteen of the eligible patients were diagnosed with SMM and 10 were diagnosed with indolent MM. Of the indolent MM patients, seven had small lytic lesions at the time of enrollment; 6 had hemoglobin less than 11gm/dL including 2 who had hemoglobin less than 10 gm/dL (9.3 gm/dL and 9.9 gm/dL). The median age was 61 years (range, 40–76). The median hemoglobin was 11.6gm/dL (range, 9.3–13.5 gm/dL). The median M protein was 3.1 gm/dL. Patient characteristics are outlined in Table I.
Table I.
Patient Characteristics
| Characteristic | All Patients | |
|---|---|---|
| No. | % | |
| Total number of eligible patients | 29 | |
| Sex | ||
| Male | 16 | 55 |
| Female | 13 | 45 |
| Smoldering/Indolent Myeloma | ||
| Smoldering Myeloma | 19 | 66 |
| Indolent Myeloma | 10 | 34 |
| Immunoglobulin heavy chain type | ||
| IgG | 24 | 83 |
| IgA | 3 | 10 |
| Light chain only (Bence Jones Protein) | 2 | 7 |
| Anemia (Hemoglobin <11 g/dL) | 6 | 21 |
| Lytic bone lesions | 7 | 24 |
| Beta 2-microglobulin >2.7 mg/L | 13 | 45 |
| Bone marrow plasma cell % ≥40% | 11 | 38 |
The median duration of therapy administered was 15 months, range 1–109 months. Three patients remain on active therapy at 93 (thalidomide dose, 50 mg; grade 1 neuropathy), 105 (thalidomide dose, 100 mg; no neuropathy), and 109 (thalidomide dose, 50 mg; grade 2 neuropathy) months, respectively.
Response
Ten patients (34%) had a partial response (PR) to the therapy; an additional 9 had a minor response (MR) for a combined PR and MR rate of 66%. Responses in M protein levels were accompanied by improvements in BM plasma cell percentages. The median time to partial response was 5 months (range 2–9 months).
Survival Analysis
The median TTP was 35 months (Figure 1); median PFS was also 35 months. There was a positive correlation between response to thalidomide and TTP. The median TTP was 61 months in those who achieved a PR, 39 months in those achieving MR, and 9 months in patients failing to achieve MR or PR, P=0.05 (log-rank); P=0.005 (Wilcoxon) (Figure 2). Corresponding median PFS durations were identical for the 3 groups. Eighteen patients have died; median overall survival from diagnosis was 86 months (Figure 3). The median survival measured from onset of symptomatic myeloma was 49 months.
Figure 1.
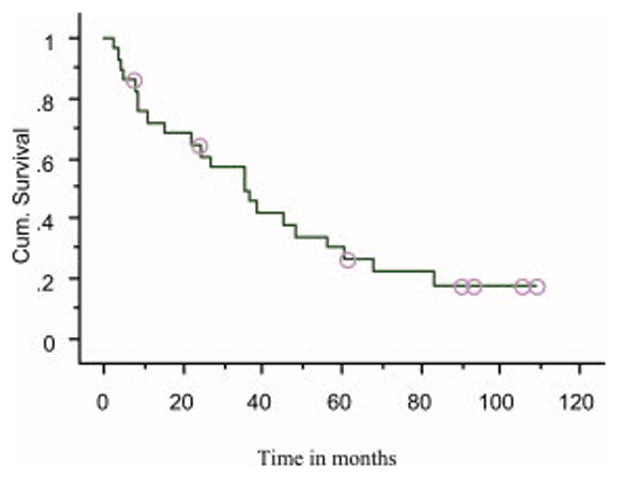
Time to progression (TTP) in patients receiving thalidomide for smoldering or indolent myeloma (median TTP = 35 months)
Figure 2.
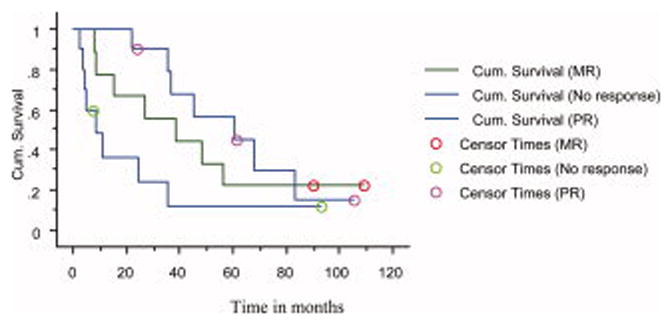
Time to Progression (TTP) by response status. Median TTP 61 months among those who achieved partial response (PR), 39 months among those who achieved minor response (MR), and 9 months among those who failed to achieve either a MR or PR, P=0.005 (Wilcoxon).
Figure 3.
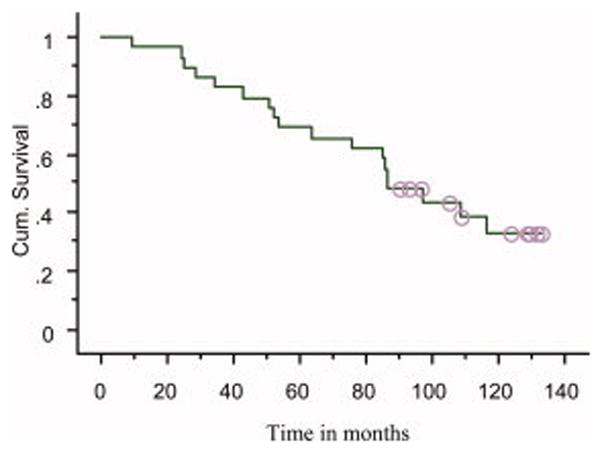
Overall survival in patients receiving thalidomide for smoldering or indolent myeloma (median survival = 86 months)
TTP was longer in patients with baseline bone marrow plasma cell percentage <40% (n=18) compared with those 40% or higher (n=11), median TTP 57 versus 27 months, respectively, P=0.03 (log-rank); P=0.16 (Wilcoxon).
Toxicities
Overall, 55% had at least one grade 3 or greater non-hematologic AE, including 16% who had at least one grade 4 toxicity. Three percent of patients had grade 3 or greater hematologic AE; no grade 4 or higher hematologic AEs were seen. The most common grade 3 toxicities experienced were neuropathy (sensory and/or motor) (14%), infection (10%), sedation (7%), and hypertension (7%). Grade 4 AEs included sinus bradycardia (7%) and infection (3%) and specifically did not include any form of peripheral neuropathy. Compared with the previous report in 2003, we saw an increase in AEs in many categories with longer duration of therapy, including neuropathy, infection, sedation, fatigue, bradycardia, ataxia, and hypothyroidism as shown in Table II.
Table II.
Toxicity Profile
| Toxicity | Percentage of patients experiencing toxicity in2003 report4 | Percentage of patients experiencing toxicity in current report |
|---|---|---|
| Skin rash | ||
| Grade 1–2 | 55 | 62 |
| Sedation | ||
| Grade 1–2 | 74 | 86 |
| Grade 3 | 6 | 7 |
| Grade 1–2 Constipation | 87 | 93 |
| Peripheral neuropathy | ||
| Grade 1–2 | 87 | 83 |
| Grade 3–4 | 3 | 14 |
| Fatigue/Weakness | ||
| Grade 1–2 | 65 | 72 |
| Grade 3 | 3 | 7 |
| Sinus bradycardia | ||
| Grade 1–2 | 23 | 28 |
| Grade 3–4 | 3 | 7 |
| Edema | ||
| Grade 1–2 | 16 | 21 |
| Grade 3 | 3 | 3 |
| Tremor | 35 | 41 |
| Grade 1–ataxia | 16 | 24 |
| Grade 3–4 Infection | 0 | 14 |
| Grade 3 hypertension | 0 | 3 |
| Hypothyroidism | 0 | 7 |
| Grade 3 hearing loss | 3 | 3 |
| Deep vein thrombosis | 3 | 3 |
Discussion
SMM makes up 10%–15% of all myeloma diagnoses overall, and 73% of patients will go on to develop symptomatic myeloma within 15 years.[1] This study was initiated shortly after the drug showed promising activity in relapsed myeloma.[12] The rationale for the trial was based on the anti-angiogenic properties of thalidomide,[13] and the hypothesis that the transition from SMM to MM is associated with increased bone marrow angiogenesis.[14] The updated data that we describe continues to be encouraging. The median TTP was 35 months among all responders, but the duration varied significantly by depth of response, with a median TTP of 61 months among those achieving a PR. In our study, there was a significant delay in TTP even in those patients who achieved a MR, therefore this is included in our response data. The effect of depth of response on TTP suggests a favorable therapeutic effect, but an alternative possibility is that response to thalidomide is an indirect prognostic factor for patients with SMM who have a more favorable underlying disease biology.
The median TTP in this study in patients treated with thalidomide is lower than the median TTP among similar patients (bone marrow plasma cells ≥10% plus either serum M spike ≥2gm/dL or urine M spike ≥400mg/day) seen in the cohort of smoldering myeloma patients studied by Kyle et al (48 months; personal communication). However, it is important to note that cohort studied by Kyle et al. specifically excluded those with indolent MM, whereas this thalidomide trial included 10 patients with indolent MM, 7 of whom had lytic disease. This makes a direct comparison between the two studies impossible, but it does help put the numbers in perspective. The patients in the cohort studied by Kyle et al with the highest tumor burden had an average TTP of 27 months,[1] and this group would more closely compare to our IMM patients.
Two other studies have evaluated the role of thalidomide in SMM. Weber et al studied 28 patients with SMM, and reported similar results with a PR rate of 36% with single-agent thalidomide.[15] The median time to response was similar (4.2 months) as in our study (5 months). Barlogie and colleagues treated 76 SMM patients with thalidomide (200 mg/day) and monthly pamidronate.[16] The PR rate was 25%. However, in contrast to our study, the patients achieving PR status had a shorter time to salvage therapy for disease progression. The reasons for the distinctly different results in this study compared to ours are not clear, but may be related to disease definition and differences in the inclusion criteria between the two studies, especially since the overall TTP was much longer in the study by Barlogie et al compared with ours.
Despite our results and those reported by others, we do not recommend the use of thalidomide for SMM. We need a better estimate of efficacy. In addition, benefits must be weighed against the many known side effects of thalidomide. There are significant adverse events associated with this regimen. In addition, there were some increased AEs since our last data was published. All of these were grade 1–2 except for one incident of grade 4 bradycardia (Table II). The emergence of new AEs, although not many, underscores the importance of continued, longer follow up of this study.
When this study was designed, thalidomide dose was escalated up to 800 mg per day in myeloma. We know now that 800 mg of thalidomide daily causes excessive toxicity. However, we employed aggressive dose-reductions, and most patients were able to decrease their dose to 50 mg to 200 mg daily without an impact on response. We would recommend any new SMM trials using thalidomide start at a daily dose of between 50 mg to 200 mg in order to decrease the number of AEs.
This study provides important information about the natural history and response to therapy of a subset of high-risk SMM patients. It also provides data on the long-term effects of thalidomide therapy. Randomized controlled trials are needed to study the impact of preventive strategies on the progression of SMM to MM and even more importantly, the impact this would have on overall survival. We are conducting a phase III trial of thalidomide plus zoledronic acid versus zoledronic acid alone in SMM. Preliminary results from a Spanish study show that lenalidomide, a newer and less toxic analog of thalidomide may be a better alternative for clinical trials in SMM, and may delay TTP compared with observation.[17] The Eastern Cooperative Oncology Group (ECOG) is expected to open a clinical trial of lenalidomide versus observation later this year.
Acknowledgments
Contract grant sponsor: National Cancer Institute, National Institutes of Health, Bethesda, MD; Contract grant numbers: CA62242, CA107476;
Footnotes
Conflict of interest statement
M.A.G has received honoraria from Celgene, Millenium, Genzyme and Amgen.
S.K. has a consultant/advisory relationship and has received research support from Celgene.
M.Q.L., A.D., T.E.W, have received research funding/grants from Celgene.
J. L. consultancy for Celgene.
R.A.K. has received honoraria from Celgene.
P.R.G. has received honoraria from Celgene and Amgen.
The remaining authors declare no conflict of interest.
References
- 1.Kyle RA, Remstein ED, Therneau TM, et al. Clinical Course and Prognosis of Smoldering (Asymptomatic) Multiple Myeloma. N Engl J Med. 2007;356:2582–2590. doi: 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- 2.Blade J, Dimopoulos M, Rosinol L, et al. Smoldering (asymptomatic) multiple myeloma: current diagnostic criteria, new predictors of outcome, and follow-up recommendations. Journal of Clinical Oncology. 2010;28:690–697. doi: 10.1200/JCO.2009.22.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajkumar SV, Gertz MA, Lacy MQ, et al. Thalidomide as initial therapy for early-stage myeloma. Leukemia. 2003;17:775–779. doi: 10.1038/sj.leu.2402866. [DOI] [PubMed] [Google Scholar]
- 5.Rajkumar SV, Dispenzieri A, Fonseca R, et al. Thalidomide for previously untreated indolent or smoldering multiple myeloma. Leukemia. 2001;15:1274–1276. doi: 10.1038/sj.leu.2402183. [DOI] [PubMed] [Google Scholar]
- 6.The International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–757. [PubMed] [Google Scholar]
- 7.Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia. 2009;23:3–9. doi: 10.1038/leu.2008.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeldis JB, Williams BA, Thomas SD, et al. S.T.E.P.S: a comprehensive program for controlling and monitoring access to thalidomide. Clinical Therapeutics. 1999;21:319–330. doi: 10.1016/s0149-2918(00)88289-2. [DOI] [PubMed] [Google Scholar]
- 9.Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–1473. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 10.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. British Journal of Haematology. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 11.Anderson KC, Kyle RA, Rajkumar SV, et al. Clinically relevant end points and new drug approvals for myeloma. Leukemia. 2007;22:231–239. doi: 10.1038/sj.leu.2405016. [DOI] [PubMed] [Google Scholar]
- 12.Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma [see comments] New England Journal of Medicine. 1999;341:1565–1571. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 13.D’Amato RJ, Loughnan MS, Flynn E, et al. Thalidomide is an inhibitor of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4082–4085. doi: 10.1073/pnas.91.9.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajkumar SV, Mesa RA, Fonseca R, et al. Bone marrow angiogenesis in 400 patients with monoclonal gammopathy of undetermined significance, multiple myeloma, and primary amyloidosis. Clin Cancer Res. 2002;8:2210–2216. [PubMed] [Google Scholar]
- 15.Weber D, Rankin K, Gavino M, et al. Thalidomide alone or with dexamethasone for previously untreated multiple myeloma. Journal of Clinical Oncology. 2003;21:16–19. doi: 10.1200/JCO.2003.03.139. [DOI] [PubMed] [Google Scholar]
- 16.Barlogie B, van Rhee F, Shaughnessy JD, Jr, et al. Seven-year median time to progression with thalidomide for smoldering myeloma: partial response identifies subset requiring earlier salvage therapy for symptomatic disease. Blood. 2008;112:3122–3125. doi: 10.1182/blood-2008-06-164228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mateos M-V, Lopez-Corral L, Hernandez MT, et al. Multicenter, Randomized, Open-Label, Phase III Trial of Lenalidomide-Dexamethasone (Len/dex) Vs Therapeutic Abstention in Smoldering Multiple Myeloma at High Risk of Progression to Symptomatic MM: Results of the First Interim Analysis. ASH Annual Meeting Abstracts; 2009. p. 614. [Google Scholar]