

Abstract
Apolipoprotein (apo) E has a storied history as a lipid transport protein. The integral association between cholesterol homeostasis and lipoprotein clearance from circulation are intimately related to apoE's function as a ligand for cell surface receptors of the low density lipoprotein receptor family. The receptor binding properties of apoE are strongly influenced by isoform specific amino acid differences as well as the lipidation state of the protein. As understanding of apoE as a structural component of circulating plasma lipoproteins has evolved, exciting developments in neurobiology have revitalized interest in apoE. The strong and enduring correlation between the apoE4 isoform and age of onset and increased risk of Alzheimer's disease has catapulted apoE to the forefront of neurobiology. Using genetic tools generated for study of apoE lipoprotein metabolism, transgenic “knock-in” and gene-disrupted mice are now favored models for study of its role in a variety of neurodegenerative diseases. Key structural knowledge of apoE and isoform specific differences is driving research activity designed to elucidate how a single amino acid change can manifest such profoundly significant pathological consequences. This review describes apoE through a lens of structure-based knowledge that leads to hypotheses that attempt to explain the functions of apoE and isoform specific effects relating to disease mechanism.
Keywords: Apolipoprotein E, cholesterol, cardiovascular disease, neurobiology, Alzheimer's disease
1. Introduction
Apolipoprotein E (apoE) is a potent modulator of plasma lipoprotein and cholesterol levels whose mode of action is mediated by interaction with members of the low-density lipoprotein (LDL) receptor family. Transgenic mice over-expressing apoE have decreased plasma cholesterol and are resistant to diet-induced atherosclerosis [1]. On the other hand, apoE-null mice manifest elevated plasma cholesterol and increased susceptibility to diet-induced atherosclerosis [2, 3]. In humans, the absence of apoE, or the presence of defective apoE, leads to type III hyperlipoproteinemia, characterized by premature atherosclerosis and accumulation of plasma cholesterol [4]. ApoE is synthesized with an 18 amino acid N-terminal signal peptide that undergoes intracellular processing before secretion of a mature 35 kDa glycoprotein containing 299 amino acids [5-7]. The protein is encoded by a 3.6 kbp, four-exon gene located on chromosome 19 [8, 9]. Plasma apoE originates predominantly from liver and, to a small but functionally significant extent, macrophages [10-12]. At the same time, apoE is expressed in a wide variety of other tissues including brain, spleen, lung, adrenal gland, ovary and kidney [13]. This broad tissue expression suggests apoE participates in additional biological processes, both related to and disparate from, lipid metabolism and cardiovascular disease. Indeed, support for this concept has emerged from studies linking apoE to innate immunity, normal brain function and a host of neurodegenerative disorders [14].
2. ApoE Structural Organization
In general, members of the class of exchangeable apolipoproteins are related by their high amphipathic α-helix content, which is critical to their function in lipoprotein particle stabilization. The opposing hydrophobic and hydrophilic faces of these helices allow exchangeable apolipoproteins to exist in alternate lipid-free and lipid-associated states and explains their activity as “detergents” capable of solubilizing lipophilic molecules [15]. Primary sequence [16, 17] and spectroscopic analyses [18], [19] reveal that apoE possesses a high content of α-helix secondary structure (∼62% in aqueous solution). Structure-function studies investigating its role in disease and cholesterol homeostasis revealed apoE is a two-domain protein [20]. Studies monitoring secondary structure content as a function of chaotrope concentration yielded a biphasic curve with transition midpoints at 0.7 and 2.5 M guanidine HCl. Limited proteolysis of full-length apoE generated 22 kDa and 10 kDa fragments that correspond to distinct N- and C-terminal domains, respectively [20]. This finding is consistent with the concept that the domains are separated by a flexible, protease sensitive loop. The C-terminal domain facilitates lipid binding and, in isolation, has lower stability and greater conformational flexibility than the N-terminal fragment. Analytical ultracentrifugation studies and C-terminal truncation analysis [21, 22] revealed that, as with other exchangeable apolipoproteins, full-length apoE forms multimeric complexes in aqueous solution [23]. Full-length apoE displays a propensity to form tetramers and this has been attributed to the C-terminus since the isolated N-terminal domain remains monomeric at concentrations up to 15 mg/mL [18]. Consistent with these observations, substitution of a limited number of bulky residues in the C-terminal domain by smaller polar residues results in a protein that is resistant to cross-linking, suggesting these residues play a role in apoE self association [24, 25].
Characterization of apoE from human subjects has revealed intriguing heterogeneity. Seminal isoelectric focusing experiments [26, 27] indicated the presence of charge variants while experiments with neuraminidase provided evidence that apoE is glycosylated [28]. Glycosyl moieties present include galactose, glucosamine, galactosamine, sialic acid, N-acetylglucosamine, and N-acetylgalactosamine [29]. Amino acid analysis identified Thr194 as the sole glycosylated residue and glycosylation was shown not to be necessary for apoE expression [30]. While some studies have linked alterations in apoE glycosylation state to disease phenotypes, the exact role of specific sugar moieties remains unknown. Hypotheses have also been advanced that propose glycosylation protects the loop region in which it resides from proteolytic cleavage [13].
Understanding of the relationship between apoE structure and function was greatly enhanced by determination of the three-dimensional structure of the N-terminal domain by X-ray crystallography [31] and, more recently, by nuclear magnetic resonance (NMR) spectroscopy [32] (Figure 1). These structures reveal four amphipathic helices that align in an up-and-down manner, forming an elongated globular helix bundle. The boundaries of the four major helices: helix 1 (residues 24-42), helix 2 (residues 54-81), helix 3 (residues 87-122) and helix 4 (residues 130-164) are augmented by helix 1′ (residues 44-53) and, in the NMR structure, helices N and C (residues 12-22 and 173-181, respectively). Although a segment of the C-terminal domain (residues 223-272) has been crystallized [33], a high-resolution structure has yet to be reported. Computer-based sequence algorithms predict residues 210-266 and 268-289 form α-helices [15, 34], with the latter believed to be involved in protein-protein interactions and/or self-association. Consistent with this prediction, recent biophysical studies of the C-terminal domain suggest that these helices are involved in close helix-helix contact, likely driving self-association of lipid-free apoE [35, 36].
Figure 1. ApoE3 NT structures by X-ray crystallography and NMR.
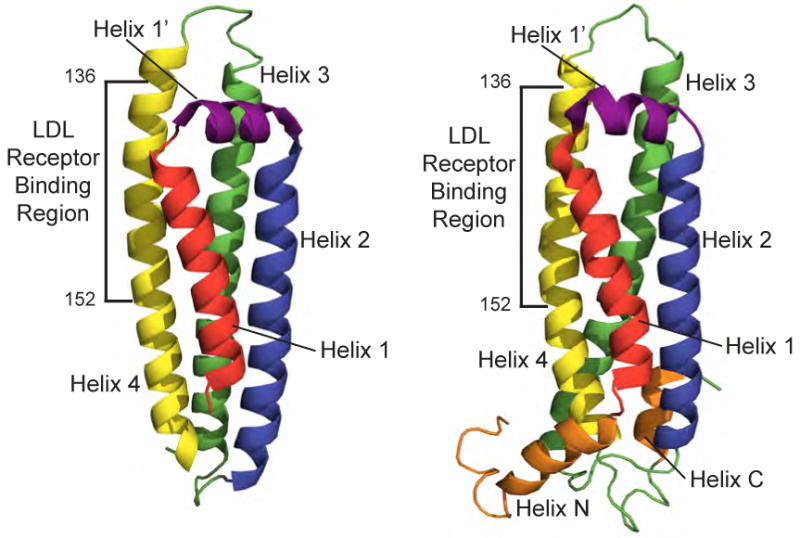
X-ray (left) and solution NMR (right) structures of the lipid-free N-terminal helix bundle of apoE3 displaying residues 23-164 and 1-183, respectively (PDB codes 1LPE [31] and 2KC3 [32]).
2.1 Isoforms
The first experiments to suggest apoE has amino acid sequence heterogeneity came from studies designed to isolate and characterize proteins found on very low-density lipoprotein (VLDL) particles by ion-exchange chromatography [37] and 2-D gel electrophoresis [38]. Speculation that amino acid sequence differences among apoE variants correlate with hyperlipoproteinemia was confirmed upon identification of the three major alleles [39, 40], ε2, ε3, and ε4. The three isoforms, apoE2, apoE3 and apoE4, differ only at positions 112 and/or 158 (Figure 2). ApoE3 contains a cysteine at position 112 and an arginine at 158 while apoE2 contains cysteine at positions 112 and 158 and apoE4 contains arginine at these sites.
Figure 2. ApoE isoform-specific differences.

Linear diagram of the apoE structural organization noting the N-terminal helical organization, functional interaction regions, isoform-specific differences at residues 112 and 158, genotypic frequencies of the human isoforms [248], and disease risk associations for the three isoforms.
Further investigation has linked apoE2 with Type III hyperlipoproteinemia and premature atherosclerosis. Interestingly, the LDL receptor binding activity of apoE2 is only ∼1% that of the parent isoform, apoE3 [41]. Despite being a rare genetic disorder, the phenotypic incidence of Type III hyperlipoproteinemia among apoE2 homozygotes (less than 5%) is even lower than predicted by genetics alone suggesting additional environmental factors are required to trigger the phenotype [42]. Studies designed to reveal the mechanism behind the reduced LDL receptor binding activity of apoE2 focused on Cys158. Following treatment of apoE2 N-terminal domain with cysteamine to convert the cysteines at positions 112 and 158 to positively charged, lysine analogs, LDL receptor binding activity increased to normal levels [43]. A mechanistic explanation for differences between apoE2 and E3 with respect to LDL receptor binding came from X-ray crystallographic comparison of the isoforms [44]. These structures revealed a salt bridge between Arg158 and Asp154 in apoE3 that is absent in apoE2. Moreover, in apoE2, an alternative salt bridge forms between Arg150 and Asp154, effectively eliminating the availability of Arg150 for interaction with the LDL receptor. In keeping with this, an alanine to arginine substitution at position 150 in apoE3 decreased receptor binding to 24% of normal [45]. Although much higher than seen with apoE2, these data suggest salt bridge interactions strongly influence receptor binding, presumably by changing the orientation or patterning of basic residues in the receptor recognition sequence (residues 136-152; see Figure 1). These authors further hypothesized that dietary factors (e.g. a high-fat diet) influencing lipoprotein size and lipid composition can, in turn, affect apoE2 Arg150 side chain conformation and, consequently, the presence or absence of the Arg150-Asp154 salt bridge [44]. Modulation of the Arg150-Asp154 salt bridge under various physiological conditions would be expected to affect LDL receptor binding activity, thus providing a possible explanation for the influence of secondary environmental factors on the occurrence of Type III hyperlipoproteinemia in apoE2 homozygotes.
While apoE3 is considered the “wild-type” isoform in humans because of its high allelic frequency and lack of strong association with a human disease phenotype, apoE4 appears to be the ancestral form since the Arg at sequence positions 112 and 158 are strongly conserved across almost all animal species possessing apoE [42]. In ε4 homozygous human carriers, the plasma concentration of apoE is lower than that for individuals possessing two ε3 alleles. Furthermore, such individuals manifest elevated plasma cholesterol and LDL as well as increased cardiovascular disease risk [46, 47]. Additionally, and more strikingly, inheritance of apoE4 is correlated with cerebral amyloid angiopathy, tauopathies, dementia with Lewy bodies, Parkinson's disease, multiple sclerosis and a higher incidence, and significantly earlier onset, of Alzheimer's disease (AD) [48]. While the molecular basis for the correlation between apoE4 and disease is not established, a unique feature of apoE4, termed “domain interaction”, may play a role. Studies have shown that arginine at position 112 causes amino acid side chain reorientation within the protein that promotes N- and C-terminal interaction via a unique salt bridge [14, 49, 50]. Domain interaction in apoE4 induces a more compact structure while the Arg at position 112 increases its molten globule like properties of this isoform [21, 51-53]. Reduced apoE4 levels in plasma, compared to apoE3, results from enhanced clearance of apoE4-containing particles [54]. Additionally, it has been noted that apoE4 displays a preference for larger lipoprotein particles, such as VLDL and chylomicron remnants, and this preference has been noted as the causative factor for the association of apoE4 with increased plasma LDL [54, 55]. Introducing human apoE4 ‘domain interaction’ into mouse apoE (by substituting Thr for Arg at position 61) results in a phenotype resembling human apoE4 subjects, including reduced abundance and binding preference for VLDL [56].
2.2 ApoE lipid particles
For the most part, biologically active apoE is associated with lipid. Indeed, lipid association is a prerequisite for apoE binding to the LDL receptor. Reconstituted HDL particles formed by combining apoE with purified phospholipids of varying fatty acid chain length and polar head group composition represent an extensively utilized model for facile reproduction of the bioactive conformation of the protein. This model system allows for the controlled manipulation and study of apoE conformational changes required for LDL receptor binding, making these particles a reliable model of apoE function in vivo. Phospholipids, including dimyristoylphosphatidylcholine (DMPC), palmitoyloleoylphosphatidylcholine or dipalmitoylphosphatidylcholine (DPPC) have been used to create nanometer scale particles wherein the apolipoprotein circumscribes the periphery of a disk-shaped phospholipid bilayer [57-59]. The exact conformation adopted by apoE in these particles remains controversial, however. Whereas some data indicate apoE aligns in such a way that the hydrophobic face of their constituent amphipathic a-helices interact with acyl chains at the edge of the phospholipid bilayer [19, 60-62] other models have been proposed. Indeed, X-ray [63], electron paramagnetic resonance [64] and recently, electron microscopic [65] data are consistent with an ellipsoidal shape for apoE-phospholipid complexes. In this model apoE a-helices align in such a way that their hydrophobic faces interact with one another while the polar faces contact phospholipid.
Regardless of the ultimate conformation adopted by apoE in lipid complexes, it is generally agreed that the protein undergoes a lipid binding-induced conformational change. On the basis of biophysical and spectroscopic studies, two models, the “open conformation” [66] and the “extended belt”, suggest possible ways in which the N-terminal helix bundle may alter its structure upon lipid association (Figure 3). In both of these models, the conformational change in apoE appears to be initiated by helix bundle opening via a “hinge” region between helices 2 and 3, permitting exposure of hydrophobic residues normally sequestered in the bundle interior. Fluorescence resonance energy transfer (FRET) studies revealed that interaction with DMPC results in increased separation between helices 1 and 3, consistent with the “hinge” hypothesis [67]. This conformational change also allows surface-exposed hydrophilic residues to retain contact with the aqueous environment and effectively substitutes helix-lipid contacts for the helix-helix interactions that stabilize the helix bundle state. The “extended belt” and “open conformation” models differ in the ultimate conformation adopted in a given reconstituted HDL particle. In the “extended belt” conformation, the hinge between helices 2 and 3 initiates further unfurling to create a fully extended helical protein that wraps around the perimeter of the disk wherein a second molecule aligns in the opposite direction to form a double belt. In contrast, the “open conformation” model preferentially retains contact between the helix 1 and 2 and helix 3 and 4 pairs wherein half-opened molecules wrap around the disk perimeter end to end [66]. Support for these models have come from a combination of FRET-based measurements [68], Fourier-transformed infrared spectroscopy [60], and tryptophan fluorescence depth quenching studies using the parallax method [62]. A similar extended conformation was reported for the C-terminal domain of apoE bound to discoidal DMPC particles [69]. An alternative, hybrid model that combines features of the “open” and “extended belt” conformations was described from FRET analysis of the NT domain of apoE3. In these experiments, intermolecular FRET was observed between helix 3 of one molecule and helix 4 of a second apoE N-terminus in which two partially extended apoE molecules interlock to encircle the disk [67]. It should be noted that an alternative “picket-fence” model, wherein anti-parallel ∼17-residue helices surround the bilayer disk and orient parallel to the lipid acyl chains, has also been advanced [19]. This model, however, is difficult to reconcile with the known helical boundaries of the lipid-free crystal structure and multiple studies that support the aforementioned conformation in which the long axis of apoE a-helices align perpendicular to the phospholipid fatty acyl chains.
Figure 3. Models of apoE helix bundle opening upon contact with lipid surfaces.

The “open” (Top panel) and “extended belt” (Bottom panel) models each permit contact of hydrophobic regions of the protein with exposed hydrophobic surface. The ultimate conformation adopted by apoE on reconstituted HDL remains unresolved.
A comprehensive study comparing particle size, apoE protein copy number and conformational parameters as they relate to changes in lipid composition and apoE isoform, has been reported [70]. These authors found that, on average, disks contained three apoE N-termini or four full-length proteins with between 200 - 250 lipid molecules per particle. While no significant differences in particle architecture were noted between apoE isoforms, differences in lipid order parameter and protein to lipid ratio were seen when comparing apoE to apoA-I [70, 71]. These observations led to the conclusion that apoE can adopt one of two predominant conformations on discoidal reconstituted HDL particles, either the canonical “belt” conformation around the disk perimeter or an alternative conformation wherein apoE helices embed horizontally within the interfacial region of the bilayer and perturb phospholipid head group organization [70].
A model of lipid-bound apoE that is distinct from previous models has emerged from studies of full-length apoE4. Fluorescence analysis using spatially sensitive probes revealed that, in DMPC reconstituted HDL, apoE4 adopts an extended conformation that loops back on itself around the periphery of the discoidal particle [72]. A similar “helical hairpin” conformation was suggested for apoE4 bound to DPPC using X-ray crystallography [63, 64]. These lipidated particles, resolved to 10 Å, display an ellipsoidal shape and contain two interlocking apoE4 molecules. In the absence of a clear delineation of secondary structure at 10 Å resolution, a predominantly helical full-length apoE4 molecule was modeled such that each extended molecule doubled back on itself and folded into a curved, horseshoe-like conformation with a 310º axis of rotation. In this model, two horseshoe shaped proteins pack into an incomplete “toroid” containing DPPC intercalated within the opposing and slightly rotated apoE molecules to form an ellipsoidal space-filling model. Support for this model was obtaining by strategic placement of electron paramagnetic resonance sensitive probes in apoE4 followed by determination of the effect of probe sequence position on side chain dynamics [73].
Structural and biophysical data on full-length apoE have led to the concept that the C-terminal domain mediates initial contact with spherical lipoprotein surfaces, effectively anchoring the N-terminal helix bundle at the particle surface [74-76]. Once localized at the lipoprotein surface, the N-terminus may either retain a receptor-inactive four-helix bundle or alter its conformation by binding to the lipid surface to adopt a receptor competent state. Thus, it may be considered that the balance between alternate conformational states of lipid associated full-length apoE will determine the extent to which apoE can mediate lipoprotein particle clearance from the plasma via interaction with LDL receptor family members. By extension, it is conceivable that isoform-specific differences could affect this balance, providing a molecular rationale for pathological consequences associated with different apoE genotypes. For example, an increased proportion of apoE4 molecules in a receptor inactive state could result in increased VLDL remnant conversion to pro-atherogenic LDL.
2.3. ApoE receptor interactions
A fundamental role of apoE is its function as a ligand for cell-surface receptors. ApoE3 is known to be a high affinity ligand for the LDL receptor, the LDL receptor related protein 1 (LRP1), apoE receptor 2 (apoER2) and the VLDL receptor. The LDL receptor is the prototype of a family of integral membrane proteins that act via ligand-activated, clathrin coated pit-mediated endocytosis to internalize plasma lipoproteins. This 839 amino acid protein is composed of five distinct regions i) an amino terminal ligand binding domain containing seven cysteine-rich LDL-A repeats, each roughly 40 amino acids in length; ii) an epidermal growth factor (EGF) precursor homology domain containing three EGF-like, cysteine-rich repeats and a β-propeller domain that mediates ligand release in the endosome via a pH-dependent conformational change preceding receptor recycling [77], iii) an O-linked sugar domain, iv) a single-pass transmembrane domain, v) and a cytoplasmic tail domain that contains an NPxY motif that facilitates receptor clustering in clathrin coated pits [78].
Studies to define in vivo ligands of the LDL receptor revealed that transgenic mice overexpressing this receptor manifest a >90% reduction in plasma apoE and apoB-100 levels, while apoA-I levels were unchanged [79]. Additional evidence emerged from studies of LDL receptor null mice wherein plasma LDL and cholesterol levels were dramatically elevated due to impaired receptor-mediated clearance of apoE and apoB containing lipoproteins [80]. By binding to the LDL receptor, apoE and apoB containing lipoproteins are cleared from plasma, thereby regulating plasma cholesterol levels [81, 82]. At neutral pH, receptor-ligand complexes are internalized into vesicles that become endosomal compartments. Subsequent pH lowering releases the lipoprotein ligand, facilitating receptor recycling and lysosomal degradation of LDL [83, 84].
As Brown and Goldstein were elucidating the LDL receptor endocytic pathway, Mahley and coworkers identified a limited, but highly conserved, sequence similarity between apoB and apoE [85, 86]. Shortly thereafter, Mahley et al. demonstrated the importance of this stretch of charged residues by showing that treatment of apoE with cyclohexanedione (an arginine-specific modifier) abolished all receptor activity [87]. From these studies, the conserved LDL receptor recognition sequence was identified. Analysis using cyanogen bromide to digest apoE at methionine residues revealed a peptide (residues 126-218) that, when complexed with DMPC, bound the LDL receptor with the same affinity as LDL [88]. This region was further delineated by abolition of receptor binding by an antibody that recognized residues 139-169 [89]. Further refinement emerged from mutational analysis. Substitution of positively charged residues at positions 142, 145, 146, and 158 for neutral amino acids markedly reduced apoE binding to the LDL receptor [13].
The region of apoE responsible for receptor recognition was further probed by generating truncation mutants and measuring receptor binding activity [90]. While apoE(1-170) and apoE(1-174) fragments retain 1% and 19% LDL receptor binding activity, respectively, apoE(1-183) possessed 85% of binding compared to full length protein. Importantly, this was the first detailed study implicating residues outside of the putative LDL receptor recognition sequence (residues 136-152) in receptor binding. Subsequent mutagenesis analysis noted the contribution of Arg172 to receptor binding activity [91]. This study confirmed the general importance of residues 170-183 by confirming that their removal reduced binding activity to 15% of full-length apoE3 levels, but strikingly, a 98% drop in binding activity was seen with a single Arg172Ala substitution mutation. Notably, an Arg172Lys substitution showed only 6% of normal activity, suggesting that arginine is required at this position to preserve the conformation necessary for receptor binding. Given that Arg172 is well outside the classical LDL receptor recognition motif and, in lipid free apoE, resides in an unstructured region beyond the boundary of helix 4, it is conceivable this segment of the protein may explain the requirement that apoE associate with lipid to be conferred with receptor recognition capability. Heteronuclear multidimensional NMR spectroscopy of an apoE-derived peptide corresponding to residues 126 – 183 [92, 93] revealed that, in the presence of trifluoroethanol or when bound to dodecylphosphocholine micelles, helix 4 extends beyond residue 165 to encompass Arg172. This hypothesis was further examined in the isolated N-terminus of apoE using site-specific electron paramagnetic resonance spectroscopy [94]. Gupta et al. showed that lipid association induced fixed secondary structure in a region of the molecule known to exist as random coil in the lipid-free state. Thus, extension of helix 4 beyond the boundary defining its lipid-free conformation may represent a key conformational change necessary for manifestation of the LDL receptor recognition properties of apoE.
In studies designed to address whether apoE binding to the LDL receptor is multivalent, the ratio of active to inactive apoE was varied on DMPC particles containing an average of four apoE molecules per particle [58]. LDL receptor binding activity was affected such that, when the number of active apoE per particle approached one, binding affinity approximated that of LDL. Other studies have shown that optimal receptor binding is achieved with spherical lipid microemulsion particles that contain at least four apoE molecules per particle [95]. This model implies that the presence of multiple apoE on discoidal and spherical particles enhances LDL receptor binding efficiency compared to particles containing a single apoB-100. In another approach, Fisher et al. employed single chain multimers of the N-terminus to show that, when bound to lipid, more than one apoE is required for high affinity binding to the LDL receptor [96]. Thus, in addition to inducing a conformational change in the structure of apoE, lipid association enhances the affinity of apoE for the LDL receptor in part by creating a multivalent ligand.
Our understanding the structural determinants of ligand binding to the LDL receptor was significantly advanced by the X-ray crystallography studies of Rudenko et al., who determined the structure of an extracellular portion of the LDL receptor at pH 5.3 [97]. In addition to confirming all known structural features of the receptor, this crystal structure gave rise to a comprehensive model to explain the mechanism of ligand release in the endosomal compartment. In this structure, LDL-A repeats 4 (residues 127-163) and 5 (residues 176-210) make contact with β propeller residues 377-642 of the EGF precursor domain. Thus, at endosomal pH, the molecule folds onto itself in a manner anticipated to result in ligand discharge. The authors suggest the strength of the interfacial interaction can be modulated as a function of pH-dependent aspartate, glutamate and histidine side chain protonation within the contact region [78, 97]. Using site-directed mutagenesis, Yamamoto et al. [98] provided evidence for a pH dependent “histidine switch” mechanism wherein ligand discharge occurs via conformational reorganization of the receptor [99]. However, other research suggests that the intramolecular contact does not drive release through a competitive mechanism and the key His residues (His190, His562, and His586) function as part of an allosteric mechanism that drives lipoprotein release [100]. Extending this work, Zhao and Michaely point out that, in addition to low pH, endosomes possess low concentrations of free calcium [101]. Using fibroblasts that express either a normal LDL receptor or a variant that is incapable of acid-dependent ligand release, these authors showed that endosomal concentrations of free calcium are sufficient to drive lipoprotein release. Thus, it is plausible that calcium-dependent and acid-dependent mechanisms cooperate to facilitate lipoprotein release from the LDL receptor.
3. Neurobiology of apoE
The importance of apoE in maintenance of cellular cholesterol homeostasis is not limited to the peripheral circulation. Indeed, its role in neurological phenomena, including neuronal plasticity, neurite outgrowth and synaptogenesis is a rapidly advancing field. While the bulk of plasma apoE originates from liver and macrophages, apoE found in the central nervous system (CNS) is produced locally. Exchange between liver and brain-derived apoE does not take place owing to the blood-brain barrier (BBB). Consistent with this, no liver derived apoE was recovered in cerebrospinal fluid (CSF) of liver transplant recipients [102]. While apoE is the predominant apolipoprotein found in the CNS, other apolipoproteins, such as apoJ, apoD, apoA-I and apoA-IV are also present. In adult brain tissue, apoE is primarily synthesized by astrocytes [103, 104], although microglia and neurons also synthesize the protein under select physiological and pathological conditions [105-107]. Under basal conditions, glial cells produce two to three times more cholesterol than neurons and also manifest elevated apoE expression. It has been demonstrated that apoE associates with lipoproteins in the brain, though astrocyte-secreted apoE-containing lipoprotein particles differ from peripheral apoE-containing lipoproteins in that they are discoidal in shape and comprised mostly of phospholipid and unesterified cholesterol [108, 109]. It is presumed that some astrocyte-secreted apoE-containing lipoproteins acquire a cholesteryl ester core on their way to the CSF since both discoidal and spherical lipoproteins have been isolated from this site [108, 110].
Accumulating evidence indicates a role for apoE in aging [111]. In this context, apoE-null mice represent a useful model for understanding its effects on natural aging. In certain studies no signs of synaptic degeneration were noted in apoE-null mice [112], with normal brain histology, an absence of neurodegenerative markers [113], normal cholinergic activity and neuronal function [114-117]. However, other studies have found that apoE-null mice develop mild to severe spatial learning and memory deficits [118, 119]. Memory impairment was accompanied by cholinergic deficits, highlighting the importance of apoE in cognition and memory [120, 121]. Studies demonstrating that apoE deficient mice are more susceptible to neurodegeneration than their wild-type counterparts implicate this protein in age-related neuropathology [122, 123].
The predominance of apoE containing lipoproteins in brain suggests it functions in cholesterol transport and clearance. In brain, cholesterol is highly abundant and is required for synapse development [124], dendrite formation [125], long term potentiation [126] and axonal guidance [127, 128]. Cholesterol delivered to neurons on apoE-containing lipoprotein particles increases synapse formation [124] by promoting biogenesis of synaptic vesicles and up-regulating the machinery necessary for their release [129, 130]. Cholesterol depletion, or a lack of cholesterol delivery, causes synaptic and dendritic spine degeneration and results in failed neurotransmission and decreased synaptic plasticity [131]. Following neuronal cell damage, cell death, traumatic brain injury or terminal differentiation, large amounts of cholesterol are lost due to membrane and myelin degeneration [132]. In response to these events, apoE is up-regulated in astrocytes and macrophages where it is presumably involved in clearance and redistribution of cholesterol and lipid debris [133, 134]. This suggests a role for apoE as a scavenger of lipophilic molecules during nerve regeneration [135, 136]. The fact that LDL receptors are up-regulated in regenerating peripheral nerves suggests enhanced lipoprotein uptake occurs during nerve growth and regeneration [137]. Moreover, it has been shown that cholesterol-containing apoE-lipoproteins secreted by astrocytes are required for synapse formation in vitro via a mechanism that is dependent upon functional apoE receptors [124, 138].
Emerging from statistical correlations examining the relationships between apoE isoforms and neurodegenerative disease progression, outcome and average age of onset, studies have focused on the differential effects of apoE3 and apoE4 on synaptic plasticity and synaptogenesis. Although both isoforms have the ability to reverse presynaptic deficits and cognitive impairment seen in apoE-null mice [139], evidence suggests apoE4 is less efficient in promoting neurological repair and maintaining proper brain function. Buttini and colleagues showed that expression of apoE3, but not apoE4, protects against neuronal damage and age-related neurodegeneration seen in apoE-null mice [140]. In contrast to apoE3-expressing mice, apoE4 mice display synaptic deficits and lower excitatory synaptic transmission, even in the absence of neuropathology [141]. In addition, apoE4 expressing mice have impaired long term potentiation, decreased numbers of synapses per neuron and reduced dendritic spine formation compared to their apoE3-expressing counterparts [142]. Significant correlative associations have emerged that suggest apoE plays a critical role in response to traumatic head injury, presumably by transporting cholesterol and lipid metabolites from the site of injury, facilitating repair. Studies examining isoform-specific effects in the context of brain injury indicate that, compared to an apoE3 cohort, apoE4 carriers display decreased recovery efficiency [143-145]. Additionally, apoE4-containing mice do not recover as efficiently from traumatic brain injury [146] and are more susceptible to cerebral ischemia [147]. Unlike under normal conditions, brain injury induces significant neuronal production of apoE, augmenting the basal production by astrocytes and microglia [148-150]. This has led to inquiry regarding the relative contribution of astrocyte versus neuron-derived apoE in the repair process. While in vitro evidence suggests apoE4 has a detrimental effect on cultured astrocytes and neurons [151-153] recent in vivo excitotoxic injury experiments have shown that expression of apoE4 by neurons preferentially causes greater cellular toxicity than does apoE4 generated by astrocytes or apoE3 generated by either neurons or astrocytes [154]. This finding supports the significant supportive role of astrocytes following injury but further suggests that apoE4 causes increased cellular burden due to its neuron specific upregulation following brain trauma.
Despite these notable injury-related pathophysiological effects of apoE4, most studies demonstrate that both apoE3- and apoE4-expressing mice perform better on cognitive tests than apoE-null mice [155]. In the case of dendritic spine morphology, the phenotype seen in apoE4-expressing mice was similar to that of apoE-null mice, suggesting an apoE4 isoform functional deficit in dendritic spine maintenance [142]. This effect, however, is age-related since an apoE4-dependent reduction in dendritic spine formation was observed only in one- to two-year old mice. This observation suggests that apoE isoform-specific effects might relate to increased risk of dementia in aged humans expressing apoE4. In older patients, with and without AD, apoE4 gene dose is inversely correlated with dendritic spine density [142]. Although a considerable number of studies implicate apoE as a significant factor in nervous system maintenance, more work is needed to address mechanistic aspects of this role. Indeed, apoE-null mice do not show gross defects in normal nerve repair and development and only manifest a neurodegenerative phenotype upon aging or injury [114, 156]. Other studies show that apoE-null mice display massive infiltration of injected dyes in brain parenchyma, indicative of BBB leakage [157-159]. This defect is selective to brain micro vessels and is exacerbated in older mice, suggesting an independent effect of aging (e.g. oxidative stress). A similar breach in BBB integrity is noted in post mortem brains of subjects with AD and cerebral amyloid angiopathy [160].
3.1 ApoE and Alzheimer's Disease
There is mounting evidence that apoE plays a central, if not direct, role in the pathogenesis of AD. The clinical symptoms of AD include progressive loss of cognition, dementia and the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles [161, 162]. ApoE4 has been shown to be a significant risk factor for development of the disease, including early and late onset non-familial [48, 163] and sporadic forms [164-166]. Among individuals that manifest late onset AD, the ε4 allele is present at a two- to three-fold higher rate compared to the general population and some studies indicate up to 65% of clinically diagnosed cases carry at least one ε4 allele [167]. Genetic and epidemiological evidence linking the allelic dose of apoE4 to premature onset and increased severity of AD continues to drive research aimed at elucidating the molecular mechanisms whereby this protein leads to neurological and neurodegenerative disease [168]. While genetic associations between apoE and AD are striking, the relationship between apoE protein expression and amyloid burden is not as straightforward. Absence of apoE in an amyloid mouse model background dramatically reduces amyloid burden without affecting amyloid-β (Aβ) peptide production [169]. This has led to the concept that apoE may contribute to conversion of Aβ to its more toxic oligomeric or fibrillar forms. However, in LDL receptor null mice, apoE levels are increased by ∼50% yet this had no effect on amyloid deposition [170]. Interestingly, apoE4 has been shown to be preferentially degraded by astrocytes, which may explain why mice display genotype-dependent effects on total apoE levels in brain and CSF (with protein levels following apoE2/2 > apoE3/3 > apoE4/4) [171] and suggests a complex mechanistic link between apoE protein levels, activity and subcellular localization and Aβ production and clearance that likely involves the coordination of multiple brain cell types.
The cholesterol transport function of apoE involves interaction with a cell surface, ATP Binding Cassette Transporter A1 (ABCA1) that promotes efflux of cellular cholesterol to acceptor proteins (e.g. apoE). In neuronal and astrocyte culture models, apoE4 is blunted in its ability to promote cholesterol efflux, presumably through a mechanism that depends on ABCA1-mediated transport [172, 173]. In ABCA1 deficient mice, apoE expression levels decrease by ∼70-80% and this is associated with decreased cholesterol efflux, poor apoE lipidation and increased amyloid burden [174-177]. On the other hand, overexpression of ABCA1 increases apoE lipidation in the CNS and decreases amyloid plaque formation [178]. These findings point to Liver-X-Receptors (LXRs), transcription factors that modulate expression of apoE and ABCA1 [179], as key regulators of brain lipid homeostasis. Indeed, deficiency of LXR-α or β increases AD pathology [180] while treatment of AD model mice with synthetic LXR agonists reduces amyloid burden and improves cognitive function [181-183]. These findings raise the intriguing possibility that LXR activation by synthetic agonists represents a potential therapeutic strategy for treatment of AD [184].
Potential reasons for the increased neuropathology of apoE4 compared to other isoforms have been investigated [111]. Analysis of the isolated N-terminal domain of the three human apoE isoforms showed that apoE4 is least resistant to chemical and heat denaturation, while apoE2 is most resistant at neutral and low pH [51-53]. Lipid binding affinity is reportedly higher for apoE4, although maximal lipid binding capacity appears equivalent among the isoforms [21, 76]. While apoE4 displays a preference for VLDL sized lipoproteins in vitro [54, 55], how this may affect brain physiology is unclear since only HDL-sized lipoproteins have been found in brain. Nevertheless, this preference has been attributed to domain interaction in apoE4, a structural feature that has also been proposed as a causative factor for cellular effects leading to AD pathology. Although the mechanism whereby domain interaction may lead to propagation of neurological defects remains elusive [151, 185, 186], Huang and colleagues documented domain interaction in living neuronal cells expressing apoE4 [187]. A novel approach to understanding the pathological significance of this phenomenon is to identify potential therapeutic small molecule inhibitors or “structure correctors” of domain interaction [188].
ApoE4 also appears to be more susceptible to proteolysis than apoE3 [189, 190] leading to speculation that apoE4 digestion products contribute to amyloid plaque formation and AD pathology, especially since these fragments have been detected in plaque from AD positive brains [191]. Furthermore, proteolysis of apoE4, and subsequent fragment accumulation in the cytosol of neurons alters cytoskeletal organization and disrupts mitochondrial energy balance. Whether this is necessary and sufficient for the progression of AD pathology remains unclear [190, 192]. Other evidence suggests the isolated N-terminal domain of apoE4 is neurotoxic [193-195], though it is not known if proteolytic cleavage occurs prior to or after interaction with Aβ. While lipidated apoE is protected from proteolysis to a greater extent than lipid-free apoE, the quantity and physiological functions of lipid-free apoE in brain is unknown [62, 196]. Offering a new perspective, Hatters et al. reported that apoE4 forms aggregates (independent of Aβ aggregation) at substantially higher rates than apoE3 or apoE2 and is more neurotoxic to cultured neuronal cells [197]. These aggregates bear an irregular protofilament-like morphology with a high α-helical content, unlike the ‘classic’ amyloid fibrils that are rich in β-sheet structures. Whether this is the cause or a consequence of amyloid neuropathology associated with AD is not known. Finally, other studies point to a role of apoE4 in potentiating Aβ-induced lysosomal leakage [152, 198] and/or activating the endoplasmic reticulum stress response, leading to increased apoptosis [199, 200].
Genetically altered mice harboring a Thr61Arg mutation in murine apoE (creating a human apoE4-like mouse apoE) results in decreased apoE levels in the brain along with synaptic and cognitive defects [201, 202]. Transgenic mice with a genetic predisposition for higher Aβ production in which murine apoE is substituted for each of the three human isoforms, develop the predicted isoform-specific differences in amyloid deposition with apoE4 > apoE3 > apoE2 [203, 204]. Interestingly, however, human isoform substituted mice manifest a delay in onset of plaque formation compared to murine apoE mice [205, 206]. Better understanding of the manner in which apoE interacts with Aβ, receptors and other binding partners in response to various conformational and lipidation states may provide further insight into its role in AD pathology.
A recent study employing mice bearing Thr61Arg apoE, to mimic human apoE4, demonstrated that domain interaction per se is associated with deficits usually noted in AD, supporting the hypothesis that apoE4 can act independent of Aβ to induce pathophysiology [207]. Furthermore, induction of domain interaction via the Thr61Arg mutation leads to endoplasmic reticulum stress and an up-regulated unfolded protein response, which in turn destines apoE for degradation. The authors proposed that endoplasmic reticulum stress results in dysfunctional astrocytes that provide sub-optimal support to neurons, which in turn respond with self-generated apoE leading to increased levels of neurotoxic fragments [193-195]. Dysfunctional astrocytes and toxic fragments likely represent early events in apoE4-associated pathophysiology independent of Aβ-related pathways (Figure 4).
Figure 4. Aβ-independent effects of apoE4 towards neurodegeneration and pathophysiology.

In healthy brain, apoE (orange) secreted from astrocytes provides support for neuronal function (Top panel). However, apoE4 increases baseline ER stress and unfolded protein response in astrocytes, slowly leading to cells that function sub-optimally (Bottom panel). As a consequence, these astrocytes are unable to provide optimal support to the neurons over a period of time. As a compensatory response, neurons generate apoE4 for self-repair, which in turn leads to increased generation of neurotoxic fragments, neurodegeneration and disease [168].
Deposition of extracellular amyloid plaque, formed by soluble and insoluble assemblies of the Aβ peptide, is a hallmark of AD and is considered one of the primary events in disease pathology. Aβ is derived from the amyloid precursor protein (APP) by sequential β- and γ-secretase-dependent intramembranous proteolysis [161]. Aβ is produced and secreted under normal metabolic conditions and can be found at high levels in normal CSF and plasma [208, 209]. Thus, disease pathology is thought to be driven by a net imbalance between Aβ clearance and production [210, 211]. Gradual increases in Aβ production lead to its oligomerization in brain interstitial fluid and within neurons [212, 213] and subsequent fibrillization to produce amyloid plaques [161]. The dominantly-inherited, familial form of AD is associated with either increased production of Aβ (most commonly caused by mutations in APP itself, the presenilin 1 and 2 gene products that form two of the necessary components of the γ-secretase complex) [214] or increased production of the longer Aβ(1-42) peptide which is more toxic than the Aβ(1-40) peptide [215]. Non-familial forms of AD have been attributed to an imbalance in the relative clearance and aggregation of Aβ [216]. The only consistently associated genetic risk factor for non-familial AD is the ε4 allele of the APOE gene. Despite this, the pathology and phenotypic display manifested by familial and non-familial forms of AD are nearly indistinguishable.
The observation that apoE is bound to Aβ in CSF prompted study of apoE as a candidate for acceleration of AD pathology [217-219]. Although a mechanism whereby apoE (and particularly apoE4) promotes AD pathology remains elusive, in vitro and in vivo evidence suggest interaction between apoE and Aβ is associated with disease progression. In vitro analysis demonstrated that Aβ and delipidated apoE4 promote fibril formation more rapidly and with higher density than those seeded with other apoE isoforms (following an aggregation rate rank order of apoE4 > apoE3 > apoE2) [163, 217, 220]. In contrast to delipidated protein, lipidated apoE has a different isoform dependent affinity for Aβ [221]. When the affinity of lipid-bound apoE was compared, using transfected eukaryotic cell lines, apoE3-Aβ complex levels were 20-fold higher than that for apoE4-Aβ complexes [222, 223]. It has further been shown that lipidated apoE3 binds Aβ two to three times more rapidly than lipidated apoE4 [222]. Studies examining the effect of apoE on neurite extension revealed that lipidated apoE3 enhances binding to Aβ and may facilitate its clearance, thereby preventing aggregation [224]. It has been shown that Aβ binds to apoE via its C-terminal domain and Aβ binding abrogates apoE lipid binding [163, 225]. These results indicate Aβ interferes with apoE function as a lipid transport protein in brain, which may contribute to AD progression by altering lipid/cholesterol homeostasis [111, 226-228]. In a neuronal cell line that overexpresses APP, apoE4 increased Aβ production to a greater extent than apoE3, [153]. This isoform-specific difference was abolished when cells were treated with small interfering RNA directed against LRP1 or upon incubation with receptor-associated protein, a known inhibitor of LRP1 function. This finding suggests apoE4-specific enhancement in Aβ production or deposition is dependent upon LRP1 function. Thus, it is conceivable that apoE4 possesses defective receptor binding activity. In vitro studies with transfected cell lines indicate apoE and its cognate receptors play a role in APP processing and Aβ production [229, 230]. A possible mechanism was suggested by studies showing that overexpression of apoE4 enhances Aβ production by promoting endocytic recycling of APP [153]. How apoE4 facilitates this recycling remains unclear. At the same time, it may be considered that clearance of Aβ is as important as its production (Figure 5). The predominant pathways for clearance of Aβ include receptor-mediated uptake by microglia and astrocytes [231-233] or LRP1 mediated transport of Aβ across the BBB [234, 235].
Figure 5. Schematic representation of the role of apoE3/E4-Aβ interaction in AD.

ApoE isoform-specific differences (red) may influence Aβ (purple) oligomerization, deposition, transport, and/or clearance mechanisms that can influence the progression of AD. Oligomerization of Aβ released from the amyloid precursor protein (APP) in neuronal membranes has been described as a causative factor for the progression of AD and may be enhanced by apoE4 compared to apoE3. Whether apoE isoform differences affect Aβ association with apoE-HDL complexes (orange and beige) remains unclear. However clearance of apoE-Aβ HDL by lipoprotein receptors (LRP1 and LDLR) appears to be promoted to a lesser extent by apoE4 than apoE3. Astrocytes play a significant role in secreting apoE-containing HDL-sized particles.
Receptor-mediated clearance of Aβ in brain likely occurs through the action of LDL receptor family members including the LDL receptor, LRP1, apoER2, SorLA/LR11 and the VLDL receptor [138]. It has been shown that both full-length and cleaved fragments of apoE [166, 191], LRP1 and other LRP1 ligands [166] colocalize, and are immunoreactive with, amyloid plaques in AD affected brain tissue. ApoE receptors have been shown to bind directly to Aβ [236] as well as through Aβ binding partner interactions, including apoE-Aβ complexes. In accordance with increased lipidated apoE3 binding to Aβ, apoE3 clears Aβ through receptor-mediated interaction more efficiently than apoE4-Aβ complexes [237]. In amyloid mouse models, expression of human apoE3 resulted in less plaque deposition than in apoE4-expressing mice [203, 238, 239]. In addition, post-mortem amyloid plaque load is increased in the brains of ε4 carriers [240, 241], suggesting efficient clearance of Aβ may impede amyloid formation, as suggested in Figure 5.
Though cellular and BBB export of Aβ is most certainly receptor-mediated, the responsible receptors remain controversial. Using real-time in situ microdialysis, Bell et al demonstrated that Aβ(1-42) passed more slowly across the BBB than Aβ(1-40), although both bind to LRP1 [242]. Association of Aβ(1-40) with lipid poor apoE slowed transport (and lipidated apoE complexed with Aβ blocked virtually all transport) across the BBB within the 30-minute time frame of the study. Using the same technique, an alternative study confirmed that lipidation of apoE, compared to lipid-poor apoE-Aβ or free peptide alone, dramatically slows transport of apoE-Aβ across the BBB [237]. Interestingly, this study also demonstrated that Aβ binding to apoE4 shifted receptor-mediated clearance from LRP1 to the VLDL receptor. Alternatively, apoE2 and apoE3 cleared Aβ through LRP1 and the LDL receptor at a significantly higher rate than apoE4-Aβ complexes. This apoE4-specific effect results in higher brain retention of apoE4-Aβ [237]. Taken together, these studies show that decreased amyloid deposition seen in apoE-null mice may be the result of enhanced transport of free Aβ across the BBB.
3.2 Tau protein and apoE
Tau protein is integrally bound to cellular microtubules and acts to stabilize these structures. Phosphorylation of tau, however, results in formation of paired helical filaments that lack microtubule stabilizing capability. Hyperphosphorylated tau is the primary component of pathological neurofibrillary tangles and is toxic to neurons. Studies demonstrating that a reduction in tau prevents Aβ-dependent cognitive impairments in an amyloid mouse model, suggests tau may be required for Aβ-induced neuronal dysfunction [243]. However, whether hyperphosphorylated tau is a primary cause of AD-associated dementia or simply a marker for the disease remains unclear since it is equally plausible that destabilization of microtubules by hyperphosphorylated tau interferes with cognition independent of, or downstream to, induction of AD. Transgenic overexpression of apoE4 in mice resulted in increased tau phosphorylation in neurons, but not astrocytes [244, 245]. However, the pathophysiological significance of this finding is unclear since neurons only express apoE following injury [105, 107], possibly implying that the contribution of apoE to tau hyperphosphorylation in neurons may be limited to conditions of excess stress or cellular damage. While it has been shown that apoE3 interacts with unphosphorylated tau more strongly than apoE4, the degree to which full-length apoE isoforms bind and associate with phosphorylated tau remains to be determined [246]. Also, it is not clear how apoE and tau, which normally partition to distinct subcellular locations, physically interact. One hypothesis is that proteolytic cleavage of apoE generates C-terminal truncation fragments that dissociate in the cytosol and interact with tau [245]. Indeed, apoE proteolytic fragments may have enhanced toxicity and intracellular activity compared to the intact protein [247].
4. Concluding Remarks
It has become increasingly clear that, in addition to its role in the maintenance and physiology of the cardiovascular system, apoE also plays a central role in healthy and pathophysiological processes in the brain. While apoE is critical for the regulation of cholesterol homeostasis in the peripheral circulation, its role in the brain appears to involve not only cholesterol transport but also intracellular exchange of metabolites between neurons and glial cells through processes that appear to be required for maintenance of healthy brain tissue. As the structural properties of apoE, its various isoforms and lipidation states continue to be defined, new appreciation is gained for its physiological and pathophysiological functions in both the brain and periphery. A concerted effort that draws upon expertise in structural biology, cell biology, animal physiology and genetic engineering will continue to enhance knowledge of the relationships that exist between apoE and complex disease processes that have an untold impact on the human condition.
Acknowledgments
The authors would like to thank Jennifer Beckstead for assistance and input in the planning and execution of the figures. Supported by grants from the California Tobacco-Related Disease Research Program (16DT-0195 to P.S.H. and 17RT-0165 to V.N.) and the National Institutes of Health (HL 64159 to R.R.).
Abbreviations
- AD
Alzheimer's disease
- apo
apolipoprotein
- FRET
fluorescence resonance energy transfer
- LDL
low-density lipoprotein
- NMR
nuclear magnetic resonance
- VLDL
very low density lipoprotein
- DMPC
dimyristoylphosphatidylcholine
- DPPC
dipalmitoylphosphotidylcholine
- CNS
central nervous system
- BBB
blood brain barrier
- CSF
cerebro spinal fluid
- Aβ
amyloid beta
- APP
amyloid precursor protein
- LXR
Liver-X-Receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shimano H, Yamada N, Katsuki M, Shimada M, Gotoda T, Harada K, et al. Overexpression of apolipoprotein E in transgenic mice: marked reduction in plasma lipoproteins except high density lipoprotein and resistance against diet-induced hypercholesterolemia. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:1750–4. doi: 10.1073/pnas.89.5.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimano H, Ohsuga J, Shimada M, Namba Y, Gotoda T, Harada K, et al. Inhibition of diet-induced atheroma formation in transgenic mice expressing apolipoprotein E in the arterial wall. The Journal of clinical investigation. 1995;95:469–76. doi: 10.1172/JCI117687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science (New York, NY. 1992;258:468–71. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 4.Ghiselli G, Schaefer EJ, Gascon P, Breser HB., Jr Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science (New York, NY. 1981;214:1239–41. doi: 10.1126/science.6795720. [DOI] [PubMed] [Google Scholar]
- 5.Zannis VI, McPherson J, Goldberger G, Karathanasis SK, Breslow JL. Synthesis, intracellular processing, and signal peptide of human apolipoprotein E. The Journal of biological chemistry. 1984;259:5495–9. [PubMed] [Google Scholar]
- 6.Rall SC, Jr, Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. The Journal of biological chemistry. 1982;257:4171–8. [PubMed] [Google Scholar]
- 7.McLean JW, Elshourbagy NA, Chang DJ, Mahley RW, Taylor JM. Human apolipoprotein E mRNA. cDNA cloning and nucleotide sequencing of a new variant. The Journal of biological chemistry. 1984;259:6498–504. [PubMed] [Google Scholar]
- 8.Olaisen B, Teisberg P, Gedde-Dahl T., Jr The locus for apolipoprotein E (apoE) is linked to the complement component C3 (C3) locus on chromosome 19 in man. Hum Genet. 1982;62:233–6. doi: 10.1007/BF00333526. [DOI] [PubMed] [Google Scholar]
- 9.Paik YK, Chang DJ, Reardon CA, Davies GE, Mahley RW, Taylor JM. Nucleotide sequence and structure of the human apolipoprotein E gene. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3445–9. doi: 10.1073/pnas.82.10.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellosta S, Mahley RW, Sanan DA, Murata J, Newland DL, Taylor JM, et al. Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E-null mice. The Journal of clinical investigation. 1995;96:2170–9. doi: 10.1172/JCI118271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazzone T. Apolipoprotein E secretion by macrophages: its potential physiological functions. Curr Opin Lipidol. 1996;7:303–7. doi: 10.1097/00041433-199610000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Tangirala RK, Pratico D, FitzGerald GA, Chun S, Tsukamoto K, Maugeais C, et al. Reduction of isoprostanes and regression of advanced atherosclerosis by apolipoprotein E. The Journal of biological chemistry. 2001;276:261–6. doi: 10.1074/jbc.M003324200. [DOI] [PubMed] [Google Scholar]
- 13.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science (New York, NY. 1988;240:622–30. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 14.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–8. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–66. [PubMed] [Google Scholar]
- 16.Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–45. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 17.Chou PY, Fasman GD. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry. 1974;13:211–22. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 18.Aggerbeck LP, Wetterau JR, Weisgraber KH, Wu CS, Lindgren FT. Human apolipoprotein E3 in aqueous solution. II. Properties of the amino- and carboxyl-terminal domains. The Journal of biological chemistry. 1988;263:6249–58. [PubMed] [Google Scholar]
- 19.De Pauw M, Vanloo B, Weisgraber K, Rosseneu M. Comparison of lipid-binding and lecithin :cholesterol acyltransferase activation of the amino- and carboxyl-terminal domains of human apolipoprotein E3. Biochemistry. 1995;34:10953–66. doi: 10.1021/bi00034a030. [DOI] [PubMed] [Google Scholar]
- 20.Wetterau JR, Aggerbeck LP, Rall SC, Jr, Weisgraber KH. Human apolipoprotein E3 in aqueous solution. I. Evidence for two structural domains. The Journal of biological chemistry. 1988;263:6240–8. [PubMed] [Google Scholar]
- 21.Sakamoto T, Tanaka M, Vedhachalam C, Nickel M, Nguyen D, Dhanasekaran P, et al. Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry. 2008;47:2968–77. doi: 10.1021/bi701923h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westerlund JA, Weisgraber KH. Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. The Journal of biological chemistry. 1993;268:15745–50. [PubMed] [Google Scholar]
- 23.Yokoyama S, Kawai Y, Tajima S, Yamamoto A. Behavior of human apolipoprotein E in aqueous solutions and at interfaces. The Journal of biological chemistry. 1985;260:16375–82. [PubMed] [Google Scholar]
- 24.Fan D, Li Q, Korando L, Jerome WG, Wang J. A monomeric human apolipoprotein E carboxyl-terminal domain. Biochemistry. 2004;43:5055–64. doi: 10.1021/bi035958w. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Vasudevan S, Sojitrawala R, Zhao W, Cui C, Xu C, et al. A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry. 2007;46:10722–32. doi: 10.1021/bi700672v. [DOI] [PubMed] [Google Scholar]
- 26.Utermann G, Langenbeck U, Beisiegel U, Weber W. Genetics of the apolipoprotein E system in man. Am J Hum Genet. 1980;32:339–47. [PMC free article] [PubMed] [Google Scholar]
- 27.Utermann G, Steinmetz A, Weber W. Genetic control of human apolipoprotein E polymorphism: comparison of one- and two-dimensional techniques of isoprotein analysis. Hum Genet. 1982;60:344–51. doi: 10.1007/BF00569216. [DOI] [PubMed] [Google Scholar]
- 28.Zannis VI, Breslow JL. Human very low density lipoprotein apolipoprotein E isoprotein polymorphism is explained by genetic variation and posttranslational modification. Biochemistry. 1981;20:1033–41. doi: 10.1021/bi00507a059. [DOI] [PubMed] [Google Scholar]
- 29.Jain RS, Quarfordt SH. The carbohydrate content of apolipoprotein E from human very low density lipoproteins. Life Sci. 1979;25:1315–23. doi: 10.1016/0024-3205(79)90397-7. [DOI] [PubMed] [Google Scholar]
- 30.Wernette-Hammond ME, Lauer SJ, Corsini A, Walker D, Taylor JM, Rall SC., Jr Glycosylation of human apolipoprotein E. The carbohydrate attachment site is threonine 194. The Journal of biological chemistry. 1989;264:9094–101. [PubMed] [Google Scholar]
- 31.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science (New York, NY. 1991;252:1817–22. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 32.Sivashanmugam A, Wang J. A unified scheme for initiation and conformational adaptation of human apolipoprotein E N-terminal domain upon lipoprotein binding and for receptor binding activity. The Journal of biological chemistry. 2009;284:14657–66. doi: 10.1074/jbc.M901012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forstner M, Peters-Libeu C, Contreras-Forrest E, Newhouse Y, Knapp M, Rupp B, et al. Carboxyl-terminal domain of human apolipoprotein E: expression, purification, and crystallization. Protein Expr Purif. 1999;17:267–72. doi: 10.1006/prep.1999.1144. [DOI] [PubMed] [Google Scholar]
- 34.Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. Amphipathic helix motif: classes and properties. Proteins. 1990;8:103–17. doi: 10.1002/prot.340080202. [DOI] [PubMed] [Google Scholar]
- 35.Choy N, Raussens V, Narayanaswami V. Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. Journal of molecular biology. 2003;334:527–39. doi: 10.1016/j.jmb.2003.09.059. [DOI] [PubMed] [Google Scholar]
- 36.Patel AB, Khumsupan P, Narayanaswami V. Pyrene fluorescence analysis offers new insights into the conformation of the lipoprotein-binding domain of human apolipoprotein E. Biochemistry. 2010;49:1766–75. doi: 10.1021/bi901902e. [DOI] [PubMed] [Google Scholar]
- 37.Shore VG, Shore B. Heterogeneity of human plasma very low density lipoproteins. Separation of species differing in protein components. Biochemistry. 1973;12:502–7. doi: 10.1021/bi00727a022. [DOI] [PubMed] [Google Scholar]
- 38.Zannis VI, Breslow JL. Characterization of a unique human apolipoprotein E variant associated with type III hyperlipoproteinemia. The Journal of biological chemistry. 1980;255:1759–62. [PubMed] [Google Scholar]
- 39.Utermann G. Apolipoprotein E polymorphism in health and disease. Am Heart J. 1987;113:433–40. doi: 10.1016/0002-8703(87)90610-7. [DOI] [PubMed] [Google Scholar]
- 40.Weisgraber KH, Rall SC, Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. The Journal of biological chemistry. 1981;256:9077–83. [PubMed] [Google Scholar]
- 41.Weisgraber KH, Innerarity TL, Mahley RW. Abnormal lipoprotein receptor-binding activity of the human E apoprotein due to cysteine-arginine interchange at a single site. The Journal of biological chemistry. 1982;257:2518–21. [PubMed] [Google Scholar]
- 42.Mahley RW, Rall SCJ. Type III hyperlipoproteinemia (Dysbetalipoproteinemia): the role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill Inc; 1995. pp. 1953–80. [Google Scholar]
- 43.Innerarity TL, Weisgraber KH, Arnold KS, Rall SC, Jr, Mahley RW. Normalization of receptor binding of apolipoprotein E2. Evidence for modulation of the binding site conformation. The Journal of biological chemistry. 1984;259:7261–7. [PubMed] [Google Scholar]
- 44.Dong LM, Parkin S, Trakhanov SD, Rupp B, Simmons T, Arnold KS, et al. Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nature structural biology. 1996;3:718–22. doi: 10.1038/nsb0896-718. [DOI] [PubMed] [Google Scholar]
- 45.Lalazar A, Weisgraber KH, Rall SC, Jr, Giladi H, Innerarity TL, Levanon AZ, et al. Site-specific mutagenesis of human apolipoprotein E. Receptor binding activity of variants with single amino acid substitutions. The Journal of biological chemistry. 1988;263:3542–5. [PubMed] [Google Scholar]
- 46.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis (Dallas, Tex. 1988;8:1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 47.Wilson PW. Relation of high-density lipoprotein subfractions and apolipoprotein E isoforms to coronary disease. Clinical chemistry. 1995;41:165–9. [PubMed] [Google Scholar]
- 48.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (New York, NY. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 49.Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. The Journal of biological chemistry. 1996;271:19053–7. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 50.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, et al. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. The Journal of biological chemistry. 1994;269:22358–65. [PubMed] [Google Scholar]
- 51.Weers PM, Narayanaswami V, Choy N, Luty R, Hicks L, Kay CM, et al. Lipid binding ability of human apolipoprotein E N-terminal domain isoforms: correlation with protein stability? Biophysical chemistry. 2003;100:481–92. doi: 10.1016/s0301-4622(02)00300-9. [DOI] [PubMed] [Google Scholar]
- 52.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, et al. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. The Journal of biological chemistry. 2002;277:50380–5. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 53.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, et al. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 2000;39:11657–66. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 54.Gregg RE, Zech LA, Schaefer EJ, Stark D, Wilson D, Brewer HB., Jr Abnormal in vivo metabolism of apolipoprotein E4 in humans. The Journal of clinical investigation. 1986;78:815–21. doi: 10.1172/JCI112645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. 1990;31:1503–11. [PubMed] [Google Scholar]
- 56.Raffai RL, Dong LM, Farese RV, Jr, Weisgraber KH. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11587–91. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Innerarity TL, Pitas RE, Mahley RW. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. The Journal of biological chemistry. 1979;254:4186–90. [PubMed] [Google Scholar]
- 58.Pitas RE, Innerarity TL, Mahley RW. Cell surface receptor binding of phospholipid. protein complexes containing different ratios of receptor-active and -inactive E apoprotein. The Journal of biological chemistry. 1980;255:5454–60. [PubMed] [Google Scholar]
- 59.Sparrow JT, Sparrow DA, Culwell AR, Gotto AM., Jr Apolipoprotein E: phospholipid binding studies with synthetic peptides containing the putative receptor binding region. Biochemistry. 1985;24:6984–8. doi: 10.1021/bi00345a035. [DOI] [PubMed] [Google Scholar]
- 60.Raussens V, Fisher CA, Goormaghtigh E, Ryan RO, Ruysschaert JM. The low density lipoprotein receptor active conformation of apolipoprotein E. Helix organization in n-terminal domain-phospholipid disc particles. The Journal of biological chemistry. 1998;273:25825–30. doi: 10.1074/jbc.273.40.25825. [DOI] [PubMed] [Google Scholar]
- 61.Lu B, Morrow JA, Weisgraber KH. Conformational reorganization of the four-helix bundle of human apolipoprotein E in binding to phospholipid. The Journal of biological chemistry. 2000;275:20775–81. doi: 10.1074/jbc.M003508200. [DOI] [PubMed] [Google Scholar]
- 62.Narayanaswami V, Maiorano JN, Dhanasekaran P, Ryan RO, Phillips MC, Lund-Katz S, et al. Helix orientation of the functional domains in apolipoprotein e in discoidal high density lipoprotein particles. The Journal of biological chemistry. 2004;279:14273–9. doi: 10.1074/jbc.M313318200. [DOI] [PubMed] [Google Scholar]
- 63.Peters-Libeu CA, Newhouse Y, Hatters DM, Weisgraber KH. Model of biologically active apolipoprotein E bound to dipalmitoylphosphatidylcholine. The Journal of biological chemistry. 2006;281:1073–9. doi: 10.1074/jbc.M510851200. [DOI] [PubMed] [Google Scholar]
- 64.Peters-Libeu CA, Newhouse Y, Hall SC, Witkowska HE, Weisgraber KH. Apolipoprotein E*dipalmitoylphosphatidylcholine particles are ellipsoidal in solution. J Lipid Res. 2007;48:1035–44. doi: 10.1194/jlr.M600545-JLR200. [DOI] [PubMed] [Google Scholar]
- 65.Zhang L, Song J, Newhouse Y, Zhang S, Weisgraber KH, Ren G. An optimized negative-staining protocol of electron microscopy for apoE4 POPC lipoprotein. J Lipid Res. 2010;51:1228–36. doi: 10.1194/jlr.D002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weisgraber KH. Apolipoprotein E: structure-function relationships. Advances in protein chemistry. 1994;45:249–302. doi: 10.1016/s0065-3233(08)60642-7. [DOI] [PubMed] [Google Scholar]
- 67.Fisher CA, Narayanaswami V, Ryan RO. The lipid-associated conformation of the low density lipoprotein receptor binding domain of human apolipoprotein E. The Journal of biological chemistry. 2000;275:33601–6. doi: 10.1074/jbc.M002643200. [DOI] [PubMed] [Google Scholar]
- 68.Fisher CA, Ryan RO. Lipid binding-induced conformational changes in the N-terminal domain of human apolipoprotein E. J Lipid Res. 1999;40:93–9. [PubMed] [Google Scholar]
- 69.Raussens V, Drury J, Forte TM, Choy N, Goormaghtigh E, Ruysschaert JM, et al. Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem J. 2005;387:747–54. doi: 10.1042/BJ20041536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneeweis LA, Koppaka V, Lund-Katz S, Phillips MC, Axelsen PH. Structural analysis of lipoprotein E particles. Biochemistry. 2005;44:12525–34. doi: 10.1021/bi050872j. [DOI] [PubMed] [Google Scholar]
- 71.Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. The Journal of biological chemistry. 2003;278:23227–32. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 72.Drury J, Narayanaswami V. Examination of lipid-bound conformation of apolipoprotein E4 by pyrene excimer fluorescence. The Journal of biological chemistry. 2005;280:14605–10. doi: 10.1074/jbc.M414019200. [DOI] [PubMed] [Google Scholar]
- 73.Hatters DM, Voss JC, Budamagunta MS, Newhouse YN, Weisgraber KH. Insight on the molecular envelope of lipid-bound apolipoprotein E from electron paramagnetic resonance spectroscopy. Journal of molecular biology. 2009;386:261–71. doi: 10.1016/j.jmb.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narayanaswami V, Ryan RO. Molecular basis of exchangeable apolipoprotein function. Biochimica et biophysica acta. 2000;1483:15–36. doi: 10.1016/s1388-1981(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 75.Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Lund-Katz S, Phillips MC. Lipid binding-induced conformational change in human apolipoprotein E. Evidence for two lipid-bound states on spherical particles. The Journal of biological chemistry. 2001;276:40949–54. doi: 10.1074/jbc.M106337200. [DOI] [PubMed] [Google Scholar]
- 76.Saito H, Dhanasekaran P, Baldwin F, Weisgraber KH, Phillips MC, Lund-Katz S. Effects of polymorphism on the lipid interaction of human apolipoprotein E. The Journal of biological chemistry. 2003;278:40723–9. doi: 10.1074/jbc.M304814200. [DOI] [PubMed] [Google Scholar]
- 77.Davis CG, Goldstein JL, Sudhof TC, Anderson RG, Russell DW, Brown MS. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature. 1987;326:760–5. doi: 10.1038/326760a0. [DOI] [PubMed] [Google Scholar]
- 78.Rudenko G, Deisenhofer J. The low-density lipoprotein receptor: ligands, debates and lore. Curr Opin Struct Biol. 2003;13:683–9. doi: 10.1016/j.sbi.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 79.Hofmann SL, Russell DW, Brown MS, Goldstein JL, Hammer RE. Overexpression of low density lipoprotein (LDL) receptor eliminates LDL from plasma in transgenic mice. Science (New York, NY. 1988;239:1277–81. doi: 10.1126/science.3344433. [DOI] [PubMed] [Google Scholar]
- 80.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. The Journal of clinical investigation. 1993;92:883–93. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. The Journal of clinical investigation. 1994;93:1885–93. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yokode M, Hammer RE, Ishibashi S, Brown MS, Goldstein JL. Diet-induced hypercholesterolemia in mice: prevention by overexpression of LDL receptors. Science (New York, NY. 1990;250:1273–5. doi: 10.1126/science.2244210. [DOI] [PubMed] [Google Scholar]
- 83.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (New York, NY. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 84.Goldstein JL, Brown MS. The LDL receptor. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:431–8. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahley RW, Weisgraber KH, Innerarity T, Brewer HB, Jr, Assmann G. Swine lipoproteins and atherosclerosis. Changes in the plasma lipoproteins and apoproteins induced by cholesterol feeding. Biochemistry. 1975;14:2817–23. doi: 10.1021/bi00684a005. [DOI] [PubMed] [Google Scholar]
- 86.Bersot TP, Mahley RW, Brown MS, Goldstein JL. Interaction of swine lipoproteins with the low density lipoprotein receptor in human fibroblasts. The Journal of biological chemistry. 1976;251:2395–8. [PubMed] [Google Scholar]
- 87.Mahley RW, Innerarity TL, Pitas RE, Weisgraber KH, Brown JH, Gross E. Inhibition of lipoprotein binding to cell surface receptors of fibroblasts following selective modification of arginyl residues in arginine-rich and B apoproteins. The Journal of biological chemistry. 1977;252:7279–87. [PubMed] [Google Scholar]
- 88.Innerarity TL, Friedlander EJ, Rall SC, Jr, Weisgraber KH, Mahley RW. The receptor-binding domain of human apolipoprotein E. Binding of apolipoprotein E fragments. The Journal of biological chemistry. 1983;258:12341–7. [PubMed] [Google Scholar]
- 89.Weisgraber KH, Innerarity TL, Harder KJ, Mahley RW, Milne RW, Marcel YL, et al. The receptor-binding domain of human apolipoprotein E. Monoclonal antibody inhibition of binding. The Journal of biological chemistry. 1983;258:12348–54. [PubMed] [Google Scholar]
- 90.Lalazar A, Mahley RW. Human apolipoprotein E. Receptor binding activity of truncated variants with carboxyl-terminal deletions. The Journal of biological chemistry. 1989;264:8447–50. [PubMed] [Google Scholar]
- 91.Morrow JA, Arnold KS, Dong J, Balestra ME, Innerarity TL, Weisgraber KH. Effect of arginine 172 on the binding of apolipoprotein E to the low density lipoprotein receptor. The Journal of biological chemistry. 2000;275:2576–80. doi: 10.1074/jbc.275.4.2576. [DOI] [PubMed] [Google Scholar]
- 92.Raussens V, Slupsky CM, Ryan RO, Sykes BD. NMR structure and dynamics of a receptor-active apolipoprotein E peptide. The Journal of biological chemistry. 2002;277:29172–80. doi: 10.1074/jbc.M204043200. [DOI] [PubMed] [Google Scholar]
- 93.Raussens V, Slupsky CM, Sykes BD, Ryan RO. Lipid-bound structure of an apolipoprotein E-derived peptide. The Journal of biological chemistry. 2003;278:25998–6006. doi: 10.1074/jbc.M301753200. [DOI] [PubMed] [Google Scholar]
- 94.Gupta V, Narayanaswami V, Budamagunta MS, Yamamato T, Voss JC, Ryan RO. Lipid-induced extension of apolipoprotein E helix 4 correlates with low density lipoprotein receptor binding ability. The Journal of biological chemistry. 2006;281:39294–9. doi: 10.1074/jbc.M608085200. [DOI] [PubMed] [Google Scholar]
- 95.Funahashi T, Yokoyama S, Yamamoto A. Association of apolipoprotein E with the low density lipoprotein receptor: demonstration of its co-operativity on lipid microemulsion particles. J Biochem. 1989;105:582–7. doi: 10.1093/oxfordjournals.jbchem.a122708. [DOI] [PubMed] [Google Scholar]
- 96.Fisher C, Abdul-Aziz D, Blacklow SC. A two-module region of the low-density lipoprotein receptor sufficient for formation of complexes with apolipoprotein E ligands. Biochemistry. 2004;43:1037–44. doi: 10.1021/bi035529y. [DOI] [PubMed] [Google Scholar]
- 97.Rudenko G, Henry L, Henderson K, Ichtchenko K, Brown MS, Goldstein JL, et al. Structure of the LDL receptor extracellular domain at endosomal pH. Science (New York, NY. 2002;298:2353–8. doi: 10.1126/science.1078124. [DOI] [PubMed] [Google Scholar]
- 98.Yamamoto T, Chen HC, Guigard E, Kay CM, Ryan RO. Molecular studies of pH-dependent ligand interactions with the low-density lipoprotein receptor. Biochemistry. 2008;47:11647–52. doi: 10.1021/bi801117t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beglova N, Blacklow SC. The LDL receptor: how acid pulls the trigger. Trends in biochemical sciences. 2005;30:309–17. doi: 10.1016/j.tibs.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 100.Zhao Z, Michaely P. The epidermal growth factor homology domain of the LDL receptor drives lipoprotein release through an allosteric mechanism involving H190, H562, and H586. The Journal of biological chemistry. 2008;283:26528–37. doi: 10.1074/jbc.M804624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao Z, Michaely P. The role of calcium in lipoprotein release by the low-density lipoprotein receptor. Biochemistry. 2009;48:7313–24. doi: 10.1021/bi900214u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Linton MF, Gish R, Hubl ST, Butler E, Esquivel C, Bry WI, et al. Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. The Journal of clinical investigation. 1991;88:270–81. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. The Journal of clinical investigation. 1985;76:1501–13. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochimica et biophysica acta. 1987;917:148–61. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 105.Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, et al. Specific regional transcription of apolipoprotein E in human brain neurons. The American journal of pathology. 1999;154:601–11. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Han SH, Einstein G, Weisgraber KH, Strittmatter WJ, Saunders AM, Pericak-Vance M, et al. Apolipoprotein E is localized to the cytoplasm of human cortical neurons: a light and electron microscopic study. Journal of neuropathology and experimental neurology. 1994;53:535–44. doi: 10.1097/00005072-199409000-00013. [DOI] [PubMed] [Google Scholar]
- 107.Aoki K, Uchihara T, Sanjo N, Nakamura A, Ikeda K, Tsuchiya K, et al. Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke. 2003;34:875–80. doi: 10.1161/01.STR.0000064320.73388.C6. [DOI] [PubMed] [Google Scholar]
- 108.Ladu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, et al. Lipoproteins in the central nervous system. Annals of the New York Academy of Sciences. 2000;903:167–75. doi: 10.1111/j.1749-6632.2000.tb06365.x. [DOI] [PubMed] [Google Scholar]
- 109.Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, et al. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (-/-), and human apoE transgenic mice. The Journal of biological chemistry. 1999;274:30001–7. doi: 10.1074/jbc.274.42.30001. [DOI] [PubMed] [Google Scholar]
- 110.Fagan AM, Holtzman DM. Astrocyte lipoproteins, effects of apoE on neuronal function, and role of apoE in amyloid-beta deposition in vivo. Microsc Res Tech. 2000;50:297–304. doi: 10.1002/1097-0029(20000815)50:4<297::AID-JEMT9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 111.de Chaves EP, Narayanaswami V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008;3:505–30. doi: 10.2217/17460875.3.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cambon K, Davies HA, Stewart MG. Synaptic loss is accompanied by an increase in synaptic area in the dentate gyrus of aged human apolipoprotein E4 transgenic mice. Neuroscience. 2000;97:685–92. doi: 10.1016/s0306-4522(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 113.Moghadasian MH, McManus BM, Nguyen LB, Shefer S, Nadji M, Godin DV, et al. Pathophysiology of apolipoprotein E deficiency in mice: relevance to apo E-related disorders in humans. FASEB J. 2001;15:2623–30. doi: 10.1096/fj.01-0463com. [DOI] [PubMed] [Google Scholar]
- 114.Anderson R, Barnes JC, Bliss TV, Cain DP, Cambon K, Davies HA, et al. Behavioural, physiological and morphological analysis of a line of apolipoprotein E knockout mouse. Neuroscience. 1998;85:93–110. doi: 10.1016/s0306-4522(97)00598-8. [DOI] [PubMed] [Google Scholar]
- 115.Anderson R, Higgins GA. Absence of central cholinergic deficits in ApoE knockout mice. Psychopharmacology (Berl) 1997;132:135–44. doi: 10.1007/s002130050329. [DOI] [PubMed] [Google Scholar]
- 116.Bronfman FC, Tesseur I, Hofker MH, Havekens LM, Van Leuven F. No evidence for cholinergic problems in apolipoprotein E knockout and apolipoprotein E4 transgenic mice. Neuroscience. 2000;97:411–8. doi: 10.1016/s0306-4522(00)00016-6. [DOI] [PubMed] [Google Scholar]
- 117.Puolivali J, Miettinen R, Pradier L, Riekkinen P., Jr Apolipoprotein E-deficient mice are not more susceptible to the biochemical and memory deficits induced by nucleus basalis lesion. Neuroscience. 2000;96:291–7. doi: 10.1016/s0306-4522(99)00545-x. [DOI] [PubMed] [Google Scholar]
- 118.Champagne D, Dupuy JB, Rochford J, Poirier J. Apolipoprotein E knockout mice display procedural deficits in the Morris water maze: analysis of learning strategies in three versions of the task. Neuroscience. 2002;114:641–54. doi: 10.1016/s0306-4522(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 119.Oitzl MS, Mulder M, Lucassen PJ, Havekes LM, Grootendorst J, de Kloet ER. Severe learning deficits in apolipoprotein E-knockout mice in a water maze task. Brain Res. 1997;752:189–96. doi: 10.1016/s0006-8993(96)01448-5. [DOI] [PubMed] [Google Scholar]
- 120.Chapman S, Sabo T, Roses AD, Michaelson DM. Reversal of presynaptic deficits of apolipoprotein E-deficient mice in human apolipoprotein E transgenic mice. Neuroscience. 2000;97:419–24. doi: 10.1016/s0306-4522(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 121.Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neuroscience letters. 1995;199:1–4. doi: 10.1016/0304-3940(95)12006-p. [DOI] [PubMed] [Google Scholar]
- 122.Walker LC, Parker CA, Lipinski WJ, Callahan MJ, Carroll RT, Gandy SE, et al. Cerebral lipid deposition in aged apolipoprotein-E-deficient mice. The American journal of pathology. 1997;151:1371–7. [PMC free article] [PubMed] [Google Scholar]
- 123.Robertson TA, Dutton NS, Martins RN, Roses AD, Kakulas BA, Papadimitriou JM. Age-related congophilic inclusions in the brains of apolipoprotein E-deficient mice. Neuroscience. 1998;82:171–80. doi: 10.1016/s0306-4522(97)00284-4. [DOI] [PubMed] [Google Scholar]
- 124.Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science (New York, NY. 2001;294:1354–7. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 125.Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 126.Koudinov AR, Koudinova NV. Cholesterol's role in synapse formation. Science (New York, NY. 2002;295:2213. doi: 10.1126/science.295.5563.2213a. [DOI] [PubMed] [Google Scholar]
- 127.de Chaves EI, Rusinol AE, Vance DE, Campenot RB, Vance JE. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. The Journal of biological chemistry. 1997;272:30766–73. doi: 10.1074/jbc.272.49.30766. [DOI] [PubMed] [Google Scholar]
- 128.Posse De Chaves EI, Vance DE, Campenot RB, Kiss RS, Vance JE. Uptake of lipoproteins for axonal growth of sympathetic neurons. The Journal of biological chemistry. 2000;275:19883–90. doi: 10.1074/jbc.275.26.19883. [DOI] [PubMed] [Google Scholar]
- 129.Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–16. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- 130.Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol. 2000;2:42–9. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- 131.Koudinov AR, Koudinova NV. Essential role for cholesterol in synaptic plasticity and neuronal degeneration. FASEB J. 2001;15:1858–60. doi: 10.1096/fj.00-0815fje. [DOI] [PubMed] [Google Scholar]
- 132.Rawlins FA, Villegas GM, Hedley-Whyte ET, Uzman BG. Fine structural localization of cholesterol-1,2- 3 H in degenerating and regenerating mouse sciatic nerve. The Journal of cell biology. 1972;52:615–25. doi: 10.1083/jcb.52.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.LeBlanc AC, Poduslo JF. Regulation of apolipoprotein E gene expression after injury of the rat sciatic nerve. J Neurosci Res. 1990;25:162–71. doi: 10.1002/jnr.490250203. [DOI] [PubMed] [Google Scholar]
- 134.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 135.Poirier J, Baccichet A, Dea D, Gauthier S. Cholesterol synthesis and lipoprotein reuptake during synaptic remodelling in hippocampus in adult rats. Neuroscience. 1993;55:81–90. doi: 10.1016/0306-4522(93)90456-p. [DOI] [PubMed] [Google Scholar]
- 136.Rawlins FA, Hedley-Whyte ET, Villegas G, Uzman BG. Reutilization of cholesterol-1,2-H3 in the regeneration of peripheral nerve. An autoradiographic study. Lab Invest. 1970;22:237–40. [PubMed] [Google Scholar]
- 137.Boyles JK, Notterpek LM, Anderson LJ. Accumulation of apolipoproteins in the regenerating and remyelinating mammalian peripheral nerve. Identification of apolipoprotein D, apolipoprotein A-IV, apolipoprotein E, and apolipoprotein A-I. The Journal of biological chemistry. 1990;265:17805–15. [PubMed] [Google Scholar]
- 138.Herz J. Apolipoprotein E receptors in the nervous system. Curr Opin Lipidol. 2009;20:190–6. doi: 10.1097/MOL.0b013e32832d3a10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Masliah E, Samuel W, Veinbergs I, Mallory M, Mante M, Saitoh T. Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res. 1997;751:307–14. doi: 10.1016/s0006-8993(96)01420-5. [DOI] [PubMed] [Google Scholar]
- 140.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, et al. Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–80. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE, et al. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis. 2005;18:390–8. doi: 10.1016/j.nbd.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 142.Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer's disease patients. Neuroscience. 2003;122:305–15. doi: 10.1016/j.neuroscience.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 143.Jordan BD. Genetic influences on outcome following traumatic brain injury. Neurochem Res. 2007;32:905–15. doi: 10.1007/s11064-006-9251-3. [DOI] [PubMed] [Google Scholar]
- 144.Sorbi S, Nacmias B, Piacentini S, Repice A, Latorraca S, Forleo P, et al. ApoE as a prognostic factor for post-traumatic coma. Nat Med. 1995;1:852. doi: 10.1038/nm0995-852. [DOI] [PubMed] [Google Scholar]
- 145.Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–71. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 146.Sabo T, Lomnitski L, Nyska A, Beni S, Maronpot RR, Shohami E, et al. Susceptibility of transgenic mice expressing human apolipoprotein E to closed head injury: the allele E3 is neuroprotective whereas E4 increases fatalities. Neuroscience. 2000;101:879–84. doi: 10.1016/s0306-4522(00)00438-3. [DOI] [PubMed] [Google Scholar]
- 147.Horsburgh K, McCulloch J, Nilsen M, Roses AD, Nicoll JA. Increased neuronal damage and apoE immunoreactivity in human apolipoprotein E, E4 isoform-specific, transgenic mice after global cerebral ischaemia. Eur J Neurosci. 2000;12:4309–17. [PubMed] [Google Scholar]
- 148.Boschert U, Merlo-Pich E, Higgins G, Roses AD, Catsicas S. Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol Dis. 1999;6:508–14. doi: 10.1006/nbdi.1999.0251. [DOI] [PubMed] [Google Scholar]
- 149.Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, et al. Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer's disease. The Journal of biological chemistry. 2004;279:3862–8. doi: 10.1074/jbc.M309475200. [DOI] [PubMed] [Google Scholar]
- 150.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–94. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci. 2004;23:189–204. doi: 10.1385/JMN:23:3:189. [DOI] [PubMed] [Google Scholar]
- 152.Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. The Journal of biological chemistry. 2002;277:21821–8. doi: 10.1074/jbc.M112109200. [DOI] [PubMed] [Google Scholar]
- 153.Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, et al. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18700–5. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Buttini M, Masliah E, Yu GQ, Palop JJ, Chang S, Bernardo A, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. The American journal of pathology. 2010;177:563–9. doi: 10.2353/ajpath.2010.090973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Veinbergs I, Mallory M, Mante M, Rockenstein E, Gilbert JR, Masliah E. Differential neurotrophic effects of apolipoprotein E in aged transgenic mice. Neuroscience letters. 1999;265:218–22. doi: 10.1016/s0304-3940(99)00243-8. [DOI] [PubMed] [Google Scholar]
- 156.Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, et al. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (-/-) mice but impaired clearance of axonal degeneration products following injury. Experimental neurology. 1998;151:314–25. doi: 10.1006/exnr.1998.6818. [DOI] [PubMed] [Google Scholar]
- 157.Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Experimental neurology. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- 158.Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–62. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- 159.Methia N, Andre P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7:810–5. [PMC free article] [PubMed] [Google Scholar]
- 160.Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–23. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 161.Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–38. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- 162.Auld DS, Kornecook TJ, Bastianetto S, Quirion R. Alzheimer's disease and the basal forebrain cholinergic system: relations to beta-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol. 2002;68:209–45. doi: 10.1016/s0301-0082(02)00079-5. [DOI] [PubMed] [Google Scholar]
- 163.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:8098–102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Frisoni GB, Govoni S, Geroldi C, Bianchetti A, Calabresi L, Franceschini G, et al. Gene dose of the epsilon 4 allele of apolipoprotein E and disease progression in sporadic late-onset Alzheimer's disease. Annals of neurology. 1995;37:596–604. doi: 10.1002/ana.410370509. [DOI] [PubMed] [Google Scholar]
- 165.Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Fraser PE, et al. Abeta1-40 but not Abeta1-42 levels in cortex correlate with apolipoprotein E epsilon4 allele dosage in sporadic Alzheimer's disease. Brain Res. 1997;748:250–2. doi: 10.1016/s0006-8993(96)01363-7. [DOI] [PubMed] [Google Scholar]
- 166.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron. 1993;11:575–80. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 167.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 168.Zhong N, Weisgraber KH. Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. The Journal of biological chemistry. 2009;284:6027–31. doi: 10.1074/jbc.R800009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 170.Fryer JD, Demattos RB, McCormick LM, O'Dell MA, Spinner ML, Bales KR, et al. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. The Journal of biological chemistry. 2005;280:25754–9. doi: 10.1074/jbc.M502143200. [DOI] [PubMed] [Google Scholar]
- 171.Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28:11445–53. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gong JS, Morita SY, Kobayashi M, Handa T, Fujita SC, Yanagisawa K, et al. Novel action of apolipoprotein E (ApoE): ApoE isoform specifically inhibits lipid-particle-mediated cholesterol release from neurons. Mol Neurodegener. 2007;2:9. doi: 10.1186/1750-1326-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Michikawa M, Fan QW, Isobe I, Yanagisawa K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74:1008–16. doi: 10.1046/j.1471-4159.2000.0741008.x. [DOI] [PubMed] [Google Scholar]
- 174.Hirsch-Reinshagen V, Maia LF, Burgess BL, Blain JF, Naus KE, McIsaac SA, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. The Journal of biological chemistry. 2005;280:43243–56. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- 175.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. The Journal of biological chemistry. 2005;280:43224–35. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 176.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. The Journal of biological chemistry. 2005;280:43236–42. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 177.Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. The Journal of biological chemistry. 2004;279:40987–93. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- 178.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. The Journal of clinical investigation. 2008;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–91. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 180.Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, et al. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:10601–6. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vanmierlo T, Rutten K, Dederen J, Bloks VW, van Vark-van der Zee LC, Kuipers F, et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 182.Eckert GP, Vardanian L, Rebeck GW, Burns MP. Regulation of central nervous system cholesterol homeostasis by the liver X receptor agonist TO-901317. Neuroscience letters. 2007;423:47–52. doi: 10.1016/j.neulet.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 183.Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–8. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 184.Fan J, Donkin J, Wellington C. Greasing the wheels of Abeta clearance in Alzheimer's disease: the role of lipids and apolipoprotein E. Biofactors. 2009;35:239–48. doi: 10.1002/biof.37. [DOI] [PubMed] [Google Scholar]
- 185.Weisgraber KH, Mahley RW. Human apolipoprotein E: the Alzheimer's disease connection. Faseb J. 1996;10:1485–94. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 186.Mahley RW, Huang Y. Apolipoprotein E: from atherosclerosis to Alzheimer's disease and beyond. Curr Opin Lipidol. 1999;10:207–17. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 187.Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. The Journal of biological chemistry. 2004;279:25511–6. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]
- 188.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5644–51. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cho HS, Hyman BT, Greenberg SM, Rebeck GW. Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Abeta aggregation. Journal of neuropathology and experimental neurology. 2001;60:342–9. doi: 10.1093/jnen/60.4.342. [DOI] [PubMed] [Google Scholar]
- 190.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8838–43. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer's disease an apolipoprotein E amyloidosis? Lancet. 1995;345:956–8. doi: 10.1016/s0140-6736(95)90701-7. [DOI] [PubMed] [Google Scholar]
- 192.Nakamura T, Watanabe A, Fujino T, Hosono T, Michikawa M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 2009;4:35. doi: 10.1186/1750-1326-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Marques MA, Owens PA, Crutcher KA. Progress toward identification of protease activity involved in proteolysis of apolipoprotein e in human brain. J Mol Neurosci. 2004;24:73–80. doi: 10.1385/JMN:24:1:073. [DOI] [PubMed] [Google Scholar]
- 194.Marques MA, Tolar M, Harmony JA, Crutcher KA. A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. Neuroreport. 1996;7:2529–32. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 195.Tolar M, Marques MA, Harmony JA, Crutcher KA. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci. 1997;17:5678–86. doi: 10.1523/JNEUROSCI.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Weisgraber KH, Roses AD, Strittmatter WJ. The role of apolipoprotein E in the nervous system. Curr Opin Lipidol. 1994;5:110–6. doi: 10.1097/00041433-199404000-00007. [DOI] [PubMed] [Google Scholar]
- 197.Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. Journal of molecular biology. 2006;361:932–44. doi: 10.1016/j.jmb.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 198.Ji ZS, Mullendorff K, Cheng IH, Miranda RD, Huang Y, Mahley RW. Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. The Journal of biological chemistry. 2006;281:2683–92. doi: 10.1074/jbc.M506646200. [DOI] [PubMed] [Google Scholar]
- 199.Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends in biochemical sciences. 2006;31:445–54. doi: 10.1016/j.tibs.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 200.Zhong N, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. The Journal of biological chemistry. 2009;284:27273–80. doi: 10.1074/jbc.M109.014464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhong N, Scearce-Levie K, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction: synaptic and cognitive deficits in mice. Alzheimers Dement. 2008;4:179–92. doi: 10.1016/j.jalz.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25:10658–63. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Carter DB, Dunn E, McKinley DD, Stratman NC, Boyle TP, Kuiper SL, et al. Human apolipoprotein E4 accelerates beta-amyloid deposition in APPsw transgenic mouse brain. Annals of neurology. 2001;50:468–75. doi: 10.1002/ana.1134. [DOI] [PubMed] [Google Scholar]
- 205.Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, et al. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's-disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1211–6. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Buttini M, Yu GQ, Shockley K, Huang Y, Jones B, Masliah E, et al. Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid beta peptides but not on plaque formation. J Neurosci. 2002;22:10539–48. doi: 10.1523/JNEUROSCI.22-24-10539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhong N, Weisgraber KH. Understanding the Basis for the Association of Apoe4 with Alzheimer's Disease: Opening the Door for Therapeutic Approaches. Curr Alzheimer Res. 2009 doi: 10.2174/156720509789207921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, et al. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–7. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 209.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–5. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 210.Hardy J. Testing times for the “amyloid cascade hypothesis”. Neurobiol Aging. 2002;23:1073–4. doi: 10.1016/s0197-4580(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 211.Golde TE. Alzheimer disease therapy: can the amyloid cascade be halted? The Journal of clinical investigation. 2003;111:11–8. doi: 10.1172/JCI17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–9. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 213.Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. The Journal of cell biology. 1998;141:1031–9. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Selkoe D, Kopan R. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565–97. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- 215.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science (New York, NY. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 216.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–51. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 217.Wisniewski T, Golabek A, Matsubara E, Ghiso J, Frangione B. Apolipoprotein E: binding to soluble Alzheimer's beta-amyloid. Biochemical and biophysical research communications. 1993;192:359–65. doi: 10.1006/bbrc.1993.1423. [DOI] [PubMed] [Google Scholar]
- 218.Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neuroscience letters. 1992;135:235–8. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- 219.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–6. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 220.Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, et al. Apolipoprotein E associates with beta amyloid peptide of Alzheimer's disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. The Journal of clinical investigation. 1994;94:860–9. doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT. Purification of apolipoprotein E attenuates isoform-specific binding to beta-amyloid. The Journal of biological chemistry. 1995;270:9039–42. doi: 10.1074/jbc.270.16.9039. [DOI] [PubMed] [Google Scholar]
- 222.Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, et al. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer's amyloid beta peptides. Biochem J. 2000;2:348. 359–65. [PMC free article] [PubMed] [Google Scholar]
- 223.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. The Journal of biological chemistry. 1994;269:23403–6. [PubMed] [Google Scholar]
- 224.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science (New York, NY. 1994;264:850–2. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 225.Tamamizu-Kato S, Cohen JK, Drake CB, Kosaraju MG, Drury J, Narayanaswami V. Interaction with amyloid beta peptide compromises the lipid binding function of apolipoprotein E. Biochemistry. 2008;47:5225–34. doi: 10.1021/bi702097s. [DOI] [PubMed] [Google Scholar]
- 226.Shobab LA, Hsiung GY, Feldman HH. Cholesterol in Alzheimer's disease. Lancet Neurol. 2005;4:841–52. doi: 10.1016/S1474-4422(05)70248-9. [DOI] [PubMed] [Google Scholar]
- 227.Poirier J. Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer's disease. Neurobiol Aging. 2005;26:355–61. doi: 10.1016/j.neurobiolaging.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 228.Fenili D, McLaurin J. Cholesterol and apoe: a target for Alzheimer's disease therapeutics. Curr Drug Targets CNS Neurol Disord. 2005;4:553–67. doi: 10.2174/156800705774322085. [DOI] [PubMed] [Google Scholar]
- 229.Cam JA, Bu G. Modulation of beta-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol Neurodegener. 2006;1:8. doi: 10.1186/1750-1326-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Bu G, Cam J, Zerbinatti C. LRP in amyloid-beta production and metabolism. Annals of the New York Academy of Sciences. 2006;1086:35–53. doi: 10.1196/annals.1377.005. [DOI] [PubMed] [Google Scholar]
- 231.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–7. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 232.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–26. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- 233.Cole GM, Ard MD. Influence of lipoproteins on microglial degradation of Alzheimer's amyloid beta-protein. Microsc Res Tech. 2000;50:316–24. doi: 10.1002/1097-0029(20000815)50:4<316::AID-JEMT11>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 234.Zlokovic BV. Clearing amyloid through the blood-brain barrier. J Neurochem. 2004;89:807–11. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]
- 235.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–8. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 236.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 237.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. The Journal of clinical investigation. 2008;118:4002–13. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, et al. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer's disease. The Journal of clinical investigation. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004;41:193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 240.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bogdanovic N, Corder E, Lannfelt L, Winblad B. APOE polymorphism and clinical duration determine regional neuropathology in Swedish APP(670, 671) mutation carriers: implications for late-onset Alzheimer's disease. J Cell Mol Med. 2002;6:199–214. doi: 10.1111/j.1582-4934.2002.tb00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–18. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science (New York, NY. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 244.Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. The American journal of pathology. 2000;156:951–64. doi: 10.1016/S0002-9440(10)64963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–34. doi: 10.1523/JNEUROSCI.4315-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, et al. Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Experimental neurology. 1994;125:163–71. doi: 10.1006/exnr.1994.1019. discussion 72-4. [DOI] [PubMed] [Google Scholar]
- 247.Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18694–9. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hill JM, Bhattacharjee PS, Neumann DM. Apolipoprotein E alleles can contribute to the pathogenesis of numerous clinical conditions including HSV-1 corneal disease. Exp Eye Res. 2007;84:801–11. doi: 10.1016/j.exer.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]