

SUMMARY
Most current treatments for epilepsy are symptomatic therapies that suppress seizures but do not affect the underlying course or prognosis of epilepsy. The need for disease-modifying or “antiepileptogenic” treatments for epilepsy is widely recognized, but no such preventative therapies have yet been established for clinical use. A rational strategy for preventing epilepsy is to target primary signaling pathways that initially trigger the numerous downstream mechanisms mediating epileptogenesis. The mammalian target of rapamycin (mTOR) pathway represents a logical candidate, because mTOR regulates multiple cellular functions that may contribute to epileptogenesis, including protein synthesis, cell growth and proliferation, and synaptic plasticity. The importance of the mTOR pathway in epileptogenesis is best illustrated by Tuberous Sclerosis Complex (TSC), one of the most common genetic causes of epilepsy. In mouse models of TSC, mTOR inhibitors prevent the development of epilepsy and underlying brain abnormalities associated with epileptogenesis. Accumulating evidence suggests that mTOR also participates in epileptogenesis due to a variety of other causes, including focal cortical dysplasia and acquired brain injuries, such as in animal models following status epilepticus or traumatic brain injury. Thus, mTOR inhibition may represent a potential antiepileptogenic therapy for diverse types of epilepsy, including both genetic and acquired epilepsies.
Keywords: seizure, epileptogenesis, epilepsy, traumatic brain injury
INTRODUCTION
Epilepsy affects about 1% of all people and leads to significant morbidity and mortality. The etiologies of epilepsy are numerous, including a variety of genetic and acquired causes. In many cases, seizures can be controlled with medications or non-medical therapies, such as epilepsy surgery, vagal nerve stimulator, or ketogenic diet. However, approximately one-third of all epilepsy patients are poorly-responsive or intractable to available therapies (Kwan and Brodie, 2006). Even in well-controlled patients, most current treatments (with the possible exception of epilepsy surgery) are primarily symptomatic therapies that suppress seizures but do not correct the underlying brain abnormalities causing epilepsy or alter the natural history and long-term prognosis of epilepsy. For example, standard seizure medications, such as phenytoin or valproate, can reduce the frequency of seizures in the immediate period following head trauma, but do not ultimately prevent the development of chronic post-traumatic epilepsy (Temkin et al., 1990, 1999; Temkin, 2001). Similarly, treatment of febrile seizures as an infant can reduce the recurrence of febrile seizures during the treatment period, but does not alter the long-term risk of epilepsy (Knudsen et al., 1996). Thus, it is now widely-recognized that completely different types of drugs are needed that are not just “anti-seizure” but are truly “antiepileptogenic” in preventing or reversing epileptogenesis, the pathophysiological changes in the brain that lead to the development of chronic epilepsy (Dichter, 2006; Loscher and Schmidt, 2006; Stefan et al., 2006). To this point, however, besides a few demonstrations of proof-of-principle that an anti-epileptogenic approach can work (Graber and Prince, 1999; Laughinghouse et al., 2008), no such anti-epileptogenic therapies have been established for clinical use
A major reason that prior attempts to develop antiepileptogenic therapies have failed is that most current medications act primarily on molecular mechanisms that mediate the end-stage symptoms of epilepsy, the seizures themselves. A more rational strategy for preventing epilepsy is to target the primary signaling pathways that initially trigger the numerous downstream cellular and molecular mechanisms mediating epileptogenesis. The mammalian target of rapamycin (mTOR) signaling pathway represents a logical candidate for such a pathway, because mTOR regulates protein synthesis and multiple other downstream cellular functions that may influence neuronal excitability and epileptogenesis. Furthermore, existing drugs are available that inhibit mTOR and could be readily tested as antiepileptogenic therapies. In particular, rapamycin, which was originally discovered as an antifungal agent in the soil of the famous Polynesian island of Rapa Nui or Easter Island, is approved for clinical use for its immunosuppressant properties, but may also have antiepileptogenic actions. In this review, evidence for the role of the mTOR signaling pathway in mechanisms of epileptogenesis and the potential utility of mTOR inhibitors as antiepileptogenic therapy will be analyzed. The genetic disease, Tuberous Sclerosis Complex (TSC), will be emphasized as a model disorder for investigating mTOR involvement in epilepsy and testing the antiepileptogenic potential of mTOR inhibitors. Furthermore, the possible contribution of mTOR to other, more common types of epilepsy, such as posttraumatic epilepsy following traumatic brain injury, will also be discussed. Overall, I propose that mTOR inhibition may represent an effective antiepileptogenic therapy for diverse types of epilepsy, including some types of both genetic and acquired epilepsies.
MTOR: A CENTRAL REGULATOR OF CELL GROWTH, PROLIFERATION & SURVIVAL
mTOR is a 290 kD serine-threonine protein kinase that is highly conserved among mammals and also has closely-related analogs in lower eukaryotes, such as Drosophila and yeast (Sarbassov et al., 2005; Sandsmark et al., 2007a). mTOR has been implicated in numerous cellular functions; in a global sense, many of these functions relate to overall cellular growth, survival, and homeostasis (Sarbassov et al., 2005; Sandsmark et al., 2007a; Tsang et al., 2007). A variety of upstream signaling pathways can regulate mTOR activity in response to different extracellular stimuli or intracellular signals, including nutrient and energy status, growth factors, and stress (Fig. 1; Bolster et al., 2002; Inoki et al., 2002; Kimura et al., 2003). In turn, mTOR responds to these upstream signals by modulating multiple downstream pathways, which mediate cellular growth, proliferation, metabolism, and survival, usually due to direct changes in the translation of relevant proteins (Burnett et al., 1998; Fingar et al., 2002). Thus, during anabolic states in the presence of nutrients, growth factors, or insulin, specific upstream regulators, such as the phosphatidylinositol-3 kinase (PI3K)/Akt (protein kinase B) pathway, activate mTOR, leading to increased protein synthesis, cellular growth, and proliferation (Inoki et al., 2002; Manning et al., 2002). In catabolic states with nutrient/energy or oxygen deprivation, other upstream pathways, such as AMP-kinase (AMPK), inhibit mTOR activity, thus decreasing protein translation and cellular growth, proliferation, and metabolism (Bolster et al., 2002; Kimura et al., 2003). Activation or inhibition of mTOR by upstream pathways is generally accomplished through opposing effects on the tuberous sclerosis gene products, hamartin and tuberin, and the small GTPase protein, Rheb (Fig. 1).
Figure 1.
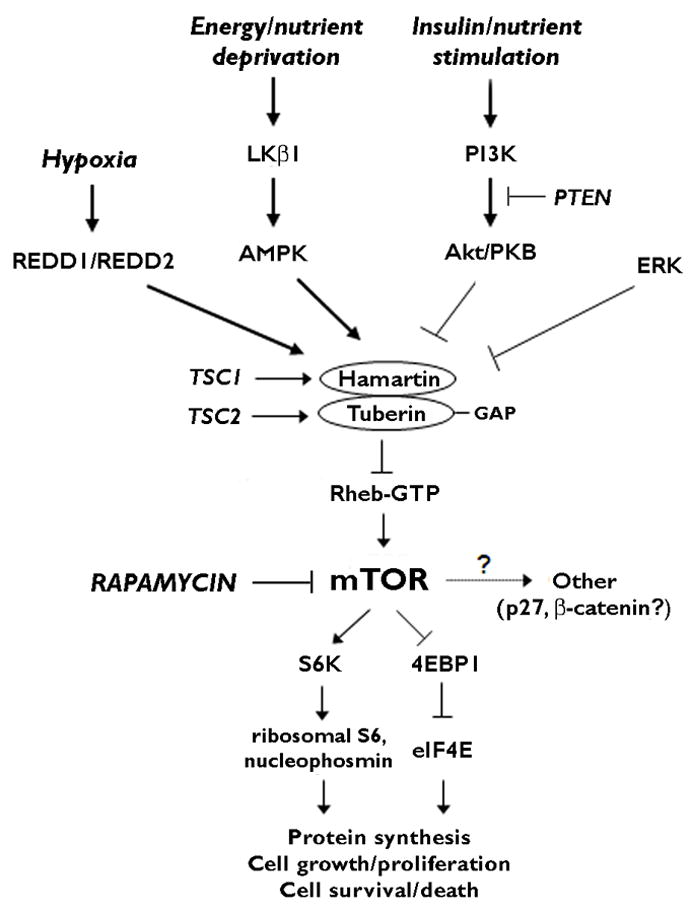
Physiological regulation of the mTOR signaling pathway. mTOR controls multiple downstream effectors, such as S6K/S6/nucleophosmin and 4EBP1/eIF4E, which regulate protein synthesis and other processes related to cellular growth, proliferation, metabolism, and survival. In response to environmental or physiological stimuli, multiple upstream pathways, primarily involving cascades of protein kinases, may either activate (PI3K/Akt, ERK) or inhibit (REDD1/REDD2, LKβ1/AMPK) mTOR via modulation of the tuberin-hamartin complex and Rheb GTPase. In anabolic states, such as with insulin, growth factor, or nutrient stimulation, PI3K/Akt and ERK activate the mTOR pathway and induce protein synthesis, cell growth and proliferation. Conversely, in catabolic states, the REDD1/REDD2 and LKβ1/AMPK pathways respond to hypoxia or energy/nutrient deprivation by inhibiting the mTOR pathway and thus slowing protein synthesis, cell growth, and metabolism. In the disease of TSC, mutation of one of the TSC genes leads to disinhibition or hyperactivation of the mTOR pathway, causing dysregulated growth and proliferation and predisposing to tumor formation. In addition to genetic mutations, acquired brain injuries may cause abnormal activation of mTOR and related pathways, which may lead to cellular and molecular changes promoting epileptogenesis (See Fig. 2). Note that this schematic figure is oversimplified for clarity, as upstream regulators, feedback loops, intermediary steps, and alternative pathways (e.g. mTORC1 vs. mTORC2) are not shown. Abbreviations: 4EBP1 – elongation factor 4E binding protein 1; AMPK – AMP-activated protein kinase; eIF4E – elongation initiation factor 4E; ERK – extracellular signal-regulated protein kinase; GAP – GTPase activating protein; mTOR – mammalian target of rapamycin; PI3K – phosphatidylinositide-3 kinase; PKB – protein kinase B (a.k.a Akt); PTEN - phosphatase and tensin homolog deleted on chromosome ten; Rheb – Ras homolog expressed in brain; S6K – ribosomal S6 kinase.
Downstream from mTOR, there are multiple pathways that mediate the effects of mTOR on protein synthesis and other cellular functions (Fig. 1). For example, mTOR activates ribosomal S6 kinase-1 (S6K1), which phosphorylates the ribosomal protein S6, promoting ribosomal biogenesis and protein translation (Chung et al., 1992; Burnett et al., 1998; Fingar et al., 2002). In addition, mTOR leads to inhibition of the elongation factor 4E binding protein 1 (4EBP1) and subsequent activation (release of inhibition) of the mRNA elongation initiation factor 4E (eIF4E), also triggering protein synthesis (Burnett et al., 1998; Fingar et al., 2002). Besides the S6K/S6 and 4EBP1/eIF4E pathways, additional mechanisms may also be stimulated by mTOR to influence protein synthesis and cell growth, such as shuttling of ribosomal subunits by the nucleolar protein, nucleophosmin (Pelletier et al., 2007; Sandsmark et al., 2007b). Furthermore, other downstream signaling elements, such as p27 and beta-catenin, may be more directly involved in regulation of cell cycle progression and proliferation, although the critical role of mTOR in triggering these pathways is less established (Kawamata et al., 1998; Soucek et al., 1998; Maki et al., 2003; Daniel et al., 2004; Jozwiak and Wlodarski, 2006). Finally, the cell signaling involving mTOR is further complicated by poorly-defined intermediate steps, multiple feedback loops, and the formation of a mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 and mTORC2 involve formation of functional complexes of mTOR bound to the regulatory proteins, raptor or rictor, respectively, which differ in their sensitivity to the mTOR inhibitor, rapamycin (Huang and Manning, 2009).
In addition to the functions of mTOR involving cellular growth and proliferation, mTOR also has other important, complex roles in regulating cell survival and cell death, especially related to the processes of autophagy, apoptosis, and immune regulation. Autophagy involves the degradation and recycling of proteins and other macromolecules and normally promotes cell survival under conditions of bioenergetic stress or catabolic states where resources are limited. However, in some situations, autophagy may also mediate an alternative (non-apoptotic, autophagic) form of programmed cell death (Type II PCD), thus revealing a dual role of autophagy in promoting cell survival and death (Shintani and Klionsky, 2004; Baehrecke, 2005; Codogno and Meijer, 2005). In anabolic states, in addition to stimulating protein synthesis, mTOR generally inhibits autophagy and thus reduces the degradation of proteins. Conversely, mTOR inhibitors, such as rapamycin, usually stimulate autophagy, with a resultant neuroprotective effect in various models of brain injury (Carloni et al., 2008; Pan et al., 2008). mTOR has similarly been implicated in participating in oxidative stress (Di Nardo et al., 2009) and apoptosis (Type I PCD), although rapamycin may have both pro- and anti-apoptotic effects under different conditions (Castedo et al., 2002; Asnaghi et al., 2004). Finally, mTOR plays a critical role in immune responses via regulation of antigen-presenting cells and T-cells, and rapamycin is used clinically as a potent immunosuppressant drug (Thomson et al., 2009). While effects of rapamycin on autophagy, apoptosis, and immune regulation may most directly translate into neuromodulatory and neuroprotective properties, these features may also contribute to antiepileptogenic effects.
The clinical and therapeutic implications of mTOR are widespread and continue to expand. Abnormal mTOR activity, leading to excessive cellular growth and proliferation, has been implicated in the pathophysiology of numerous human cancers, including both sporadic, isolated tumors of specific organs and multiorgan, genetic tumor syndromes (Guertin and Sabatini, 2007). In many of these cases, specific mutations of some component of the mTOR signaling pathway have been documented, resulting in hyperactivation of mTOR or its downstream effectors. Besides cancer, dysregulation of mTOR has also been implicated in a number of other diseases, involving metabolic or environmental derangements, such as diabetes, obesity, cardiovascular disease, and neurodegenerative disorders (Tsang et al., 2007). From a translational standpoint, the potential role of mTOR in the pathophysiology of these various disorders has immediate therapeutic implications, as a number of clinically available drugs exist that inhibit mTOR, including the prototype, rapamycin. Thus, rapamycin and other mTOR inhibitors are already being tested in clinical trials for various cancers and other diseases (Dancey, 2006; Tsang et al., 2007).
Overall, mTOR appears to serve as a master switch responding to different physiological and pathological stimuli to maintain homeostasis by regulating cellular growth, proliferation, and survival. mTOR is increasingly recognized to be involved in a large spectrum of physiological functions under normal conditions and to be dysregulated in a diverse group of diseases. mTOR inhibitors, such as rapamycin, are being considered or are already in clinical trials for a number of these diseases. Based on the physiological and pathophysiological properties of mTOR, it is reasonable to hypothesize that mTOR signaling could be involved in mechanisms of epileptogenesis. In the remainder of this review, evidence for the role of mTOR in epileptogenesis and the potential utility of mTOR inhibitors as anti-epileptogenic agents will be analyzed in different types of epilepsies, starting with the genetic epilepsy, Tuberous Sclerosis Complex, and other tumor- or cortical malformation-associated epilepsies, and extending to more common, acquired epilepsies.
TUBEROUS SCLEROSIS COMPLEX: A MODEL GENETIC DISORDER OF MTOR INVOLVEMENT IN EPILEPSY
Epilepsy in TSC
Tuberous Sclerosis Complex (TSC) is an autosomal dominant disease that affects ~1 in 6000 people and represents one of the most common genetic causes of epilepsy (Sparagana and Roach, 2000; Kwiatkowski, 2003; Crino et al., 2006). TSC is caused by mutation of one of two genes, the TSC1 and TSC2 genes, and is characterized by benign tumor or hamartoma formation in multiple organs, such as the brain, skin, eyes, kidneys, heart and lungs. While many of the TSC-related lesions may be minimally-symptomatic, neurological manifestations in TSC are quite common and usually represent the most disabling problems of the disease, including epilepsy, developmental delay/cognitive deficits, and autism. Epilepsy is particularly common, affecting up to 80–90% of individuals with TSC and is often refractory to available treatment options (Webb et al., 1991; Sparagana et al., 2003; Holmes et al., 2007). Seizures are usually thought to originate within or around hamartamous lesions or tubers in the cortex, but this is often debated and the specific mechanisms of seizure generation in TSC are poorly defined (Holmes et al., 2007; Wong, 2008).
Recently, it has become apparent that TSC may represent a model disease for developing antiepileptogenic approaches for several reasons. First of all, patients are often diagnosed with TSC prior to the onset of epilepsy, due to non-neurological findings or a positive family history; since TSC patients are at high risk for subsequently developing epilepsy, this subgroup of patients represent an appropriate population to attempt a preventative, antiepileptogenic therapy. Other TSC patients present initially with infantile spasms, which later transitions into chronic, intractable epilepsy; initiation of an anti-epileptogenic therapy may be appropriate in TSC patients who first develop infantile spasms. Finally, critical for developing an antiepileptogenic approach, recent insights into the molecular defects and signaling abnormalities due to mutation of the TSC genes, especially in the mTOR signaling pathway, provide potential novel therapeutic targets for interrupting epileptogenesis at an early, primary mechanistic stage.
mTOR pathway signaling in tumorigenesis in TSC
The major clinical features of TSC involve tumor or hamartoma formation in multiple organs, which reflects a primary abnormality in cellular growth and proliferation. Over the past several years, tremendous progress has been made in identifying the defective cell signaling mechanisms by which TSC gene mutations lead to tumorigenesis (Fig. 1). A major discovery was that the TSC1 and TSC2 gene products, hamartin and tuberin, act as a complex that inhibits the mTOR signaling pathway (Gao et al., 2002; Inoki et al. 2002; Tee et al., 2002; El-Hasemite et al., 2003). The tuberin-hamartin complex is a GTPase activating protein (GAP) that most directly inhibits the small GTPase-protein, Ras homolog expressed in brain (Rheb) (Inoki et al., 2003; Zhang et al., 2003; Saucedo et al., 2003; Stocker et al., 2003). As Rheb, in turn, activates mTOR, the TSC genes function to dampen down mTOR signaling pathway activity, and, given the role of mTOR in stimulating cell growth and proliferation, normally prevent excessive growth and proliferation. Thus, in the disease of TSC, mutation of either the TSC1 or TSC2 gene leads to a disinhibition or increase in Rheb activity, and subsequent hyperactivation of mTOR and its downstream pathways, which ultimately results in increased cell growth and tumor formation. Conveniently, the use of rapamycin and other mTOR inhibitors provides the opportunity to test experimentally the critical role of the mTOR pathway in tumorigenesis, and if effective, could be translated directly into clinical trials of TSC patients (Crino, 2008). In fact, mTOR inhibitors have been shown to slow tumor growth in mouse models of TSC (Lee et al., 2005) and recent clinical trials have reported encouraging results for tumors in TSC patients (Franz et al., 2006; Bissler et al., 2008).
mTOR pathway signaling in epileptogenesis in TSC
While the mechanistic link between mTOR activation and tumorigenesis in TSC is firmly established, it is reasonable to hypothesize that the mTOR signaling pathway could also be involved in mechanisms of epileptogenesis (Fig. 2). First of all, abnormal cell growth and proliferation due to mTOR hyperactivation in TSC could indirectly affect the excitability of neuronal circuits and promote seizures. In addition to effects on cell growth and proliferation, mTOR has been implicated in regulating a number of essential neuronal functions, such as neurotransmitter receptor and ion channel expression, neuronal structure and synaptic plasticity (Tang et al., 2002; Jaworski et al., 2005; Kumar et al., 2005; Raab-Graham et al., 2006; Wang et al., 2006). Thus, abnormal mTOR signaling in TSC could directly influence a variety of downstream mechanisms of epileptogenesis, especially those dependent on protein synthesis, such as alterations in neurotransmitter receptor and ion channel expression and neuronal and synaptic organization. Although mechanisms of epileptogenesis in TSC are still incompletely understood, recent progress has been made in defining specific abnormalities in neurons and glia, including alterations in mTOR signaling, that promote epilepsy in TSC (Holmes et al., 2007; Wong, 2008). Several mouse models of TSC have been generated with abnormal neurological phenotypes, which are responsive to mTOR inhibition (Ehninger et al., 2008; Meikle et al., 2008; Zeng et al., 2008; Zhou et al., 2009). In particular, rapamycin has been found to be effective in preventing or reversing a variety of molecular, cellular, and histological abnormalities in these mouse models that likely relate to epileptogenesis, including megencephaly, neuronal hypertrophy, astrocytosis, and impaired myelination (Meikle et al., 2008; Zeng et al., 2008). The role of abnormal mTOR signaling in the development of epilepsy itself has been delineated in most detail in knock-out mice involving inactivation of the Tsc1 gene primarily in glial fibrillary acidic protein (GFAP)-positive cells, which typically develop seizures around 4 weeks of age (Erbayat-Altay et al, 2007). Early rapamycin treatment started prior to the onset of seizures prevents the subsequent development of epilepsy in these mice, whereas late treatment reduces seizure-frequency in mice that already have epilepsy (Zeng et al, 2008). An important limitation, however, is that after rapamycin treatment is stopped, the neurological phenotype of these mice subsequently develops with a delay of several weeks, including the histopathological abnormalities and epilepsy (Zeng et al., 2008). In addition to beneficial effects on epilepsy, remarkably rapamycin has also been shown to reverse learning deficits in another TSC mouse model (Ehninger et al., 2008).
Figure 2.

mTOR may be a central signaling pathway for coordinating multiple mechanisms of epileptogenesis due to diverse causes of epilepsy. The mTOR signaling pathway may be abnormally activated by a variety of genetic defects or acquired injuries, including upstream TSC or PTEN gene mutations, status epilepticus, or traumatic brain injury. In turn, mTOR hyperactivation may trigger multiple downstream mechanisms of epileptogenesis via regulation of protein synthesis and other cellular processes, such as expression of ion channels, apoptosis, autophagy, axonal sprouting, and neurogenesis. Inhibition of mTOR by rapamycin may represent an effective treatment for preventing epileptogenesis in many types of epilepsy involving abnormal mTOR activation.
The reported effects of rapamycin in mouse models of TSC are of potentially high significance because they appear to be most consistent with antiepileptogenic, not simply seizure-suppressing, effects. First of all, in contrast to standard seizure medications that inhibit seizures by directly decreasing neuronal activity, other studies have found no direct effect of rapamycin on neuronal excitability (Daoud et al., 2007; Ruegg et al., 2007). Furthermore, while available seizure medications dampen neuronal excitability primarily by binding immediately and directly to ion channels and neurotransmitter receptors, rapamycin interrupts the more gradual, progressive neuropathological and cellular processes mediating epileptogenesis in these models (Meikle et al., 2008; Zeng et al., 2008). Although these findings are encouraging in supporting an antiepileptogenic action of rapamycin in TSC, continued, long-term treatment with rapamycin was necessary to maintain the beneficial effects. Once rapamycin is stopped, mTOR hyperactivation due to the underlying chronic genetic defect in the Tsc1 gene is no longer suppressed and can proceed to trigger epileptogenesis.
TSC patients may represent good candidates for attempting an antiepileptogenic approach with rapamycin, particularly those who present initially with infantile spasms or are diagnosed with TSC prior to the onset of epilepsy but have a high tuber load. As epilepsy in TSC often appears to follow a progressive, intractable course (Holmes et al. 2007), TSC patients who already have epilepsy may also benefit from mTOR inhibitors as potential disease-modifying therapy. Anecdotal reports already suggest that mTOR inhibitors reduce seizures in patients with TSC (Muncy et al, 2009). However, controlled clinical trials are necessary to establish the efficacy and indications of rapamycin for epilepsy in TSC, as well as its safety. Given the abundance of normal physiological functions of mTOR, there are concerns about the adverse effects of rapamycin on growth and development, especially for chronic, long-term use as might be necessary for a genetic disease. Nevertheless, based on both basic science and preliminary clinical data, mTOR inhibitors show great promise as antiepileptogenic therapy for TSC.
MTOR SIGNALING ABNORMALITIES IN EPILEPSIES ASSOCIATED WITH OTHER TUMOR SYNDROMES OR CORTICAL MALFORMATIONS
TSC is often viewed as a model system that may have relevance or applications to many disorders, given the multi-organ involvement and the widespread functions of the cell signaling pathways in TSC. In particular, abnormalities in the mTOR signaling pathway that are central to the pathogenesis of TSC may also be important for epilepsy in other, pathologically-related tumor syndromes or malformations of cortical development. For example, dysmorphic neurons and cytomegalic cells from pathological specimens of focal cortical dysplasia obtained from intractable epilepsy patients exhibit abnormal activation of molecules in the mTOR signaling pathways (Baybis et al., 2004; Miyata et al., 2004; Ljungberg et al., 2006; Schick et al., 2007). Similarly, gangliomas, low-grade brain tumors that share pathological features with focal cortical dysplasia and TSC and are highly epileptogenic, also display molecular markers of mTOR hyperactivation (Samadani et al., 2007). These studies suggest that abnormal mTOR signaling may contribute to epilepsy in other neoplastic or developmental brain lesions related to TSC. However, it is difficult to prove a pathophysiological role of mTOR in epileptogenesis in these entities based purely on human pathological studies.
Animal models have provided more direct evidence that increased mTOR signaling may be involved in other tumor syndromes or malformations of cortical development. Patients with mutations in the PTEN (phosphatase and tensin homolog deleted on chromosome ten) gene may exhibit a range of phenotypes, including macrocephaly, mental retardation, epilepsy, isolated cancers, and tumor/hamartoma syndromes. As PTEN is an inhibitor of the PI3K/Akt pathway upstream from mTOR, it is perhaps not surprising that various PTEN knock-out mice exhibit neurological features that closely resemble the TSC mouse models, such as megencephaly, neuronal hypertrophy, and seizures. Furthermore, similar to the TSC knock-out mice, rapamycin decrease seizures in these PTEN mouse models (Kwon et al., 2003; Ljundberg et al., 2009; Zhou et al., 2009), as well as improving social deficits suggestive of autism (Zhou et al., 2009). Although single gene mutations are relatively rare causes of epilepsy in general, the findings from these genetic diseases involving PTEN or TSC mutations demonstrate “proof-of-principle” that mTOR inhibition may represent a rational, effective antiepileptogenic strategy for a variety of neurological disorders associated with mTOR hyperactivation, including ganglioglioma, focal cortical dysplasia, and possibly also acquired brain injuries.
MTOR SIGNALING ABNORMALITIES IN MODELS OF ACQUIRED EPILEPSY DUE TO BRAIN INJURY
In addition to specific genetic and developmental epilepsies, the mTOR signaling pathway represents a logical mechanism for triggering epileptogenesis in acquired epilepsies. In the classic concept of epileptogenesis in acquired epilepsies, an initial precipitating injury, such as head trauma, stroke, or an episode of status epilepticus, causes the subsequent development of spontaneous, recurrent seizures. The time interval between the original brain insult and the clinical presentation of the first seizure can often span months to years and is referred to as the latent period of epileptogenesis. During this latent period, numerous abnormalities in the molecular, cellular, and network properties of the brain develop in response to the injury and are responsible for ultimately producing an epileptic state, such as alterations in expression of neurotransmitter receptors and ion channels, apoptosis/autophagy, oxidative/ER stress, axonal sprouting, and neurogenesis (Dudek et al., 2002). Many of these cellular processes mediating epileptogenesis involve protein synthesis and other cellular processes that could, at least partially, utilize the same mTOR-dependent or related pathways that are deregulated in TSC. Thus, a general hypothesis is that mTOR is a central signaling pathway that is activated by a variety of acquired brain injuries and genetic defects and then triggers multiple downstream mechanisms of epileptogenesis (Fig. 2).
Independent of epilepsy per se, the broader role of mTOR inhibitors as neuroprotective agents for acquired brain injuries or neurodegenerative disorders has already been considered in some detail (Zemke et al., 2007; Swiech et al., 2008). As discussed above, mTOR plays an important, complex role in pathophysiological processes and determinants of cell survival and death, including apoptosis, autophagy, oxidative stress, and immune responses. As such, mTOR inhibitors have been found to have neuroprotective properties in various models of brain injury and neurological diseases, such as neonatal hypoxia-ischemia (Carloni et al., 2008), traumatic brain injury (Erlich et al., 2007), toxin-induced neurodegeneration (Pan et al., 2008), and Huntington’s disease models (Ravikumar et al., 2004). While prevention of neuronal death could potentially contribute to antiepileptogenic effects of mTOR inhibitors in response to brain injury, mTOR inhibition could theoretically also counteract injury-induced epileptogenesis via other downstream effects on neurotransmitter receptor/ion channel expression, neuronal structure, synaptic plasticity, and neurogenesis (Tang et al., 2002; Jaworski et al., 2005; Kumar et al., 2005; Raab-Graham et al., 2006; Wang et al., 2006; Buckmaster et al., 2009).
In light of these theoretical considerations, the potential involvement of mTOR in epileptogenesis due to brain injuries has just recently started to be investigated. In a popular rodent model of acquired limbic epilepsy, a single injection of the neurotoxin kainate causes an initial episode of status epilepticus, followed by a latent period, during which multiple cellular and molecular signs of brain injury evolve and may contribute to epileptogenesis, such as changes in ion channel expression, neuronal death, neurogenesis, synaptic reorganization and axonal sprouting (Dudek et al., 2002). After this latent period of epileptogenesis, the animals develop spontaneous epilepsy. Recent studies have found that the mTOR signaling pathway is abnormally activated by kainate status epilepticus, (Shacka et al., 2007; Zeng et al., 2009), as well as in the closely-related pilocarpine model (Buckmaster et al., 2009). In the kainate model, there were two phases of transient mTOR activation: an acute phase lasting several hours that occurred during the immediate period of status epilepticus and was seen in both neocortex and hippocampus; then, after a couple days, a second, longer phase of mTOR activation arose lasting several weeks in hippocampus only, which seemed to correlate with the period of epileptogenesis in hippocampus (Zeng et al., 2009). Rapamycin pretreatment for the three days prior to kainate status epilepticus was able to block both phases of mTOR activation and correspondingly decreased the subsequent frequency of spontaneous seizures, as well as reduced neuronal death, neurogenesis, and mossy fiber sprouting. In addition, rapamycin treatment initiated within a day after the resolution of kainate status epilepticus could block the late phase of mTOR activation and reduce subsequent development of mossy fiber sprouting and epilepsy, but had no effect on neuronal death and neurogenesis (Zeng et al., 2009). Similarly, in the pilocarpine model, chronic focal infusion of rapamycin into hippocampus reduced mossy fiber sprouting (Buckmaster et al., 2009), although the effects on epilepsy were not investigated.
Similar to the studies in the TSC models, the effects of rapamycin in status epilepticus-induced models of epilepsy appear to be most consistent with an antiepileptogenic, not simply seizure-suppressing, action. Rapamycin had no effect on the acute properties of kainate-induced status epilepticus (Zeng et al., 2009). The efficacy of a brief period of rapamycin treatment prior to status epilepticus is most clearly indicative of an antiepileptogenic effect. Unlike TSC which involves chronic mTOR hyperactivation due to a genetic defect and thus may require indefinite treatment, it is possible that in acquired epilepsies short-term suppression of acute, transient mTOR activation around the time of the injury may be sufficient to prevent activation of epileptogenesis over the long-term. However, cessation of rapamycin treatment in the pilocarpine model led to a recurrence of mossy fiber sprouting (Buckmaster et al., 2009), suggesting that pilocarpine status epilepticus also induces a chronic mTOR activation. Additional studies are needed to determine whether there may be a therapeutic window for mTOR inhibition during the latent period to prevent epileptogenesis following acquired brain injury.
In terms of specific downstream mechanisms, mossy fiber sprouting, neuronal death, and neurogenesis have all been implicated in mediating epileptogenesis following status epilepticus (Dudek et al., 2002) and reversal of these processes may, at least partially, account for the antiepileptogenic effects of rapamycin. However, other mTOR-dependent mechanisms of epileptogenesis could also be involved, such as immune modulation, inflammatory responses, autophagy, and oxidative stress. Thus, additional studies are needed to determine the critical downstream effectors that mediate the antiepileptogenic effects of mTOR inhibition in response to the initial brain injury. Similarly, while in TSC there is a direct link between the TSC genes and mTOR, the signaling pathways upstream from mTOR that are initially activated in the kainate model are unknown. Given the massive glutamate receptor activation that occurs with kainate status epilepticus, a rationale hypothesis might involve initial stimulation of ionotrophic or metabotropic glutamate receptors with subsequent activation of the PI3K/Akt pathway (Zhu et al., 2002; Sutton and Chandler, 2002; Hou and Klann, 2004; Lenz and Avruch et al., 2005; Gong et al., 2006). As pharmacological modulators of PI3K and other upstream signaling molecules exist, identification of the upstream pathways that trigger mTOR activation in response to brain injury might lead to other antiepileptogenic therapies that complement mTOR inhibitors.
Besides status epilepticus-induced brain injury, recent studies have begun to investigate the role of mTOR in epileptogenesis in response to other brain injuries. Preliminary studies suggest that mTOR activation could be involved in epileptogenesis following neonatal hypoxia (Zhou et al., 2008). Similarly, the mTOR signaling pathway is abnormally activated by traumatic brain injury (TBI) and rapamycin has neuroprotective effects in animal models of TBI (Chen et al., 2007; Erlich et al., 2007), but the specific effects of rapamycin in preventing posttraumatic epilepsy has not yet been reported. Interestingly, however, curcumin, a herbal remedy with potential anti-cancer and anti-oxidant properties, has been shown to retard epileptogenesis in a model of posttraumatic epilepsy (Jyoti et al., 2009). Although curcumin may have multiple mechanisms of action, it has recently been shown to be a potent mTOR inhibitor (Beevers et al., 2009), which could account for possible antiepileptogenic effects of this compound. As TBI represents one of the most visible and plausible targets for an antiepileptogenic approach in people, confirmation of an antiepileptogenic effect of mTOR inhibitors for posttraumatic epilepsy could have enormous clinical applications and impact.
FUTURE DIRECTIONS
The critical need to develop antiepileptogenic, rather than just seizure-suppressing, treatments has been recognized for some time, but progress in translating potential disease-modifying therapies for epilepsy into the clinical arena has been disappointingly slow. Although much basic and clinical research remains to be done in order to understand the full potential of mTOR inhibitors as antiepileptogenic therapy, there is much promise that this therapeutic strategy may actually become clinically useful for epilepsy patients.
Presently, TSC represents the most obvious application for mTOR inhibition that is closest to clinical use for epilepsy. Uncontrolled clinical trials already indicate that mTOR inhibitors are effective in slowing tumor growth in TSC (Franz et al., 2006; Bissler et al., 2008), and comparable controlled trials are in planning or under way. Although only anecdotal cases of rapamycin use for epilepsy in TSC patients have been reported to this point (Muncy et al., 2009), hopefully more complete clinical data on epilepsy will soon become available from ongoing or future clinical trials of mTOR inhibitors in TSC. Furthermore, while initial studies of epilepsy drugs logically test patients with established epilepsy, the ultimate goal will be to complete preventative, antiepileptogenic drug trials in TSC patients, which are practically much more difficult to design and conduct. Fortunately, many patients with TSC may represent appropriate candidates for an antiepileptogenic drug trial, such as those presenting with infantile spasms or pre- or minimally-symptomatic patients with a high tuber load.
In addition to clinical data on efficacy for epilepsy, there is a need for more information on the safety of mTOR inhibitors, especially for long-term use. While rapamycin is FDA approved as an immunosuppressant drug, there are both known and theoretical risks of mTOR inhibitors. Infections likely related to immunosuppression have been the main serious adverse effects reported for rapamycin (Bissler et al., 2008). Furthermore, as most of the beneficial effects of rapamycin appear to reverse upon discontinuation of the drug in both animal models and the clinical trials (Bissler et al., 2008; Zeng et al., 2008; Buckmaster et al., 2009), mTOR inhibitors may represent a life-long treatment for TSC patients. Given the numerous physiological functions of mTOR, the possible adverse effects of long-term use of rapamycin on normal growth, development, and learning, especially in children, need to be studied in more detail. The key to balancing the efficacy and safety of mTOR inhibition may lie in carefully titrating the dose to prevent excessive pathological mTOR activation but still allow normal physiological mTOR activity.
As TSC and other genetic diseases that directly affect mTOR are relatively rare, perhaps the potential impact and application of mTOR inhibitors are largest for acquired, symptomatic epilepsies due to various brain injuries. In this regard, the field is still in its infancy, with primarily animal models suggesting possible neuroprotective or antiepileptogenic effects of mTOR inhibition in response to various brain injuries. While the pathophysiological role of mTOR in TSC is relatively well understood, much remains to be done on the basic science level to establish whether mTOR is truly a critical component mediating epileptogenesis in different animal models of acquired epilepsies, such as in TBI models. Assuming mTOR plays an important role in epileptogenesis in some of these models, additional work will be needed to identify upstream and downstream mTOR-related pathways and mechanisms activated by brain injury. Furthermore, the presence of a critical therapeutic window between the initial precipitating injury and the subsequent development of epilepsy needs to be examined in animal models, as well as the minimal effective duration of treatment following the injury. Unlike TSC which may require life-long treatment, it is reasonable to expect that antiepileptogenic therapy may be effective when applied for a limited time following an acute brain injury. However, similar to TSC, the risks of inhibiting mTOR following brain injury needs to be assessed, as many mTOR-dependent processes could be compensatory and beneficial for recovery from brain injury. If the evidence from pre-clinical studies is favorable, the possibility of clinical trials of mTOR inhibitors for acquired epilepsies in people could be considered. Designing preventative or antiepileptogenic drug trials in people is again difficult, but there is a strong precedent and clinical interest for studies of patients with TBI (Temkin et al., 1990, 1999). While there will likely be a number of problems and pitfalls in the process, based on recent progress with mTOR and epilepsy, this is a promising time to pursue the development of antiepileptogenic therapies.
Acknowledgments
The author receives funding from the National Institutes of Health (K02 NS045583, R01 NS056872), the Tuberous Sclerosis Alliance, and Citizens United for Research in Epilepsy, and has no other conflicts of interest to disclose. The author has read the Journal’s position on issues involved in ethical publication and affirms that this report is consistent with those guidelines.
References
- Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res. 2004;50:545–549. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- Beevers CS, Chen L, Liu L, Luo Y, Webster NJG, Huang S. Curcumin disrupts the mammalian target of rapamycin-raptor complex. Cancer Res. 2009;69:1000–1008. doi: 10.1158/0008-5472.CAN-08-2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salibury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p79 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Castedo M, Ferri KF, Kroemer G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell Death Differ. 2002;9:99–100. doi: 10.1038/sj.cdd.4400978. [DOI] [PubMed] [Google Scholar]
- Chen S, Atkins CM, Liu CL, Alonso OF, Dietrich WD, Hu BR. Alterations in mammalian target of rapamycin signaling pathways after traumatic brain injury. J Cereb Blood Flow Metab. 2007;27:939–949. doi: 10.1038/sj.jcbfm.9600393. [DOI] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks grown-dependent activation of and signaling by the 70 kD S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12:1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- Crino PB. Do we have a cure for tuberous sclerosis complex? Epilepsy Curr. 2008;8:159–162. doi: 10.1111/j.1535-7511.2008.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Dancey JE. Therapeutic targets: mTOR and related pathways. Cancer Biol Ther. 2006;5:1065–1073. doi: 10.4161/cbt.5.9.3175. [DOI] [PubMed] [Google Scholar]
- Daniel C, Pippin J, Shankland SJ, Hugo C. The rapamycin derivative RAD inhibits mesangial cell migration through the CDK-inhibitor p27KIP1. Lab Invest. 2004;84:588–596. doi: 10.1038/labinvest.3700078. [DOI] [PubMed] [Google Scholar]
- Daoud D, Scheld HH, Speckmann EJ, Gorji A. Rapamycin: brain excitability studied in vitro. Epilepsia. 2007;48:834–836. doi: 10.1111/j.1528-1167.2006.00976.x. [DOI] [PubMed] [Google Scholar]
- Dichter MA. Models of epileptogenesis in adult animals available for antiepileptogenesis drug screening. Epilepsy Res. 2006;68:31–35. doi: 10.1016/j.eplepsyres.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, Sahin M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29:5926–5937. doi: 10.1523/JNEUROSCI.0778-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Hellier JL, Williams PA, Ferraro DJ, Staley KJ. The course of cellular alterations with the development of spontaneous seizures after status epilepticus. Prog Brain Res. 2002;135:53–65. doi: 10.1016/S0079-6123(02)35007-6. [DOI] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shiyansky C, Zhou Y, Li W, Kwaitkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signaling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- Erbayat-Altay E, Zeng LH, Xu L, Gutmann D, Wong M. The natural history and treatment of epilepsy in a murine model of tuberous sclerosis. Epilepsia. 2007;48:1470–1476. doi: 10.1111/j.1528-1167.2007.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Fingar DC, Salarma S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Stafstrom CE the Tuberous Sclerosis Study Group. Tuberous Sclerosis Complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48:617–630. doi: 10.1111/j.1528-1167.2007.01035.x. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositidide-3′ kinase’Akt’mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozwiak J, Wlodarski P. Hamartin and tuberin modulate gene transcription via β-catenin. J Neurooncol. 2006;79:229–234. doi: 10.1007/s11060-006-9134-0. [DOI] [PubMed] [Google Scholar]
- Jyoti A, Sethi P, Sharma D. Curcumin protects against electrobehavioral progression of seizures in the iron-induced experimental model of epileptogenesis. Epilepsy Behav. 2009;14:300–308. doi: 10.1016/j.yebeh.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Kawamata S, Sakaida H, Hori T, Maeda M, Uchiyama T. The upregulation of p27Kip1 by rapamycin results in G1 arrest in exponentially growing T-cell lines. Blood. 1998;91:561–569. [PubMed] [Google Scholar]
- Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, Kemp BE, Witters LA, Mimura O, Yonezawa K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signaling pathway. Genes Cell. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- Knudsen FU, Paerregaard A, Andersen R, Andresen J. Long-term outcome of prophylaxis for febrile convulsions. Arch Dis Child. 1996;74:13–18. doi: 10.1136/adc.74.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Refractory epilepsy: mechanisms and solutions. Expert Rev Neurother. 2006:397–406. doi: 10.1586/14737175.6.3.397. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ. Tuberous Sclerosis: from tubers to mTOR. Ann Hum Genet. 2003;67:87–96. doi: 10.1046/j.1469-1809.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Baker SJ. mTOR is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughinghouse C, Wang F, Phadke A, Mission J, Agarwal RK, Englot DJ, Motelow J, Nerseyan H, Waxman SG, Levin AR. Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia. 2008;49:400–409. doi: 10.1111/j.1528-1167.2007.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, Sun Y, Schmidt K, Albert MS, El-Hashemite N, Lader AS, Onda H, Zhang H, Kwiatokowski DJ, Dabora SL. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- Lenz G, Avruch J. Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem. 2005;280:38121–38124. doi: 10.1074/jbc.C500363200. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2 doi: 10.1242/dmm.002386. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. New horizons in the development of antiepileptic drugs: innovative strategies. Epilepsy Res. 2006;69:183–272. doi: 10.1016/j.eplepsyres.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki BC, Takemaru KI, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J Biol Chem. 2003;278:5947–5951. doi: 10.1074/jbc.C200473200. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee Ar, Loqsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata H, Chiang ACY, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008;32:16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Pelletier CL, Maggi LB, Jr, Scheidenhelm DK, Gutmann DH, Weber JD. Tsc1 sets the rate of ribosome export, and protein synthesis through nucleophosmin translation. Cancer Res. 2007;67:1609–1617. doi: 10.1158/0008-5472.CAN-06-2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Graham KF, Haddick PC, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314:144–148. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Ruegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadani U, Judkins A, Alkpalu A, Aronica E, Crino PB. Differential gene expression in ganglioglioma. Epilepsia. 2007;48:646–653. doi: 10.1111/j.1528-1167.2007.00925.x. [DOI] [PubMed] [Google Scholar]
- Sandsmark DK, Pelletier C, Weber JD, Gutmann DH. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol Histopathol. 2007a;22:895–903. doi: 10.14670/HH-22.895. [DOI] [PubMed] [Google Scholar]
- Sandsmark DK, Zhang H, Hegedus B, Pelletier CL, Weber JD, Gutmann DH. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res. 2007b;67:4790–4799. doi: 10.1158/0008-5472.CAN-06-4470. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Cur Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signaling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J, Schoch S, Becker AJ. Differential Pi3K-pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol. 2007;17:165–173. doi: 10.1111/j.1750-3639.2007.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414:57–60. doi: 10.1016/j.neulet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek T, Yeung RS, Hengstschlager M. Inactivation of the cyclin-dependent kinase inhibitor p27-Kip1 upon loss of the tuberous sclerosis complex gene-2. Proc Natl Acad Sci USA. 1998;95:15653–15658. doi: 10.1073/pnas.95.26.15653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparagana SP, Delgado MR, Batchelor LL, Roach ES. Seizure remission and antiepileptic drug discontinuation in children with tuberous sclerosis complex. Arch Neurol. 2003;60:1286–1989. doi: 10.1001/archneur.60.9.1286. [DOI] [PubMed] [Google Scholar]
- Sparagana S, Roach ES. Tuberous sclerosis complex. Cur Opin Neurol. 2000;13:115–119. doi: 10.1097/00019052-200004000-00001. [DOI] [PubMed] [Google Scholar]
- Stefan H, Lopes da Silva FH, Loscher W, Schmidt D, Perucca E, Brodie MJ, Boon PA, Theodore WH, Moshe SL. Epileptogenesis and rational therapeutic strategies. Acta Neurol Scand. 2006;113:139–155. doi: 10.1111/j.1600-0404.2005.00561.x. [DOI] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–565. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwaitkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and –2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515–524. doi: 10.1046/j.1528-1157.2001.28900.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Anderson GD, Wilensky AJ, Holmes MD, Cohen W, Newell DW, Nelson P, Awan A, Winn HR. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg. 1999;91:593–600. doi: 10.3171/jns.1999.91.4.0593. [DOI] [PubMed] [Google Scholar]
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nature Rev Immuno. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang CK, Qi H, Liu LF, Zheng XFS. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Disc Today. 2007;12:112–124. doi: 10.1016/j.drudis.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Wang Y, Barbaro MF, Baraban SC. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett. 2006;401:35–39. doi: 10.1016/j.neulet.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Webb DW, Fryer AE, Osborne JP. On the incidence of fits and mental retardation in tuberous sclerosis. J Med Genet. 1991;28:395–397. doi: 10.1136/jmg.28.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development involving abnormal glioneuronal proliferation. Epilepsia. 2008;49:8–21. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemke D, Azhar S, Majid A. The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug News Perspect. 2007;20:495–499. doi: 10.1358/dnp.2007.20.8.1157618. [DOI] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumor suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lan VJ, Fitzgerald E, Talos DM, Jensen FE. Seizure-induced upregulation of the mammalian target of rapamycin (mTOR) signaling pathway in the developing non-TSC rat brain. Epilepsia. 2008;49(Suppl7):325. [Google Scholar]
- Zhu D, Lipsky RH, Marini AM. Coactivation of the phosphatidylinositol-3-kinase/Akt signaling pathway by N-methyl-D-aspartate and TrkB receptors in cerebellar granule cell neurons. Amino Acids. 2002;23:11–17. doi: 10.1007/s00726-001-0103-9. [DOI] [PubMed] [Google Scholar]