

Abstract
Background
SLC19A3 (solute carrier family 19, member 3) is a thiamin transporter with 12 transmembrane domains. Homozygous or compound heterozygous mutations in SLC19A3 cause two distinct clinical phenotypes, biotin-responsive basal ganglia disease and Wernicke's-like encephalopathy. Biotin and/or thiamin are effective therapies for both diseases.
Methods
We conducted on the detailed clinical, brain MRI and molecular genetic analysis of four Japanese patients in a Japanese pedigree who presented with epileptic spasms in early infancy, severe psychomotor retardation, and characteristic brain MRI findings of progressive brain atrophy and bilateral thalami and basal ganglia lesions.
Results
Genome-wide linkage analysis revealed a disease locus at chromosome 2q35-37, which enabled identification of the causative mutation in the gene SLC19A3. A pathogenic homozygous mutation (c.958G > C, [p.E320Q]) in SLC19A3 was identified in all four patients and their parents were heterozygous for the mutation. Administration of a high dose of biotin for one year improved neither the neurological symptoms nor the brain MRI findings in one patient.
Conclusion
Our cases broaden the phenotypic spectrum of disorders associated with SLC19A3 mutations and highlight the potential benefit of biotin and/or thiamin treatments and the need to assess the clinical efficacy of these treatments.
Background
The SLC19 (solute carrier family 19) gene family (comprising SLC19A1, SLC19A2, and SLC19A3) is responsible for the uptake of water-soluble vitamins into cells. SLC19A1 was first identified as a gene that could restore reduced folate carrier activity and methotrexate sensitivity in methotrexate-resistant human breast cancer cells [1]. SLC19A2 was isolated as the gene responsible for thiamin-responsive megaloblastic anemia syndrome (TRMA; MIM 249270) [2-4] and was identified as a thiamin transporter [5]. SLC19A3 was cloned based on its homology with SLC19A1 and SLC19A2, and it was identified as a second thiamin transporter [6-8]. There are significant structural similarities among the three SLC19 proteins at the amino-acid-sequence level (> 39% identity). All three SLC19 genes encode transporter proteins composed of 12 transmembrane domains, and all are expressed ubiquitously [9]. Two apparently unrelated autosomal recessive disease phenotypes, biotin-responsive basal ganglia disease (BBGD; MIM 607483) [10,11] and Wernicke's-like encephalopathy (MIM 606152) [12], are associated with mutations of SLC19A3. We herein report on a study of four patients that presented with epileptic spasms in early infancy, severe psychomotor retardation, and characteristic brain MRI findings of progressive brain atrophy and bilateral thalami and basal ganglia lesions. We found that these phenotypes were caused by a homozygous SLC19A3 mutation and discuss the phenotype-genotype correlation of the disorders associated with SLC19A3 mutations.
Methods
Case Presentation
The experiments were conducted after approval by the institutional review boards at the Institute for Developmental Research, Aichi Human Service Center and Brain Research Institute, Niigata University. Written informed consent was obtained from the parents of the four patients participating in this study.
Patient V-2
The proband was an 18-year-old Japanese boy born to healthy consanguineous parents (Figure 1): his maternal grandmother and father are first cousins. Notably, his three cousins (Figure 1 patients V-3, V-4, and V-6) had very similar clinical courses and features. This patient was born at a gestational age of 38 weeks without complications. At 1 month of age, he presented an opisthotonic posture, and at 2.5 months of age, epileptic spasms appeared. His EEG demonstrated multifocal spikes but no hypsarrhythmia; thus, he was diagnosed with atypical infantile spasms without hypsarrhythmia. A complete blood count, biochemical analyses for a wide range of metabolic disorders and an immunohistochemical analysis of skeletal muscle biopsies were normal. Two courses of ACTH therapy were performed but were only transiently effective. At 1 year of age, intractable tonic seizures appeared, and he showed severe mental retardation and spastic quadriplegia with an increased jaw-jerk reflex and hyperreflexia in the extremities, including positive Babinski sign. Involuntary movement was not observed. Brain MRI at 4 months of age revealed symmetrical abnormal intensities in the bilateral thalami and basal ganglia on T1- and T2-weighted images (Figure 2A and 2B). Profound brain atrophy progressed at 1 year (Figure 2C, 2D, 2E and 2F). At 12 years of age, gastrostomy was performed because of swallowing difficulties. At present, he is bedridden with contractures of the extremities and has several tonic seizures daily.
Figure 1.
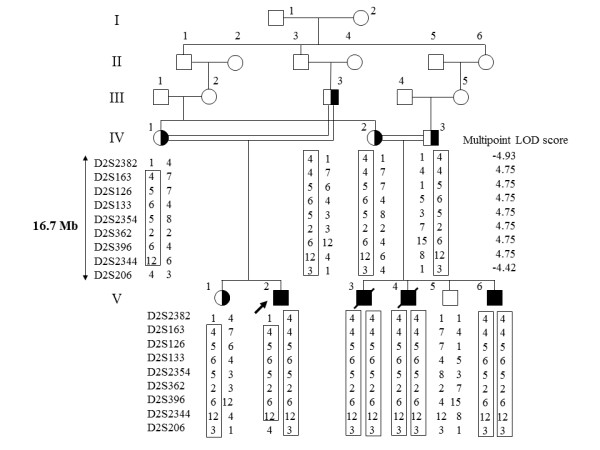
Pedigree and haplotypes in the linkage region. Squares and circles indicate males and females, respectively. The proband and affected individuals are indicated by an arrow and closed symbols, respectively. Only informative markers that define the genetic breakpoints are shown. The maximum LOD score was in the region between D2S163 to D2S2344.
Figure 2.

MRIs of patients V-2, V-3, V-4 and V-6 at various ages. Areas of abnormal intensity in the thalami and basal ganglia (arrowheads) were distinct at 4 months in V-2 (A, B). The sagittal and axial views of T1- and T2-weighted images of V-2 at 1 year showed severe brain atrophy (D, E, F). The sagittal and axial views of T1- and T2-weighted images of V-3 at 7 years, 5 months (G, H, I), V-4 at 6 years, 3 months (J, K, L), and V-6 at 3 years, 5 months (M, N, O) showed severe brain atrophy, including cerebellar atrophy (I, L, O) and abnormal intensity areas in the thalami and basal ganglia.
Patient V-3 and Patient V-4
Two other patients who are cousins of patient V-2 and born to parents who are second cousins also had quite similar clinical course and brain MRI findings (Figure 2G, H, I, J, K, L and Table 1). Patients V-3 and V-4 died of respiratory failure at 12 years and 3 months and 9 years and 7 months of age, respectively.
Table 1.
Clinical and brain MRI findings of the present patients, BBGD and Wernicke's-like encephalopathy caused by SLC19A3 mutations
| Patients | V-2 | V-3 | V-4 | V-6 | BBGD | Wernicke's-like | |
|---|---|---|---|---|---|---|---|
| Present age | 18Y | 12Y(dead) | 9Y(dead) | 6Y | 9-33Y | ND | |
| Sex | male | male | male | male | male, female | male | |
| Consanguinity | + | + | + | + | +/- | - | |
| Neurological findings | |||||||
| bad temper | 1M | 2M | 2M | 2M | ND | ND | |
| head control | - | - | - | - | + | + | |
| opisthotonic posture | 1M | - | 2M | 11M | ++ | - | |
| mental retardation | ++ | ++ | ++ | ++ | ++ (without treatment) | - | |
| dysphagia | ++ | ++ | ++ | + | + | - | |
| pyramidal signs | ++ | ++ | ++ | ++ | ++ | - | |
| quadriplegia | ++ | ++ | ++ | ++ | ++ | - | |
| dystonia | - | - | - | - | ++ | - | |
| cogwheel rigidity | - | - | - | - | ++ | - | |
| ophthalmoplegia | - | - | - | - | + | ++ | |
| nystagmus | - | - | - | - | - | ++ | |
| ataxia | - | - | - | - | - | ++ | |
| Seizure | |||||||
| onset | 2M | 3M | 2M | 11M | childhood | second decade | |
| type | ES→SGE | ES→SGE | ES→SGE | ES→PE | PE, SGE | PE | |
| Gastrostomy | 12Y | 7Y | 6Y | - | - | - | |
| Tracheotomy | - | 4Y | 4Y | - | - | - | |
| Pathologic brain MRI findings | 11M | 7Y | 6Y | 14M | 3-33Y | 36Y | |
| cerebral atrophy | ++ | ++ | ++ | ++ | - | - | |
| cerebellar atrophy | ++ | + | ++ | + | - | - | |
| brain stem atrophy | + | - | - | - | - | - | |
| Abnormal signals in | |||||||
| thalamus | + | + | + | + | - | + | |
| caudate nuclei | + | + | + | + | + | - | |
| putamen | + | + | + | + | + | - | |
| cortical & subcortical region | - | - | - | - | + | - | |
| periaqueductal region | - | - | - | - | - | + | |
| Treatment | NP | NP | NP | biotin: ineffective | biotin (and thiamin): effective | thiamin: effective | |
Y = year(s); M = month(s); ES = epileptic spasm; SGE = symptomatic generalized epilepsy; PE = partial epilepsy; (+) = presence of clinical signs, + to ++ increasing severity; (-) = minus sign; ND = not described; NP = not performed. Typical clinical and brain MRI findings of BBGD [10,16] and Wernicke's-like encephalopathy [12] are summarized.
Patinet V-6
Patient V-6 was born at 40 weeks gestation without complications. He is the fourth child and the younger brother of patients V-3 and V-4. At 11 months of age, he began experiencing epileptic spasms in clusters. His EEG demonstrated anterior dominant irregular diffuse spikes and waves, and polyspikes and waves; however, hypsarrhythmia was not observed. He was diagnosed with atypical infantile spasms. At 1 year of age, he was admitted to our hospital for treatment for epilepsy. He was able to eat weaning food; however, he did not exhibit head control and was unable to roll over. Neurological examination revealed generalized hypotonia, and deep tendon reflexes of the extremities were not elevated. ACTH therapy was transiently effective in alleviating the attacks. Brain MRI at 14 months of age demonstrated severe cortical atrophy and abnormal intensities in the bilateral thalami and basal ganglia. Physical examination at 2 years of age demonstrated severe psychomotor retardation; he was not able to sit without support and was also unable to roll over. Neurological examination demonstrated generalized hypotonia with spasticity of limbs and severe mental retardation. Involuntary movement was not observed. Because he was identified as homozygous for a SLC19A3 mutation, patient V-6 had taken biotin (5 mg/kg/day) for one year beginning at 19 months of age with informed consent from his parents; however, his neurological symptoms and brain MRI findings did not improve. Brain MRI at 3 years and 5 months of age demonstrated diffuse brain atrophy and abnormal intensities in the bilateral thalami, caudate nuclei and putamen on T1- and T2-weighted images (Figure 2M, 2N and 2O). Currently, Patient V-6, at 6 years of age, is bedridden. The clinical features and brain MRI findings of the four patients are summarized in Table 1.
Genome-Wide Linkage Analysis
We performed a genome-wide linkage analysis in ten family members related to the four neurological patients using 763 microsatellite markers (Applied Biosystems, Foster City, CA) to cover the all autosomes with an average interval of 4.6 cM. Multi-point LOD (logarithm of odds) scores were calculated using the Allegro program for all loci under the assumption of autosomal recessive inheritance and a disease frequency of 0.001 [13]. Haplotypes were constructed manually to minimize the number of recombination events.
Genetic Analyses
Genomic DNA was isolated from white blood cells by phenol/chloroform extraction. PCR-amplified DNA fragments were isolated with a QIAEX II Gel Extraction Kit (QIAGEN, Valencia, CA) and purified using polyethylene glycol (PEG 6000) precipitation. Isolated PCR products were cloned and sequenced with an SQ5500E DNA sequencer (HITACHI, Tokyo, Japan), or were sequenced directly with specific inner primers or the same primers used for PCR [14].
Construction of Expression Vectors
First-strand cDNA was synthesized by reverse transcription of 10 μg of total RNA from normal or the proband's lymphoblastoid cells using a First-Strand cDNA Synthesis Kit (GE Healthcare, Tokyo, Japan) with specific antisense primer (A1: 5'-GTTGCGTCTAGATTAGAGTTTTGTTGAC-3', XbaI site is underlined) in a reaction volume of 15 μl. The cDNA products were amplified with LA-Taq DNA Polymerase (TAKARA BIO INC, Otsu, Japan) using the specific primers (S1: 5'-AACAGACACTCCCTTCTGAATTCATG-3' and A1, EcoRI site is underlined). PCR was performed in a total volume of 20 μl for 36 cycles as follows: denaturation at 94°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 90 s. The 1,535-bp PCR products were subcloned into the EcoRI/XbaI sites of the pCI-neo vector, a mammalian expression vector driven by a cytomegalovirus (CMV) promoter and enhancer, to generate pCI-neo-SLC19A3(WT) and pCI-neo-SLC19A3(E320Q).
Thiamin Transport Assay
We performed a thiamin transport assay according to the protocol of Ashokkumar et al. [15]. Briefly, a 1.0 μg aliquot of the SLC19A3 expression vector (pCI-neo-SLC19A3(WT), pCI-neo-SLC19A3(E320Q)) and the control vector (pCI-neo) were transfected into HEK293 cells in 24-well plates using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA). Two days after transfection, the HEK293 cells were washed two times with pre-warmed phosphate-buffered saline (PBS) and incubated for 10 min at 37°C in 200 μl of assay buffer (20 mM HEPES (pH 7.4), 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM sucrose) containing 0.1 μM of [3H]-labeled thiamin (14.8 kBq:0.74TBq/mmol) with or without 0.1 mM unlabeled thiamin. Thiamin transport was terminated by the addition of 0.5 ml of ice-cold PBS followed by immediate aspiration. Cells were rinsed two times with 0.5 ml of ice-cold PBS and lysed with 200 μl of 0.2 M NaOH for 30 min at 65°C. A 50-μl aliquot of lysate was spotted onto Whatman 3 MM chromatography paper (GE Healthcare), and radioactivity counts were conducted using a liquid scintillation counter. Protein concentrations were estimated using an Advanced Protein Assay Kit (Cytoskeleton Inc. Denver, CO) according to the manufacturer's instructions.
Results
The Disease Maps to Chromosome 2q35-37
The patients' characteristic clinical features without obvious extrapyramidal symptoms, rapid clinical course and brain MRI findings did not lead us to diagnosis a specific disease. However, we conducted a genome-wide linkage analysis that revealed a disease locus in the region from D2S2382 to D2S206 on 2q35-37 (~16.7 Mb) with a maximum multipoint LOD score of 4.75 (Figure 1).
Identification of Homozygous SLC19A3 Mutation
SLC19A3 gene maps in the region, and then we identified a homozygous mutation (c.958G > C, [p.E320Q]) in exon 3 of SLC19A3 in all patients, while their parents were heterozygous for the mutation (Figure 3A and 3B).
Figure 3.
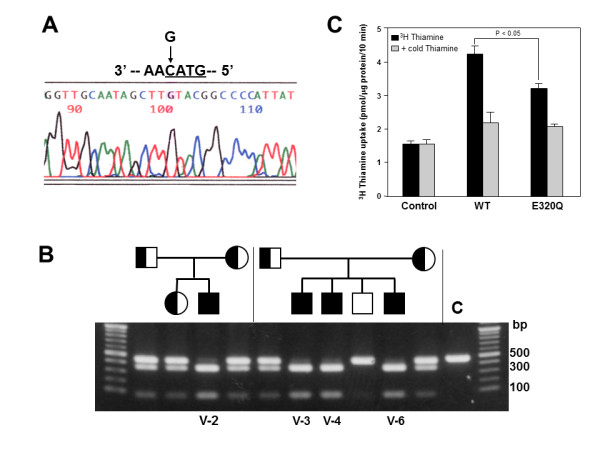
Identification and characterization of a disease mutation (c.958G > C, [p.E320Q]). (A) The sequence analyses showed a G to C substitution at nucleotide position 958 in Exon 3 of SLC19A3, resulting in a substitution of glutamic acid at codon 320 for glutamine (c.958G > C, [p.E320Q]), indicated by an arrow. (B) RsaI (GTAC)-digested PCR products were run through a 1.2% agarose gel. Digestion of products containing the E320Q mutation resulted in 357- and 86-bp fragments. All patients (V-2, V-3, V-4 and V-6) were homozygous for the mutation, and their parents were heterozygous for the mutation. (C) [3H]-thiamin uptake by HEK293 cells expressing wild-type or E320Q SLC19A3. Two days after transfection, cells were incubated with 0.1 μM of [3H]-labeled thiamin (14.8 kBq:0.74TBq/mmol) for 10 min with or without 0.1 mM unlabeled thiamin. Incorporated [3H]-thiamin was measured using a liquid scintillation counter. Results are expressed as mean ± SE of specific uptake values from three independent experiments.
Determination of thiamin transport activity of mutant SLC19A3 Proteins in HEK293 cells
To address the effect of the E320Q mutation on thiamin transport activity, we transfected wild-type and mutant SLC19A3 expression constructs into HEK293 cells. We then analyzed the uptake of [3H]-labeled thiamin into these cells. We found that the E320Q mutation decreased thiamin uptake to 63% of that of the wild-type protein (Figure 3C), corroborating the findings of Kono et al. [12] using Chinese hamster ovary (CHO) cells.
Discussion
In the present study, we demonstrated that a homozygous mutation (E320Q) in SLC19A3 is associated with epileptic spasms in early infancy, severe psychomotor retardation and characteristic MRI findings, including progressive brain atrophy and bilateral thalami and basal ganglia lesions. It should be noted that four patients in this family showed quite similar clinical presentations, strongly arguing for the distinct genotype-phenotype correlation associated with the homozygous E320Q mutation. Two apparently unrelated disease phenotypes, BBGD (MIM 607483) [10,11] and Wernicke's-like encephalopathy (MIM 606152) [12], have been shown to be associated with mutations of SLC19A3. Patients of BBGD with homozygous mutations (G23V, T422A) in SLC19A3 showed childhood-onset encephalopathy, characterized by epilepsy, confusion, external ophthalmoplegia, dysarthria, dysphagia, dystonia, rigidity and quadriparesis. In these patients, the administration of high doses of biotin (5-10 mg/kg/day) early in the progression of the disorder eliminated their symptoms, whereas thiamin was ineffective [10,11]. Recently, compound heterozygous mutations (E320Q, K44E) in SLC19A3 were shown to cause Wernicke's-like encephalopathy, which is characterized by acute onset of epilepsy, ataxia, nystagmus and ophthalmoplegia in the second decade of life, and symptoms are effectively alleviated with thiamin treatment [12].
Despite sharing one mutant allele (E320Q) with the known causative mutations in Wernicke's-like encephalopathy, our patients clearly differed in age at onset, brain MRI findings and symptoms, raising the possibility that the functions altered by the mutant SLC19A3 with K44E are substantially different (Table 1). Although both the BBGD phenotype and our patients showed similar bilateral basal ganglia lesions in the brain and encephalopathy that are fatal unless treated, the clinical presentations of our patients are distinct from the previously reported BBGD phenotype in the following ways: (1) epileptic spasms appeared in infancy in all of our patients, in contrast to the epilepsy onset in childhood for patients with BBGD; (2) MRI findings in our patients are characterized by progressive brain atrophy and additional lesions in the bilateral thalami that are not observed in patients with BBGD; (3) our patients did not display obvious dystonia and cogwheel rigidity reflecting dysfunctions in basal ganglia; (4) administration of a high dose of biotin for one year improved neither the neurological symptoms nor the brain MRI findings in Patient V-6, though it is unclear whether administration of biotin at early stages of the disorder (e.g., when epileptic spasms first appeared) improved the subsequent clinical trajectory (Table 1). Thus our cases broaden the phenotypic spectrum of disorders associated with mutations in SLC19A3.
Recently, two novel mutations, both of which created premature stop codons, were identified in BBGD patients [16]. Transfection studies demonstrated that G23V and T422A mutations identified in the BBGD phenotype lead to mutant SLC19A3 that is nonfunctional for thiamin uptake activity [8]. These findings suggest that loss-of-function mutations in SLC19A3 are associated with the BBGD phenotype. In contrast to these loss-of-function mutations, we demonstrated that mutant SLC19A3 with E320Q possesses some thiamin uptake activity that is approximately 60% of wild-type SLC19A3 in CHO cells [12] and HEK293 cells (Figure 3C). These findings alone seem difficult to account for severe clinical presentations in our cases. Interestingly, a previous report showed that the negative charge at position E320 was conserved in other transporters (SLC19A1, SLC19A2) and was crucial for the formation of a salt bridge with a conserved, positively-charged residue, K380 [8]. The homozygous mutation of E320 may, therefore, destabilize the protein conformation and bring with more deleterious effect, including gain-of-toxic function and/or the dramatically changed protein structure.
In this study, the efficacy of high doses of biotin and/or thiamin for the clinical phenotypes of our patients has not been determined. This indicates that we have not determined whether our patients are phenotypic variation of "biotin-responsive" basal ganglia disease or "biotin-unresponsive" another type of a disease. Therefore, accumulation of genetic analysis and clinical courses from more patients who undergo biotin and/or thiamin treatment as well as further laboratory studies employing cellular or mouse models will be needed to better characterize the clinical phenotypes and biochemical characteristics of the homozygous E320Q mutation in SLC19A3.
Conclusions
The present study indicated that four Japanese patients in a Japanese pedigree who presented with epileptic spasms in early infancy, severe psychomotor retardation, and characteristic brain MRI findings of progressive brain atrophy and bilateral thalami and basal ganglia lesions were caused by homozygous mutation (c.958G > C, [p.E320Q]) in SLC19A3. Our cases broaden the phenotypic spectrum of disorders associated with SLC19A3 mutations and highlight the potential benefit of biotin and/or thiamin treatments and the need to assess the clinical efficacy of these treatments.
Abbreviations
MRI: Magnetic Resonance Imaging; EEG: Electroencephalogram; ACTH: Adrenocorticotropic hormone
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KY participated in planning and design of the study, performed gene sequencing analysis and interpretation of the data and drafted and finalized the manuscript. KM, MS, TK, NI examined the clinical features of each patients and contributed to acquisition of materials, interpretation of data and drafted the manuscript. KH and RK performed genome-wide linkage analysis and contributed to interpretation of the data and drafted the manuscript. KN performed transient expression study of wild-type and mutant SLC19A3 proteins and contributed to interpretation of data and drafted the manuscript. YY performed gene sequencing analysis and interpretation of the data and drafted the manuscript. ST conducted genome-wide linkage analysis and contributed to interpretation of data, drafted and finalized the manuscript. NW organized this study, performed interpretation of the data and drafted and finalized the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Kenichiro Yamada, Email: ktyamada@inst-hsc.jp.
Kiyokuni Miura, Email: kiyokuni@s2.dion.ne.jp.
Kenju Hara, Email: kenju2007@yahoo.co.jp.
Motomasa Suzuki, Email: moto-su@tb4.so-net.ne.jp.
Keiko Nakanishi, Email: nakanishi@inst-hsc.jp.
Toshiyuki Kumagai, Email: ay4t-kmgi@asahi-net.or.jp.
Naoko Ishihara, Email: naokois@med.nagoya-u.ac.jp.
Yasukazu Yamada, Email: yasyam@inst-hsc.jp.
Ryozo Kuwano, Email: ryosun@bri.niigata-u.ac.jp.
Shoji Tsuji, Email: tsuji@m.u-tokyo.ac.jp.
Nobuaki Wakamatsu, Email: nwaka@inst-hsc.jp.
Acknowledgements
We are grateful to the patients who participated in this study and to their families. We appreciate useful discussions with Dr. J. Natsume (Department of Pediatrics, Nagoya University) about the patients. This work was supported by the Uehara Memorial Foundation (to N.W.) and The Ministry of Education, Culture, Sports, Science and Technology of Japan (to K.Y. and N.W.).
References
- Dixon KH, Lampher BC, Chiu J, Kelley K, Cowan KH. A novel cDNA restores reduced folate carrier activity and methotrexate sensitivity to transport deficient cells. J Biol Chem. 1994;269:17–20. [PubMed] [Google Scholar]
- Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, Szargel R, McDonald L, Shalata A, Nosaka K, Gregory S, Cohen N. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22:300–304. doi: 10.1038/10372. [DOI] [PubMed] [Google Scholar]
- Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. The gene mutated in thiamine-responsive anaemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat Genet. 1999;22:305–308. doi: 10.1038/10379. [DOI] [PubMed] [Google Scholar]
- Diaz GA, Banikazemi M, Oishi K, Desnick RJ, Gelb BD. Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anaemia syndrome. Nat Genet. 1999;22:309–312. doi: 10.1038/10385. [DOI] [PubMed] [Google Scholar]
- Dutta B, Huang W, Molero M, Kekuda R, Leibach FH, Devoe LD, Ganapathy V, Prasad PD. Cloning of the human thiamine transporter, a member of the folate transporter family. J Biol Chem. 1999;274:31925–31929. doi: 10.1074/jbc.274.45.31925. [DOI] [PubMed] [Google Scholar]
- Eudy JD, Spiegelstein O, Barber RC, Wlodarczyk BJ, Talbot J, Finnell RH. Identification and characterization of the human and mouse SLC19A3 gene: a novel member of the reduced folate family of micronutrient transporter genes. Mol Genet Metab. 2000;71:581–590. doi: 10.1006/mgme.2000.3112. [DOI] [PubMed] [Google Scholar]
- Rajgopal A, Edmondnson A, Goldman ID, Zhao R. SLC19A3 encodes a second thiamine transporter ThTr2. Biochim Biophys Acta. 2001;1537:175–178. doi: 10.1016/s0925-4439(01)00073-4. [DOI] [PubMed] [Google Scholar]
- Subramanian VS, Marchant JS, Said HM. Biotin-responsive basal ganglia disease-linked mutations inhibit thiamine transport via hTHTR2: biotin is not a substrate for hTHTR2. Am J Physiol Renal Physiol. 2006;291(5):C851–C859. doi: 10.1152/ajpcell.00105.2006. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Smith SB, Prasad PD. SLC19: the folate/thiamine transporter family. Pflugers Arch. 2004;447:641–646. doi: 10.1007/s00424-003-1068-1. [DOI] [PubMed] [Google Scholar]
- Ozand PT, Gascon GG, Al Essa M, Joshi S, Jishi E, Bakheet S, Al Watban J, Al-Kawi MZ, Dabbagh O. Biotin-responsive basal ganglia disease: a novel entity. Brain. 1998;121:1267–1279. doi: 10.1093/brain/121.7.1267. [DOI] [PubMed] [Google Scholar]
- Zeng WQ, Al-Yamani E, Acierno JS Jr, Slaugenhaupt S, Gillis T, MacDonald ME, Ozand PT, Gusella JF. Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet. 2005;77:16–26. doi: 10.1086/431216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono S, Miyajima H, Yoshida K, Togawa A, Shirakawa K, Suzuki H. Mutations in a thiamine-transporter gene and Wernicke's-like encephalopathy. N Engl J Med. 2009;360:1792–1794. doi: 10.1056/NEJMc0809100. [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Goto H, Suzumori K, Adachi R, Ogasawara N. Molecular analysis of five independent Japanese mutant genes responsible for hypoxanthine guanine phosphoribosyltransferase (HPRT) deficiency. Hum Genet. 1992;90:379–384. doi: 10.1007/BF00220463. [DOI] [PubMed] [Google Scholar]
- Ashokkumar B, Vaziri ND, Said HM. Thiamin uptake by the human-derived renal epithelial (HEK-293) cells: cellular and molecular mechanisms. Am J Physiol Renal Physiol. 2006;291(4):F796–F805. doi: 10.1152/ajprenal.00078.2006. [DOI] [PubMed] [Google Scholar]
- Debs R, Depienne C, Rastetter A, Bellanger A, Degos B, Galanaud D, Keren B, Lyon-Caen O, Brice A, Sedel F. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol. 2010;67:126–30. doi: 10.1001/archneurol.2009.293. [DOI] [PubMed] [Google Scholar]