

Abstract
Identifying molecular mechanisms or therapeutic targets is typically based on large-scale cellular analysis that measures the abundance of mRNA or protein; however, abundance does not necessarily correlate with activity. We report a method for direct large-scale quantification of active pathways that employs a cellular array with parallel gene delivery of constructs that report pathway activity. The reporter constructs encode luciferase, whose expression is influenced by binding of transcription factors (TFs), which are the downstream targets of signaling pathways. Luciferase levels are quantified by bioluminescence imaging (BLI), which allows for rapid, non-invasive measurements. Activity profiles by BLI of 32 TFs were robust, consistent, and reproducible, and correlated with standard cell lysis techniques. The array identified five TFs with differential activity during phorbol-12-myristate-13-acetate (PMA)-induced differentiation of breast cancer cells. A system for rapid, large-scale, BLI quantification of pathway activity provides an enabling technology for mechanistic studies of cellular responses and processes.
Keywords: cellular array, transcription factor activity, bioluminescence imaging
Introduction
Activities of signaling pathways within the intracellular network determine cellular response and signaling pathways with aberrant activity underlying many diseases (Barnes, 2006; Latchman, 1996; Scherzer et al., 2008; Strano et al., 2007). Methods to identify active pathways within cells could be used to molecularly dissect the basis for normal and diseased states and disease progression. Numerous signal transduction pathways interact to form a complex network, whose net output determines the cellular response. The outputs of the network or termination points of signaling pathways are transcription factors (TFs), which directly modulate gene expression and are powerful regulators of cell function (Nebert, 2002; Orphanides and Reinberg, 2002). The potential of TFs to regulate cellular responses was recently evidenced by the induction of pluripotent stem cells from adult fibroblasts through the expression of four TFs (Takahashi and Yamanaka, 2006). TF activity is defined by its ability to activate or suppress transcription as a result of sequence-specific binding to regions that directly influence the expression of genes. The activities of TFs are commonly measured using TF reporter vectors, which contain a TF-specific binding enhancer element upstream of a promoter that drives the expression of a reporter protein such as firefly luciferase. Measuring the activity of a TF through the reporter protein thus reflects the net activity of the upstream signaling pathway.
Analysis of the complex signaling network requires large-scale cell-based assays to capture the numerous signals within the cell that determine the response. To measure the TF activity, a cell-based assay is essential to maintain the physiological context of the components of the cellular pathways and the TF, thus accounting for any post-translational modifications and changes in cellular localization. Post-translational processes such as ubiquitination, phosphorylation, acetylation, and methylation can influence the net activity of a signaling pathway and subsequent activation of a TF (Zhang and Reinberg, 2001). TF activity also requires nuclear localization, as many TFs are found in the cytoplasm and translocate upon activation. This regulation of TF activity by post-translational modification or cellular compartmentalization cannot be directly assessed by techniques that quantify the copy number of mRNA or proteins, such as microarray or proteomic techniques (Erfle et al., 2007). In contrast, TF reporter vectors directly measure binding and transcriptional activity, which results in the production of a reporter protein that can be easily measured. Normalized for the extent of transfection, these reporter constructs have the ability to provide a quantitative measure of TF activity. However, these constructs are typically applied on a focused number of pathways, which provides only a limited view of the active signals within the cell (Paech et al., 1997).
In this report, we describe a system that employs cell-based arrays for large-scale, rapid, non-invasive, and quantitative analysis of TF activity. A two-plasmid system was employed, with each position within the cell array receiving the same constitutively active normalization plasmid encoding for Renilla luciferase and a distinct TF reporter vector driving the expression of firefly luciferase. Luciferase levels were quantified by bioluminescence imaging (BLI), which provides a high-throughput platform with rapid data acquisition. BLI has accurately reported TF activity (Pannier et al., 2007), and this report employs this technique to quantify the activity of multiple TFs in parallel. By eliminating the need to lyse cells, BLI provided a non-invasive method of measuring luciferase levels while preserving the viability of the cell population and removes a labor-intensive step present in most traditional TF assays. Initial studies validated the measurement of multiple TFs with BLI through correlation to values based on standard techniques using cell lysates. Subsequent experiments measured the activity for 32 TFs and investigated the array-to-array and day-to-day consistency of the system. Finally, the system was applied to capture the differential TF activity profile during cell differentiation. The cellular array developed in this report represents a robust and reliable method for the large-scale measurement of TF activity that may ultimately be employed to investigate mechanisms of disease progression, or identify targets for therapeutic intervention.
Materials and Methods
Cells and Plasmids
The ER-positive MCF-7:WS8 mammary carcinoma cells, clonally derived from MCF-7 cells, were used for the study (Jiang et al., 1992). Cells were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 100 μM non-essential amino acids, 100U antibiotics/antimycotic, 1 mM l-glutamine, and 6 ng/mL insulin and maintained at 37°C in a humidified 5% CO2 atmosphere (Pannier et al., 2007). All media and media components were purchased from Invitrogen (Carlsbad, CA). Plasmids were purified from bacteria cultures using Qiagen (Valencia, CA) reagents and stored at −20°C in Tris–EDTA buffer. TF-specific reporter plasmids were obtained from Panomics (Fremont, CA). The reporter plasmids contain multiple copies of a cis-acting enhancer element containing TF-specific binding sites that are located upstream of a minimal TATA box promoter (TA) driving the expression of firefly luciferase. A second plasmid used for normalization contains a minimal TA promoter driving the expression of Renilla luciferase (Ariazi et al., 2007).
Transfection in Plates
DNA complexes were formed following manufacturers instructions using Lipofectamine LTX (Invitrogen) with a DNA/lipid ratio of 1:2 in serum-free Opti-MEM media. After 20 min, lipoplexes in volumes of 100 or 20 μL were added to 96- and 384-well plates, respectively, to create a DNA concentration of 0.75 μg/well in a 96-well plate, and 0.075 μg/ well in a 384-well plate. The plates were incubated at 37°C for 2 h to allow for the immobilization of complexes on the surface and facilitate a hybrid of surface and bolus DNA delivery. During incubation, a cell suspension of MCF-7:WS8 cells was prepared in 2× supplemented media (RPMI-1640 with 20% FBS, 200 μM non-essential amino acids, 200 U antibiotics/antimycotic, 2 mM l-glutamine, and 12 ng/mL insulin). The cell solution was added to the wells (for a total of 9 × 103 cells/well in the 384-well plate and 3.5 × 104 cells/well in the 96-well plate), and the plates were incubated for 24 h. Transfection efficiency in 384- and 96-well plates was not significantly different (Supplementary Fig. 1).
Quantification of TF Activity by Lysis
Dual luciferase assays on cell lysates were performed to determine TF activity, which later served as the reference to validate the BLI. Complexes containing TF-specific firefly luciferase reporter plasmids and the Renilla luciferase normalization plasmid in a 3:1 ratio were formed and deposited into a 96-well plate as described in the previous section. Twenty-four hours after transfection, cells were harvested, lysed, and assayed for firefly and Renilla luciferase activity using the Dual-Luciferase Reporter assay system (Promega, Madison, WI). The dual-luciferase assays were carried out using BioTek® Synergy™ two multi-mode microplate reader. Each TF activity experiment contained a TA vector control that acted as a negative control. The TA vector control is a plasmid encoding for firefly luciferase driven by a minimal TA promoter and lacking a TF-specific enhancer element along with the Renilla normalization plasmid. The lysed cell relative ratio was calculated by dividing the firefly luciferase luminescence signal by the Renilla signal followed by normalization to the ratio of firefly to Renilla luciferase of the TA vector control.
Quantification of TF Activity by BLI
TF binding activity was also measured using live cell BLI within 384-well plates. TF activity arrays were formed as previously described using TF-specific firefly luciferase reporter plasmids and the Renilla luciferase normalization plasmid in a 3:1 ratio within 384-well plates. Arrays were analyzed for TF activity by imaging at 24 h after transfection. Prior to imaging, the plates and all reagents were warmed to 37°C. To image the Renilla activity, the substrate ViviRen (Promega) was first diluted to 100 μM in serum containing media and then added to each well to achieve a final concentration of 20 μM. The light flux was captured at 5 min after substrate addition with an exposure time of 1 min. Immediately following Renilla signal image acquisition, the firefly luciferase substrate d-luciferin (Molecular Therapeutics, Inc., Ann Arbor, MI, 5 mM in PBS) was added to each well to achieve a final concentration of 1 mM d-luciferin per well. The plate was then imaged again 6 min after d-luciferin addition for a total exposure time of 1 min. Light emission from the array was quantified using an IVIS imaging system (Xenogen Corp., Alameda, CA). The activity from BLI is referred to as the normalized TF activity, was calculated by dividing the firefly luciferase luminescence signal by the Renilla signal, and normalized to the ratio of firefly luciferase to Renilla luciferase of the TA vector control. Prior to quantitative analysis of BLI results, we validated that there was negligible crossing of the luminescence signal between wells. Empty wells positioned next to wells with luminescence that exceeded the maximal values for our system had 98.0 ± 0.5% lower photon emissions and were not statistically different from the Renilla substrate background.
PMA Stimulation
Phorbol-12-myristate-13-acetate (PMA) was used to stimulate multiple signal transduction pathways. The MCF-7:WS8 cells used in the induction experiments were cultured in estrogen-free media (substituting phenol red-free RPMI-1740 and dextran-coated charcoal-stripped FBS in the media) for 3 days prior to the experiment. Live cell TF activity arrays were formed as previously described. Eighteen hours after cell seeding, the culture media were replaced with serum-free media containing 20 ng/mL PMA or serum-free media containing the equivalent volume of vehicle control, 100% ethanol. Arrays were imaged as previously described 6 h after the media change. For analysis, the results of two arrays from each culture condition were pooled giving n equal to 8 for each TF.
Statistical Analysis
We have used two different methods to assess differences between conditions, across arrays, and across experimental days: an empirical hierarchical Bayesian method (EHBM) and a Monte Carlo bootstrap method (MCBM; Lonnstedt and Speed, 2002; Smyth, 2004).
Empirical Hierarchical Bayesian Method
The Renilla and the firefly photon flux for each condition were compared to substrate background using a one-tailed two-sample t-test (n = 4) to determine their significance level relative to background. Reporters that were significantly above Renilla and firefly photon flux compared to the wells containing no DNA were considered to be above the background level at a significant level, α = 0.05. TF activity (ratio between firefly and the Renilla photon flux) was normalized to the corresponding activity of the TA vector control. Normalized TF activity relative to the control was transformed using a variance stabilization normalization (VSN) procedure (Bolstad et al., 2003) to assure the independence of the mean and the variance of the measurement.
We employed an empirical hierarchical model developed by Lonnstedt and Speed (2002) with empirical Bayesian hyperparameters that are calculated using Smyth’s method (2004). Normality of the individual TF was confirmed by Shapiro’s test and the homogeneity of variance by the Bartlett and Flinger methods independently. EBHM was employed to (a) assess that the normalized TF activity was above the TA vector control activity, (b) determine the differences between arrays, and (c) compare the effect of the treatment in the TF activities of the cell. The EBHM has two main assumptions: (a) measurements of activity for individual TFs are normal or approximately normal and (b) the variance and the mean of each sample are independent. Analysis of the normalized TF activity was analyzed to determine if the mean and variance of all experiments were highly correlated (Supplementary Fig. 2). The independence of the mean and the variance is assured by the VSN transformation. The mean and variance of all the experiments are highly correlated. The normality of each individual TF for all the arrays is determined using a Shapiro test, after the corresponding transformation. All the TFs are normal, with a few exceptions that may be due to outliers as the non-normality of the identified TFs do not occur in all the experiments and conditions. The data of all the experiments have homogeneity of variance according to the Bartlet and Flinger (data not shown). Therefore, EBHM might be employed to analyze the results from the array experiments.
Array reproducibility was established by array correlations and by an EBHM. Array correlations were performed using linear regression between all the normalized TF activities relative to the control. An EBHM was used to compare the individual normalized TF activity relative to the control between pairs of arrays. All the comparisons of the individual moderated t-tests are summarized by a moderated F-test. The final P-values are corrected by the false discovery rate (FDR) procedure (Benjamini et al., 2001) to reduce the number of false positives. Results from the same condition performed in multiple experiments were combined using a modified meta-analysis procedure (Zaykin et al., 2002). Note that only the repeats with the same directionality are group together to avoid false positives. A more conservative method (Pyne et al., 2006) based on a corrected significant level, α′, is also presented to further reduce the presence of false positives. α′ is defined as α′ = α2/4L2,where α is the original significant level and L is the number of experiments to be combined.
Monte Carlo Bootstrap
As a complement to the above-mentioned analysis, a more rigorous resampling-based inference method was also used to analyze the array. Unlike parametric tests, MCBM is not sensitive to assumptions of normality (Efron and Tibshirani, 1993). MCBM was used for comparison of the live cell relative ratio for each TF condition to the TA vector control, across arrays and across experiments (Do and Hall, 1991; Hall and Wilson, 1991). For multiple comparisons across arrays within an experiment, the average mean difference of the normalized TF activity was calculated and used as the test statistic to quantify differences between arrays. The significance of the statistic was assessed using the bootstrap method where the distribution of the statistic under the null hypothesis was estimated by 10,000 cycles of Monte Carlo simulations of sampling with replacement of the pooled values for the live cell relative ratio from all arrays, and calculating values for the average delta mean. Significance for the array-to-array and day-to-day comparison was assessed by counting the number of P-values for the individual conditions that were less than α and using the binomial distribution to calculate how likely this count is under the null hypothesis. To account for multiple comparisons, the FDR approach was used to modify α and ensure the probability of false positives to be <5% (Benjamini and Hochberg, 1995; Saville, 1990).
Results
Analysis of TF Activity Assay
Lysed cell assays, the standard approach to luciferase assays, were initially performed to quantify the activities of six TFs through delivery of TF reporter plasmids. Lipoplexes were deposited in 96-well plates to transfect MCF7:WS8 cells and report on the activities of AP1, ER, NFκB, SP1, E2F1, and GATA3 in conjunction with the Renilla luciferase normalization plasmid. Relative to the TA vector control, which lacks the specific TF binding site, the lysed cell relative ratio of AP1, ER, NFκB, SP1, and E2F1 was all significantly increased (Fig. 1). The dual plasmid system normalized for transfection variability and produced quantitative measurements of the TF activity for the six TFs. The TF activity profile provided a reference for the analysis of TF activity using BLI.
Figure 1.
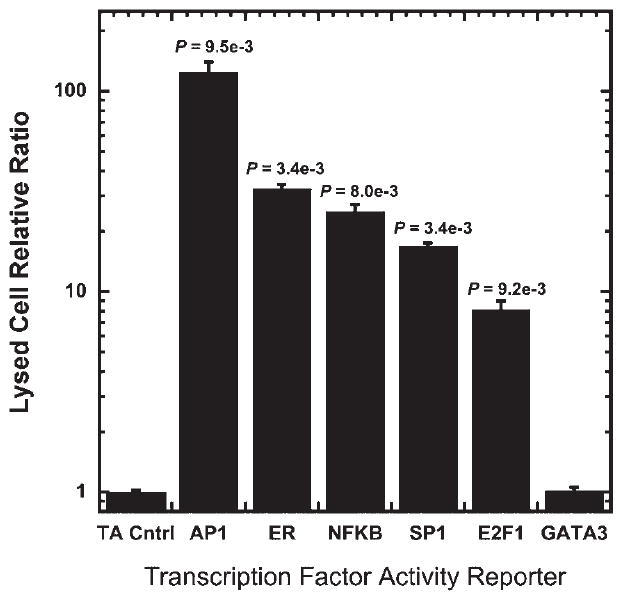
Dual-luciferase reporter assay to measure TF activity. MCF-7:WS8 cells were transfected in parallel with reporter constructs containing enhancer elements for specific TFs (AP1, ER, NFκB, SP1, E2F1, and GATA3) and TA vector control in combination with a Renilla luciferase reporter to normalize for transfection. Cells were lysed 24 h after transfection and TF activitywas quantified using a dual-luciferase assay. Values are means ± SD of three independent experiments done in triplicate.
BLI following dual plasmid delivery was subsequently investigated for rapid, non-invasive data acquisition. This imaging approach minimizes post-transfection processing, which reduces variability and facilitates the use of smaller well formats that can reduce reagent usage by one-tenth. Cell arrays were produced using the same six TF reporter plasmids employed for quantification by cell lysis. The transfected cell arrays were imaged and values for photon flux were determined for Renilla (Fig. 2a and b) and firefly luciferase (Fig. 2c and d). All wells had Renilla luciferase expression significantly above the substrate background of a control without DNA, indicating substantial transfection within each well. The raw output for the photon emission for firefly luciferase (Fig. 2d) was divided by the photon emission from Renilla luciferase (Fig. 2b) to normalize light emission. This normalized light emission for each TF was divided by the light emission from the TA vector control condition to produce the normalized TF activity (Fig. 2e). Relative to the TA control, the normalized activity was significantly increased for AP1, ER, NFκB, SP1, and E2F1. The normalized TF activities measured by the lysed and live cell assays identified the same TFs as being significantly active and were highly correlated (Fig. 2f, R2 = 0.999). BLI was thus used for all subsequent arrays.
Figure 2.
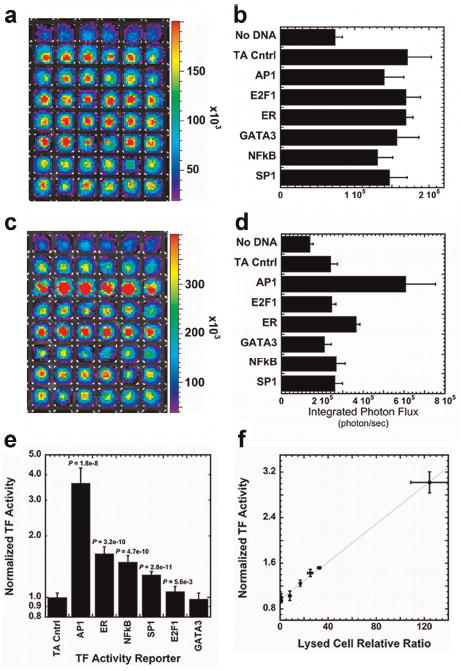
Dual-luciferase reporter constructs to measure TF activity. Dual-luciferase levels were analyzed using bioluminescence imaging 24 h after transfection by first imaging for Renilla luciferase (a and b) and then imaging each array for firefly luciferase (c and d). a and c: Pseudo-color mapping of photon flux localized within wells of the array were quantified to determine average photon fluxes for each well (b and d). e: The raw output for the firefly photon flux of each well was normalized by the photon emission from Renilla luciferase to produce the normalized light emission. f: The TF activity profile for MCF7:WS8 cells using lysed and live cell assays were directly compared using linear regression. The TF activity results were highly correlated (R2 = 0.999). [Color figure can be seen in the online version of this article, available at wileyonlinelibrary.com.]
Large-Scale TF Activity Analysis
The array was subsequently expanded to quantify the activity for a larger library of plasmids and the array was evaluated for repeatability and robustness. Arrays with 32 TF activity reporter plasmids (Supplementary Table I) and 2 internal controls were formed within a 384-well plate with four replicates of each condition. A total of six arrays were formed and imaged in two independent experiments, and the resulting TF activity profiles were analyzed to determine the TFs with activity above the TA vector control, the consistency between arrays, and the consistency between days. To reduce false negatives resulting from poor transfection, wells that do not have a photon flux above the substrate background, either Renilla or firefly luciferase, were excluded from analysis. The Renilla and firefly luciferase light emissions of the reporter genes were significantly above the substrate background for 98% of wells (Fig. 3a and b) indicating consistently sufficient levels of transfection. Analysis of the multiple arrays formed on different days indicated that the TF activity arrays had high reproducibility, with correlation coefficients ≥0.95 (Fig. 3c). A Bayesian analysis and an MCBM similarly indicated no significant difference between arrays within an experiment or between experiments (Supplementary Tables II and III), further confirming reproducibility of results and demonstrating the independence of the method of analysis. Active TFs within the cell were identified by comparing the normalized TF activity and the normalized TA vector control. At a highly conservative alpha (α′ = 0.00015), the Bayesian method identified seven TFs (AP1, CRE, ER, HIF-1, NFκB, p53, and SP1) with activities greater than the TA vector control (Fig. 4 and Table I). However, with more relaxed constraints the array identified two additional TFs with activity over control (GATA4 and E2F1). Monte Carlo methods confirmed the results of the Bayesian methods (Supplementary Table IV).
Figure 3.
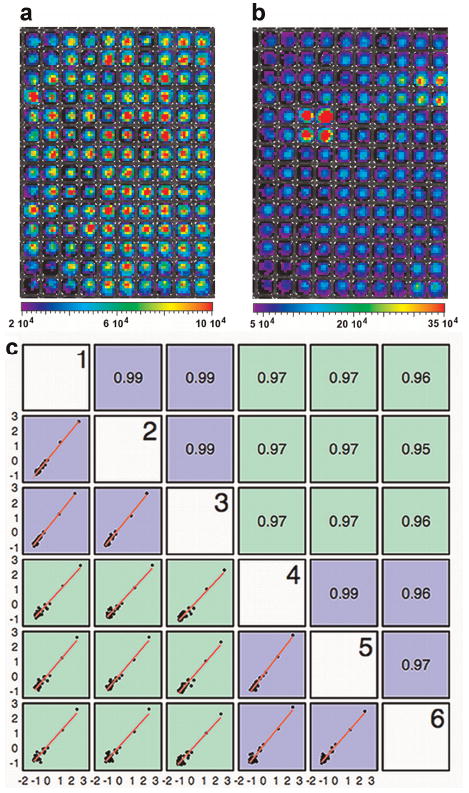
Transfected cell array for the large-scale analysis of the activity of 32 TFs using BLI. Pseudo-colored image of light emission within wells of a 384-well plate upon subsequent addition of substrates for Renilla luciferase (a) and firefly luciferase (b). c: Linear regression comparison between arrays within and across experiments. TF activity profiles between six arrays formed in two independent experiments are highly correlated within experiments (blue comparisons) and across experiments (green comparisons) with a minimum correlation coefficient of 0.95. [Color figure can be seen in the online version of this article, available at wileyonlinelibrary.com.]
Figure 4.
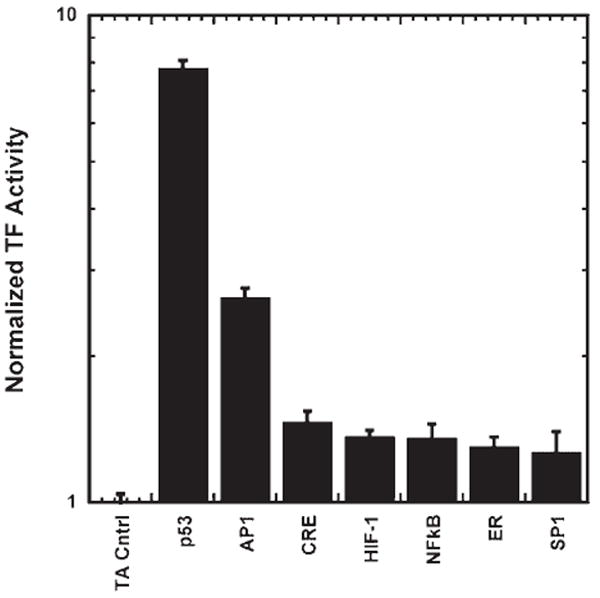
Transfected cell array for the large-scale analysis of the activity of 32 TFs using BLI. The normalized TF activity profile identified seven TFs with activities significantly higher than the TA vector control.
Table I.
Summary of normalized TF activity for large-scale activity array and statistical significance.
| TF | TF activity |
P-value | |
|---|---|---|---|
| Average | Standard deviation | ||
| Control | 1.000 | 0.074 | 1 |
| p53 | 7.487 | 1.214 | 1.79E−92 |
| AP1 | 2.620 | 0.280 | 1.69E−65 |
| CRE | 1.442 | 0.150 | 5.54E−24 |
| HIF-1 | 1.307 | 0.094 | 2.61E−19 |
| NFκB | 1.291 | 0.106 | 8.13E−18 |
| ER | 1.284 | 0.135 | 4.57E−13 |
| SP1 | 1.259 | 0.138 | 1.27E−11 |
| GATA4 | 1.144 | 0.078 | 2.03E−04 |
| SRF | 1.107 | 0.116 | 1.04E−01 |
| E2F1 | 1.101 | 0.151 | 1.85E−02 |
| AP2 | 1.085 | 0.117 | 1.00E+00 |
| AP4 | 1.071 | 0.110 | 1.00E+00 |
| GATA | 1.047 | 0.089 | 1.00E+00 |
| IRF-1 | 1.045 | 0.049 | 1.00E+00 |
| SRE | 1.044 | 0.150 | 1.00E+00 |
| VDR | 1.034 | 0.082 | 1.00E+00 |
| GAS | 1.030 | 0.083 | 1.00E+00 |
| NFAT | 1.018 | 0.112 | 1.00E+00 |
| GATA1/2 | 1.017 | 0.082 | 1.00E+00 |
| Smad 3/4 | 1.016 | 0.110 | 1.00E+00 |
| YY1 | 1.013 | 0.090 | 1.00E+00 |
| AR | 0.991 | 0.072 | 1.00E+00 |
| AP3 | 0.988 | 0.089 | 1.00E+00 |
| GR | 0.986 | 0.082 | 1.00E+00 |
| PR | 0.977 | 0.132 | 1.00E+00 |
| HSE | 0.977 | 0.068 | 1.00E+00 |
| Smad | 0.976 | 0.111 | 1.00E+00 |
| MEF-3 | 0.973 | 0.092 | 1.00E+00 |
| MEF-2 | 0.970 | 0.077 | 1.00E+00 |
| MEF-1 | 0.957 | 0.063 | 1.00E+00 |
| STAT4 | 0.950 | 0.076 | 1.00E+00 |
| GATA3 | 0.950 | 0.083 | 1.00E+00 |
TFs in bold are considered to have statistically significant higher activity than the TA vector control using Bayesian-based comparisons and an FDR corrected α′ = 0.00015.
Identification of Changes in TF Activity
The potential of the array to detect changes in the TF activity profile was subsequently investigated by performing cultures with and without PMA stimulation, which inhibits proliferation within MCF-7:WS8 cells, resulting from a specific block within the G1 cell-cycle phase to induce a more differentiated phenotype (Guilbaud et al., 1990; Valette et al., 1987). Five TFs (SRF, CRE, NFκB, AP1, and p53) had significant changes in activity upon PMA treatment (20 ng/mL) relative to the vehicle control (Fig. 5 and Supplementary Table IV). As expected, several of the differentially activated TFs have been reported to play key roles in modulation of cell cycle, and cellular differentiation and proliferation. The activity of p53, a tumor suppressor protein, is reduced with PMA and activities of SRF, CRE, AP1, and NFκB were all increased with PMA treatment; these observations are consistent with the previously published data (Abdel-Mageed and Agrawal, 1998; Adiseshaiah et al., 2005; Bale and Dorsa, 1998; Garcia-Rodriguez and Rao, 1998; Guilbaud et al., 1990; Hermanson et al., 2002; Hill et al., 1994; Liu et al., 1999; Ma et al., 2009; Siebenlist et al., 1994).
Figure 5.
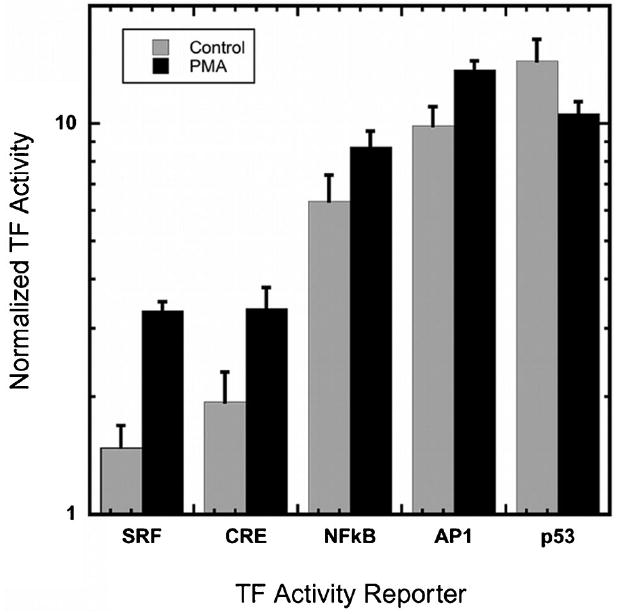
Changes in normalized TF activity profile of MCF7 cells upon induction with PMA. Of the 32 TFs in the array, 5 TFs had differential activity in response to PMA treatment, 4 TFs had increased activity upon PMA induction over vehicle delivery, SRF (+124%), CRE (+73%), NFκB (+38%), and AP1 (+39%), and 1 TF decreased in activity as compared to vehicle delivery (−26%).
Discussion
This report presents a system for rapid, non-invasive, and large-scale quantification of the activities of TFs using a combination of gene delivery of TF reporter constructs and BLI. A critical distinction of this system is that the measurement of TF activity within the cell can indicate that the signaling pathways upstream of the TF are active, and that the effectors within the pathway are present in the appropriate compartments and have undergone the necessary post-translational modifications. While genomics and proteomics approaches are being employed to infer pathway activity (Hoheisel, 2006), this inference is hindered by the complexity of promoter regions on genes, assumptions that mRNA and protein levels correlate to activity, and is computationally intensive. Rather than inferring activity, the cellular arrays reported herein directly measured TF activity with great sensitivity and minimal variability. Importantly, we applied this technology for simultaneous, large-scale analysis of TF activity, which can capture the breadth of active pathways within the cell.
This technology combines parallel transient transfection, BLI, and the use of luciferase reporter vectors to quantify TF activity. Large-scale pathway activity has previously been investigated by creating cell lines stably expressing a GFP reporter construct, which required both the generation and maintenance of multiple cell lines (King et al., 2007). By using transient transfection, the array presented herein may increase the flexibility of the system by allowing for the application of the array to any easily transfected cell line. Difficult to transfect cell lines or primary cells may require viral vectors to determine the TF activity profile. Relative to fluorescence imaging, BLI has the potential for substantially lower and more consistent background, which could provide for greater sensitivity. Finally, the reporter gene luciferase provides signal amplification by virtue of its enzymatic activity, which may also increase sensitivity.
The TF activity array employs parallel gene delivery for the determination of TF activity, with two constructs delivered to enable quantitative analysis. The inclusion of the Renilla luciferase normalization vector within each condition allows normalization of the firefly TF reporter signal to account for varying transfection and can identify wells with insufficient transfection that can be removed from analysis, thus reducing false positives. Importantly, all plasmids within the array contain the same minimal TA promoter, which is also important for proper normalization and should allow for comparisons between multiple cell lines. The TF activity profile of MCF7:WS8 cells was analyzed using statistical techniques common to microarray analysis and also non-parametric resampling-based inference methods, both of which supported the reproducibility of the system not only between arrays but also across experimental days. This consistency in array output is beneficial for reducing the number of experiments and minimizing false positives and negatives common to large-scale analysis of biological processes. The potential for false positives and negatives must be evaluated within any large-scale assay. For the statistical analysis of the array, methods that reduce the risk for false positives were chosen, knowing that this may increase the risk of false negatives. False negatives can occur for TFs that produce luminescence that is near background for the substrate. This luminescence assay is not as sensitive as some protein binding methods, such as electrophoresis mobility shift assay or chromatin immunoprecipitation. Consequently, the strength of the array is identifying multiple TF that are active (i.e., positive) rather than ones that are not (i.e., negative). Consistent with other high-throughput approaches (e.g., microarrays), results of the array must be validated before drawing conclusions about the biological system. The identification of false negatives is challenging for most high-throughput technologies, including microarrays. In this report, the array determined that 7 of 32 (22%) TFs were significantly active within the MCF7:WS8 cells (in basal media conditions). The TFs identified are known for their roles in cell-cycle regulation, differentiation, cell survival, apoptosis, and differentiation, and therefore are of interest in the study of cancer cells. Many TFs are not universally expressed or only respond to specific stimuli, which was consistent with our results that only a fraction of the TFs had activity above background under basal conditions. Transfection can stimulate pathways associated with inflammation, which can activate the associated TFs. This stimulation of these pathways by transfection must be considered when analyzing the cellular response.
The array also captured the TFs with altered activity in response to a factor that induces differentiation of MCF-7 cells (Guilbaud et al., 1990; Valette et al., 1987). Pharmacological activators such as PMA are commonly used to target individual signaling pathways, yet the broad impact of these small molecules on the signaling network, has not been well characterized. The TF activity array identified four TFs (AP1, CRE, NFκB, SRF) with increased activity and one TF with decreased activity (p53) at 6 h after PMA stimulation. These TFs have previously been linked to PMA activation individually (Abdel-Mageed and Agrawal, 1998; Adiseshaiah et al., 2005; Bale and Dorsa, 1998; Garcia-Rodriguez and Rao, 1998; Guilbaud et al., 1990; Hermanson et al., 2002; Hill et al., 1994; Liu et al., 1999; Ma et al., 2009), yet the ability to capture changes associated with several pathways in a single experiment represents the power of the TF activity array. This ability to measure the response of numerous TFs will enable fundamental studies, such as the specificity of drug actions or mechanisms of drug resistance to identify the regulatory processes that are differentially active between normal and disease phenotypes.
BLI of luciferase expression was the enabling tool for large-scale analysis of TF activity. Previously, Romanov et al. (2008) had employed reporter constructs with specific processing tags that, upon amplification, enabled quantification of transcript levels by capillary electrophoresis. Herein, we combined BLI with the reporter protein luciferase, which provides great sensitivity as a result of enzymatic amplification, allowing detection of TFs with low activity (Corish and Tyler-Smith, 1999; Ignowski and Schaffer, 2004). Live cell BLI captured the output of all wells simultaneously, rather than sequentially as with microplate readers that measure one well at a time, allowing for rapid data acquisition by eliminating the need for the cell lysis step. Relative to approaches based on cell lysis, the ability to monitor TF activity by BLI may enable TF profiles to be determined dynamically within the same cell population in a single well of the array. The imaging of both Renilla and firefly luciferase within a single array can be accomplished in approximately 16 min, which includes time for substrate addition and incubation, plate loading, and imaging, and dynamic imaging would need to account for this manipulation of the plate. Importantly, we demonstrated that BLI of TF activity produced results that are highly correlated with standard lysed cell assays. The rapid data acquisition combined with minimal post-transfection processing enabled by BLI will ultimately enable extension of the array to a larger library of reporter constructs. Since array-to-array variability is low, the number of reporter constructs analyzed with this approach is not expected to be limiting.
In summary, we report a dual-plasmid TF activity array to provide a rapid, non-invasive, large-scale quantification of TF activity that can detect the impact of perturbations to the culture environment. BLI produces comparable results to the traditional cell lysis approach, yet requires less handling of the sample is rapid and thus has relatively low data acquisition time. Furthermore, this method allows for a smaller plate format thus requiring fewer cells and can be employed to monitor numerous wells simultaneously. The activities of 32 TFs were assessed and the outputs of multiple arrays in different experiments were consistent and reproducible between arrays and experimental day. Finally, the array captured changes in the TF activity profile of MCF-7:WS8 breast cancer cells initiated by PMA activation. The TF activity array was able to monitor changes in multiple pathways in parallel, allowing for broad analysis of the overall interplay of signaling within the cells. Ultimately, transfected cell arrays have the potential to provide a rapid characterization of TF activity profiles of different cellular phenotypes and to detect alterations in TF activity within a changing environment. This system, which can connect environmental changes to TF activity patterns, may be a fundamental tool to investigate molecular mechanisms underlying diseases such as cancer and may provide a means to identify signaling pathways that could be targeted for therapeutic intervention.
Supplementary Material
Acknowledgments
Support for this research was provided by grants from the NIH (R21 006520 and T32 GM008449). This work was also funded by the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust. We would like to thank Anne Cooley, Eric Ariazi, and V. Craig Jordan for their technical contributions.
Contract grant sponsor: NIH
Contract grant number: R21 006520; T32 GM008449
Contract grant sponsor: The Searle Funds at The Chicago Community Trust
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Abdel-Mageed AB, Agrawal KC. Activation of nuclear factor kappaB: Potential role in metallothionein-mediated mitogenic response. Cancer Res. 1998;58(11):2335–2338. [PubMed] [Google Scholar]
- Adiseshaiah P, Peddakama S, Zhang Q, Kalvakolanu DV, Reddy SP. Mitogen regulated induction of FRA-1 proto-oncogene is controlled by the transcription factors binding to both serum and TPA response elements. Oncogene. 2005;24(26):4193–4205. doi: 10.1038/sj.onc.1208583. [DOI] [PubMed] [Google Scholar]
- Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogenrelated receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res. 2007;5(1):71–85. doi: 10.1158/1541-7786.MCR-06-0227. [DOI] [PubMed] [Google Scholar]
- Bale TL, Dorsa DM. NGF, cyclic AMP, and phorbol esters regulate oxytocin receptor gene transcription in SK-N-SH and MCF7 cells. Brain Res Mol Brain Res. 1998;53(1–2):130–137. doi: 10.1016/s0169-328x(97)00287-8. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Transcription factors in airway diseases. Lab Invest. 2006;86(9):867–872. doi: 10.1038/labinvest.3700456. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300. [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12(12):1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- Do KA, Hall P. On importance resampling for the bootstrap. Biometrika. 1991;78(1):161–167. [Google Scholar]
- Efron B, Tibshirani R. An introduction to the bootstrap. London: Chapman & Hall; 1993. [Google Scholar]
- Erfle H, Neumann B, Liebel U, Rogers P, Held M, Walter T, Ellenberg J, Pepperkok R. Reverse transfection on cell arrays for high content screening microscopy. Nat Protoc. 2007;2(2):392–399. doi: 10.1038/nprot.2006.483. [DOI] [PubMed] [Google Scholar]
- Garcia-Rodriguez C, Rao A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP) J Exp Med. 1998;187(12):2031–2036. doi: 10.1084/jem.187.12.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud NF, Gas N, Dupont MA, Valette A. Effects of differentiation- inducing agents on maturation of human MCF-7 breast cancer cells. J Cell Physiol. 1990;145(1):162–172. doi: 10.1002/jcp.1041450122. [DOI] [PubMed] [Google Scholar]
- Hall P, Wilson SR. Two guidelines for bootstrap hypothesis-testing. Biometrics. 1991;47(2):757–762. [Google Scholar]
- Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: Multiple modes of modification. Trends Endocrinol Metab. 2002;13(2):55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R. Serum-regulated transcription by serum-response-factor (Srf)—A novel role for the DNA-binding domain. EMBO J. 1994;13(22):5421–5432. doi: 10.1002/j.1460-2075.1994.tb06877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoheisel JD. Microarray technology: Beyond transcript profiling and genotype analysis. Nat Rev Genet. 2006;7(3):200–210. doi: 10.1038/nrg1809. [DOI] [PubMed] [Google Scholar]
- Ignowski JM, Schaffer DV. Kinetic analysis and modeling of firefly luciferase as a quantitative reporter gene in live mammalian cells. Biotechnol Bioeng. 2004;86(7):827–834. doi: 10.1002/bit.20059. [DOI] [PubMed] [Google Scholar]
- Jiang SY, Wolf DM, Yingling JM, Chang C, Jordan VC. An estrogenreceptor positive Mcf-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90(1):77–86. doi: 10.1016/0303-7207(92)90104-e. [DOI] [PubMed] [Google Scholar]
- King KR, Wang S, Irimia D, Jayaraman A, Toner M, Yarmush ML. A high-throughput microfluidic real-time gene expression living cell array. Lab Chip. 2007;7(1):77–85. doi: 10.1039/b612516f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman DS. Mechanisms of disease—Transcription-factor mutations and disease. N Engl J Med. 1996;334(1):28–33. doi: 10.1056/NEJM199601043340108. [DOI] [PubMed] [Google Scholar]
- Liu J, Li C, Ahlborn TE, Spence MJ, Meng L, Boxer LM. The expression of p53 tumor suppressor gene in breast cancer cells is down-regulated by cytokine oncostatin M. Cell Growth Differ. 1999;10(10):677–683. [PubMed] [Google Scholar]
- Lonnstedt I, Speed T. Replicated microarray data. Stat Sin. 2002;12:31–46. [Google Scholar]
- Ma G, Tabanca N, Husnu Can, Baser K, Kirimer N, Pasco DS, Khan IA, Khan SI. Inhibition of NF-kappaB-mediated transcription and induction of apoptosis in human breast cancer cells by epoxypseudoisoeugenol-2-methyl butyrate. Cancer Chemother Pharmacol. 2009;63(4):673–680. doi: 10.1007/s00280-008-0784-9. [DOI] [PubMed] [Google Scholar]
- Nebert DW. Transcription factors and cancer: An overview. Toxicology. 2002;181–182:131–141. doi: 10.1016/s0300-483x(02)00269-x. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108(4):439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277(5331):1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- Pannier AK, Ariazi EA, Bellis AD, Bengali Z, Jordan VC, Shea LD. Bioluminescence imaging for assessment and normalization in transfected cell arrays. Biotechnol Bioeng. 2007;98(2):486–497. doi: 10.1002/bit.21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S, Futcher B, Skiena S. Meta-analysis based on control of false discovery rate: Combining yeast ChIP-chip datasets. Bioinformatics. 2006;22(20):2516–2522. doi: 10.1093/bioinformatics/btl439. [DOI] [PubMed] [Google Scholar]
- Romanov S, Medvedev A, Gambarian M, Poltoratskaya N, Moeser M, Medvedeva L, Diatchenko L, Makarov S. Homogeneous reporter system enables quantitative functional assessment of multiple transcription factors. Nat Methods. 2008;5(3):253–260. doi: 10.1038/nmeth.1186. [DOI] [PubMed] [Google Scholar]
- Saville DJ. Multiple comparison procedures—The practical solution. Am Stat. 1990;44(2):174–180. [Google Scholar]
- Scherzer CR, Grass JA, Liao ZX, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci USA. 2008;105(31):10907–10912. doi: 10.1073/pnas.0802437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebenlist U, Franzoso G, Brown K. Structure regulation and function of Nf-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:article 3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- Strano S, Dell’Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G. Mutant p53: An oncogenic transcription factor. Oncogene. 2007;26(15):2212–2219. doi: 10.1038/sj.onc.1210296. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Valette A, Gas N, Jozan S, Roubinet F, Dupont MA, Bayard F. Influence of 12-O-tetradecanoylphorbol-13-acetate on proliferation and maturation of human-breast carcinoma-cells (Mcf-7)—Relationship to cell-cycle events. Cancer Res. 1987;47(6):1615–1620. [PubMed] [Google Scholar]
- Zaykin DV, Zhivotovsky LA, Westfall PH, Weir BS. Truncated product method for combining P-values. Genet Epidemiol. 2002;22(2):170–185. doi: 10.1002/gepi.0042. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Reinberg D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15(18):2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.