

Abstract
Background
Freshwater planarians are an attractive model for regeneration and stem cell research and have become a promising tool in the field of regenerative medicine. With the availability of a sequenced planarian genome, the recent application of modern genetic and high-throughput tools has resulted in revitalized interest in these animals, long known for their amazing regenerative capabilities, which enable them to regrow even a new head after decapitation. However, a detailed description of the planarian transcriptome is essential for future investigation into regenerative processes using planarians as a model system.
Results
In order to complement and improve existing gene annotations, we used a 454 pyrosequencing approach to analyze the transcriptome of the planarian species Schmidtea mediterranea Altogether, 598,435 454-sequencing reads, with an average length of 327 bp, were assembled together with the ~10,000 sequences of the S. mediterranea UniGene set using different similarity cutoffs. The assembly was then mapped onto the current genome data. Remarkably, our Smed454 dataset contains more than 3 million novel transcribed nucleotides sequenced for the first time. A descriptive analysis of planarian splice sites was conducted on those Smed454 contigs that mapped univocally to the current genome assembly. Sequence analysis allowed us to identify genes encoding putative proteins with defined structural properties, such as transmembrane domains. Moreover, we annotated the Smed454 dataset using Gene Ontology, and identified putative homologues of several gene families that may play a key role during regeneration, such as neurotransmitter and hormone receptors, homeobox-containing genes, and genes related to eye function.
Conclusions
We report the first planarian transcript dataset, Smed454, as an open resource tool that can be accessed via a web interface. Smed454 contains significant novel sequence information about most expressed genes of S. mediterranea. Analysis of the annotated data promises to contribute to identification of gene families poorly characterized at a functional level. The Smed454 transcriptome data will assist in the molecular characterization of S. mediterranea as a model organism, which will be useful to a broad scientific community.
Background
One of the challenges that medical research must address in the near future is to understand why some animals are able to regenerate complex structures, including eyes and even whole bodies, from small body fragments, while others are not. With the recent emergence of the field of regenerative medicine, the future biomedical ramifications of the study of animal regeneration are obvious.
Freshwater planarians are a classic model for studying the fascinating process of regeneration [1-4] because they are capable of re-building a complete organism from almost any small body fragment. This is made possible by a unique population of adult somatic stem cells called neoblasts. During regeneration and constant homeostatic cell turnover, neoblasts differentiate into all cell types, including germ cells in sexual species [5,6]. In recent years, several studies have begun to unravel the mechanisms by which regeneration is regulated at the molecular level. For example, different genes have been shown to play pivotal roles in axon guidance and neurogenesis [7], the regulation of neoblast proliferation and differentiation [8,9], and the re-establishment and maintenance of the anteroposterior (AP) and dorsoventral (DV) body axes [10]. Schmidtea mediterranea and Dugesia japonica are the two planarian species most often used in regeneration studies. There are about 78,000 ESTs (Expressed Sequence Tags) for S. mediterranea in NCBI generated in different projects [11,12]. Those sequences were clustered to produce a set of 10,000 putative mRNAs which are available from the NCBI Unigene database [13]. The S. mediterranea genome has also been sequenced and assembled [14] at the Genome Sequencing Center at Washington University in St. Louis (WUSL, USA) after approval of a white paper [15]. However, because of this genome's internal complexity (67% A+T, [16]) and the lack of a BAC library, its completeness and assembly still needs improvement. A step towards this end was taken when the S. mediterranea genome and EST information were integrated and approximately 30,000 genes were predicted using an annotation pipeline called MAKER[16]. Those gene models, together with ~9,000 mRNAs generated using next-generation sequencing technology, were mapped on the planarian genome and used to improve the assembly [17]. The current assembly contains 43,673 contigs. These are accessible, together with the MAKER annotation data, in the S. mediterranea genome database (SmedGD; [17]).
In order to expand our knowledge of the planarian transcriptome and to provide a new tool that can be used to improve the S. mediterranea genome annotation, we generated a new transcriptome dataset using 454 pyrosequencing technology [18]. The Smed454 dataset can be freely accessed via a website, and the complete sequence data can be downloaded by anyone from there. Mapping of the Smed454 ESTs onto the genome scaffolds shows that the Smed454 dataset contains more than 3 million nucleotides sequenced de novo. In addition, this mapping extends and connects currently fragmented genomic contigs. Finally, GO annotation of the Smed454 dataset assigns candidate functions to those sequences and facilitates their grouping into distinct gene families. In this way, whole gene families can be analyzed for putative roles in planarian regeneration. Thus we are confident that the Smed454 dataset will improve our understanding of how planarian regeneration works at the molecular level.
Results and Discussion
Construction and sequencing of the Smed454 dataset
In order to obtain the most representative set of planarian genes expressed under different physiological conditions, total RNA was isolated from a mixture of non-irradiated and irradiated intact and regenerating planarians (see Methods). We used planarians regenerating both head and tail to identify the genes specifically expressed in a tissue-specific manner. Similarly, planarians at different stages of regeneration were used in order to isolate genes with different temporal expression profiles. Irradiation destroys planarian neoblasts within 1-2 days, and the animals die within a few weeks because they cannot sustain normal cell turnover. By including irradiated animals, potential transcripts specifically expressed under those conditions will be contained in the 454 dataset.
Using 454 pyrosequencing, 601,439 sequencing reads with an average length of 327 bp were obtained. After sequence cleaning to remove vector contamination, the remaining 598,435 sequences were assembled using different cut-off values for sequence similarity (90%, 95% and 98%). In addition, our 454 sequence reads were assembled together with the ~10,000 S. mediterranea UniGene set available at NCBI, using the 90% similarity criteria. This last set, which was used in most of the analyses reported, is referred to as the 90e set. Table 1 summarizes the number of contigs and singletons obtained in each of those assemblies. The similarities between the three assemblies (90, 98 and 90e) are illustrated in Figure 1 a Venn diagram which shows that 72.68% of the raw sequencing reads were integrated into contigs common to all three assemblies, and 20.51% of the sequencing reads make up a shared pool of single sequencing reads (singletons). Therefore, differences between the assemblies can be explained by differential inclusion corresponding to 6.81% of the sequencing reads.
Table 1.
Summary of sequence statistics for each assembly.
| SET | Contigs | Singletons | TOTAL SEQs | GC% | LENGTHs [min/median/max/avg] |
|||
|---|---|---|---|---|---|---|---|---|
| 90 | 52,885 | 137,213 | 190, 098 | 35.130 | 20 | 354 | 6812 | 355.78 |
| 95 | 52,501 | 137,077 | 189,578 | --- | --- | --- | --- | --- |
| 98 | 52,321 | 137,353 | 189,674 | 35.127 | 20 | 354 | 6812 | 355.82 |
| 90e | 53,867 | 138,766 | 192,633 | 35.108 | 20 | 358 | 7918 | 364.81 |
Set names are related to the corresponding homology level cutoff value (90e stands for 90% similarity including the set of NCBI Unigene ESTs). Contigs are the result of at least two sequencing reads, and singletons of only one read. GC content is the average value for all sequences. Sequence lengths are shown as minimum, median, maximum and average values in nucleotides for each set.
Figure 1.

Overlap-analysis of Smed454 assemblies. Comparison of the 454-sequencing reads taken into account to build each Smed454 dataset. Venn-diagram numbers in plain format correspond to singleton reads, while numbers in bold correspond to sequencing reads that were assembled into Contigs. About 4,000 raw reads where split into two or more fragments, due to quality clipping. However, only distinct raw read identifiers, after removing the fragment suffix, were used to produce this figure. sgl: total number of singletons for that assembly; ctg: total number of contigs; uni: number of NCBI Unigene sequences not assembled into a contig (90e only); sqs: total number of sequences for a given dataset.
Average GC content and sequence length and their respective distributions were similar for all three assemblies (Table 1 and Figure 2). GC content is distributed around 35%, the expected value for coding sequences in this species. The 90e length distribution shape was slightly shifted towards larger sequences. This shift was mainly due to a set of long sequences (> 800 bp) from the NCBI Unigene ESTs included in this assembly. This causal relationship was evident in the comparison of the following four subsets of sequences from the 90e set (lightblue violin plots on Figure 2 right panel): singletons (136,271), contigs that do not contain UniGene ESTs (46,958), contigs including Unigene ESTs (6,909), and finally, Unigene ESTs not assembled into a contig (2,495).
Figure 2.

GC content and length distributions of different assemblies. Violin plots show the distribution of frequencies of a given variable in different datasets using a density kernel estimator [71]. White marks on the violin plots indicate the median value for a given variable, and the red points indicate the mean. The thick line marks the 25/75% inter-quartile range. GC content (left panel) distribution is quite similar in all the datasets, with higher frequencies around 35%. Nucleotide length (right panel) highlights the major differences between un-assembled (NCBI ESTs and the 454 raw reads) and assembled (NCBI UniGene, 90, 98 and 90e) sets. The last four plots (light blue) show the length distribution for the component subsets of the 90e assembly.
Mapping the 90e assembly onto the genome
The 90e assembly (192,633 sequences, 70,274,612 bases in total, average length of 365 bp per sequence) was aligned to scaffolds from the S. mediterranea WUSL genome assembly, version 3.1 [14] (43,294 sequences, 901,626,601 bases in total, average length of 20,8 kilobases per scaffold). Figure 3 shows all possible high-scoring segment pair (HSP) relationships between those two sequence sets. From almost 30 million initial HSPs, around 7 million were selected using a combination of thresholds, as described in the Methods section. Discarding singleton sequences in a second round of filtering further reduced the number of HSPs to 5 million, and HSP coverage dropped from 25.36% and 77.24%, for scaffolds and 90e respectively, to 10.57% and 37.93%. However, when the total nucleotide length was considered only for the contigs (56,363 sequences, 32,518,399 bases in total, with an average of 577 bp per sequence), HSP coverage for 90e rose to 81.97%. This means that most of the significant HSP hits are retained after the second round of filtering. In total, 8,831 contigs from 90e did not map to the genomic contigs (3,242,054 bp that are completely novel and also transcribed, see column A in Figure 3). Conversely, 5,138 genomic contigs did not match a sequence from 90e (column B). Of the 90e contigs, 322 extended a genomic sequence from the left (column C) and 3,051 from the right (column J). The largest intergenic distance was 42,209 bp, with an average value of 1,102 bp (column H). The largest intron was estimated to be about 9,300 bp, the average length being 238 bp (column E). Finally, there were 20,504 HSPs connecting different genomic sequences via 8,604 different 90e contigs (column I). Of the 8,831 90e contigs not found on the genome, 3,480 had a BLAST hit to the NCBI NR protein database (39.41%), and, of those, 2,401 had a hit to a protein with GO annotation (27.19%). After discarding abundant actin-like sequences (1,503), ATP/ADP transporter proteins (722) and sequences matching bacterial, protozoan or fungal genes (1,234), 71 90e contigs remained as new sequences not mapping on the genome (see Additional File 1).
Figure 3.

Distribution of different HSP types from 90e over genome sequences. The top table shows the total number of similarity hits, while the bottom table classifies the hits into different types of HSPs: A) 454 contigs not mapping to a genomic sequence; B) genomic contigs not mapping to a 454 contig; C and J) 454 contigs with an unmapped sequence on the left and right, respectively; D) missing sequence on 454 contigs corresponding to a putative gap in the assembly; E) contiguous HSPs on 454 contigs related to a genomic intron; F) co-linear unmapped sequences on both sequence sets; G) contiguous overlapping HSPs defining a larger similarity segment; H) unaligned genomic sequences between HSPs of two different 454 contigs, which can be interpreted as putative intergenic sequences; I) HSPs on 454 contigs supporting a pair of genomic contigs, which could then be merged into a larger genomic scaffold. All columns show HSP numbers--the '#HSPs' row--except for A and B, which correspond to number of sequences.
In order to validate exonic structures, 6,226 90e contigs mapping 1-to-1 over genome sequences were selected. After re-aligning the 90e/genomic sequence pairs, 4,739 contained at least one putative intron (see the corresponding splice sites boundaries in Additional File 2). In total 8,609 introns were retrieved from the genomic contigs. Figure 4 shows the number of introns per 90e contig, as well as the length distribution for those introns. Pictograms summarize the nucleotide frequencies for the donor and acceptor splice sites, both for the U2 (canonical) and U12 (non-cannonical) introns. The splice sites patterns resemble those from other metazoan [19], taking into account that the genome of S.mediterranea is A/T-rich [16].
Figure 4.

Analysis of intronic features and splice sites on a set of 90e contigs. A) Distribution of the number of putative introns per 90e contig. B) Length distribution of putative introns. C) Pictograms summarizing the consensus donor and acceptor splice sites for the predicted introns. n corresponds to the number of intron sequences used to compute the nucleotide position weight matrices for the pictograms. Light grey shadowed regions correspond to the commonly used signal lengths for gene-finding, while dark grey ones define the nucleotide boundaries of the introns. Numbers below pictograms are the bit-scores that describe the information content per position.
Also, 50 randomly picked 90e contigs that either mapped or did not map to the genome were validated by RT-PCR (see Additional File 3 containing a list of the selected 90e contigs, as well as information on the primers used to amplify them). Additionally, 20 out of those 50 genes were further validated by sequencing. Finally, to further confirm the quality and coverage of the sequences from the 90e dataset, the S. mediterranea genes already annotated in NCBI GenBank [20] were compared with those sequences. After discarding 18 S and 28 S ribosomal RNA genes and alpha-tubulins, 124 known genes were aligned to the 90e sequences. In total, 108 of these genes had at least one significant similarity hit with one 90e sequence, and two matched 5 sequences from 90e. On average, the known genes had co-linear similarity hits against 1.32 different Smed454 sequences. Minimum and average similarities were 8.35% and 85.34% respectively, and 71 sequences had more than 95% similarity. Mean coverage dropped to 77.63% when each hit was considered separately. A summary of these similarity analyses is shown in Additional File 4.
Browsing the Smed454 dataset
In order to make the Smed454 dataset useful and accessible to the planarian and non-planarian communities, a public database is available via web [21]. The web site allows users to view contig assemblies along with their read alignments, and to perform BLAST searches against assembled sequences. The BLAST option in the home page menu (1 in Figure 5) allows the user to BLAST sequences of interest against the 90, 98, and 90e databases (1.2 in Figure 5). Both nucleotide (BLASTN) and protein (BLASTP) searches can be performed (1.1 in Figure 5). Clicking on the Search button (1.3 in Figure 5) brings up a new window displaying a list of hits. When a score value is selected (1.4 in Figure 5), the alignment between the query sequence and the Smed454 hit is shown. The site also offers the option of downloading Smed454 sequences of interest (1.5 in Figure 5). The contig or singleton accession number can be browsed directly from the main home page (2 in Figure 5). When the user searches for a specific contig, a new window appears showing the alignment of all the sequencing reads assembled in that contig. At the bottom of that window, the result of a pre-computed BLAST on the contig consensus sequence is displayed. When a contig, singleton or read name is selected (2.1 in Figure 5), a new window will display the requested sequence. All raw and assembled sequence data are available from that web site too.
Figure 5.

The content of the Smed454 web site. Screenshots of the pages that facilitate access to the three sequence assemblies (90, 98 and 90e), including the page displaying alignments of raw reads. A BLAST interface, adapted from NBCI's toolkit, is also available for querying the sequences from the datasets. The web site is available at http://planarian.bio.ub.es/datasets/454/
Functional annotation of 90e sequences
In order to characterize the gene families that can be found on Smed454, we annotated the three datasets; we will focus on 90e dataset here. In total, 42.42% of the sequences had a similarity hit with at least one protein sequence in the NCBI NR protein database [20]. Of these, almost two-thirds had 250 or more hits (see Figure 6), but the BLASTX output was limited to a maximum of 250 hits per 90e sequence owing to the large number of HSPs reported by BLAST for some of them. The Gene Ontology (GO) [22] database was used to computationally annotate all the sequences (see Additional File 5 for 90, 98, and 90e datasets) by mapping onto them the functional codes already assigned to known proteins from NCBI NR. Many of these sequence hits matched to a short ATP-binding domain, in most cases corresponding to proteins of the actins family. Consequently, that functional class, which was also anomalously over-represented, was discarded from the total number of annotations for the 90e set, as shown in Table 2.
Figure 6.
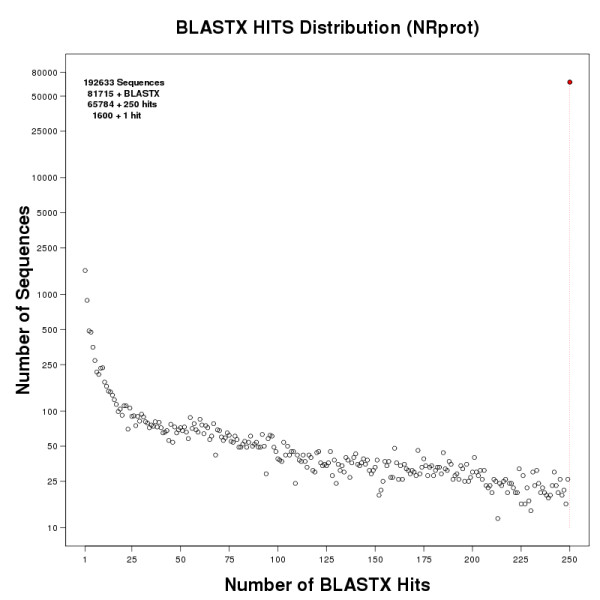
Distribution of BLASTXhits of 90e sequences against NCBI NRprot. Sequences from the 90e dataset were compared against the NCBI NR protein database using BLASTX. The figure shows the distribution of the number of sequences binned by the number of HSPs they had. Y-axis in log scale.
Table 2.
Gene Ontology annotation for 90e set sequences.
| GO | Molecular Function | Count | Freq% | GO | Biological Process | Count | Freq% | GO | Cellular Component | Count | Freq% |
| GO:0000166 | nucleotide binding | 54,823 | --- | --- | --- | --- | --- | GO:0043229 | intracellular organelle | 60,817 | --- |
| --- | unannotated | 9,709 | --- | --- | unannotated | 62,834 | --- | --- | unannotated | 11,131 | --- |
| GO:0016787 | hydrolase activity | 5,197 | 35.669 | GO:0043170 | macromolecule metabolic process | 5,793 | 35.610 | GO:0043234 | protein complex | 2,918 | 40.788 |
| GO:0016740 | transferase activity | 2,030 | 13.933 | GO:0022607 | cellular component assembly | 2,182 | 13.413 | GO:0044424 | intracellular part | 2,314 | 32.346 |
| GO:0043167 | ion binding | 1,323 | 9.080 | GO:0006810 | transport | 1,213 | 7.456 | GO:0031982 | vesicle | 819 | 11.448 |
| GO:0003735 | structural constituent of ribosome | 874 | 5.999 | GO:0006950 | response to stress | 1,070 | 6.577 | GO:0044425 | membrane part | 469 | 6.556 |
| GO:0005488 | binding | 761 | 5.223 | GO:0050789 | regulation of biological process | 1,012 | 6.221 | GO:0016020 | membrane | 210 | 2.935 |
| GO:0016491 | oxidoreductase activity | 703 | 4.825 | GO:0006807 | nitrogen compound metabolic process | 722 | 4.438 | GO:0005622 | intracellular | 111 | 1.552 |
| GO:0022857 | transmembrane transporter activity | 678 | 4.653 | GO:0048869 | cellular developmental process | 655 | 4.026 | GO:0044446 | intracellular organelle part | 91 | 1.272 |
| GO:0030235 | nitric-oxide synthase regulator activity | 597 | 4.097 | GO:0065009 | regulation of molecular function | 622 | 3.823 | GO:0005576 | extracellular region | 63 | 0.881 |
| GO:0043176 | amine binding | 580 | 3.981 | GO:0009056 | catabolic process | 507 | 3.117 | GO:0045211 | postsynaptic membrane | 21 | 0.294 |
| GO:0005515 | protein binding | 532 | 3.651 | GO:0044419 | interspecies interaction between organisms | 280 | 1.721 | GO:0044420 | extracellular matrix part | 19 | 0.266 |
| GO:0003676 | nucleic acid binding | 401 | 2.752 | GO:0055114 | oxidation reduction | 236 | 1.451 | GO:0043233 | organelle lumen | 18 | 0.252 |
| GO:0005215 | transporter activity | 387 | 2.656 | GO:0065008 | regulation of biological quality | 206 | 1.266 | GO:0031012 | extracellular matrix | 16 | 0.224 |
| GO:0016829 | lyase activity | 71 | 0.487 | GO:0048856 | anatomical structure development | 193 | 1.186 | GO:0042597 | periplasmic space | 15 | 0.210 |
| GO:0016853 | isomerase activity | 55 | 0.377 | GO:0051649 | establishment of localization in cell | 183 | 1.125 | GO:0000267 | cell fraction | 15 | 0.210 |
| GO:0048037 | cofactor binding | 52 | 0.357 | GO:0044237 | cellular metabolic process | 182 | 1.119 | GO:0044462 | external encapsulating structure part | 11 | 0.154 |
| GO:0016874 | ligase activity | 49 | 0.336 | GO:0023060 | signal transmission | 150 | 0.922 | GO:0031975 | envelope | 8 | 0.112 |
| GO:0004871 | signal transducer activity | 45 | 0.309 | GO:0048870 | cell motility | 141 | 0.867 | GO:0005615 | extracellular space | 7 | 0.098 |
| GO:0003824 | catalytic activity | 32 | 0.220 | GO:0008152 | metabolic process | 139 | 0.854 | GO:0009986 | cell surface | 6 | 0.084 |
| GO:0060589 | nucleoside-triphosphatase regulator activity | 32 | 0.220 | GO:0023033 | signaling pathway | 107 | 0.658 | GO:0043204 | perikaryon | 5 | 0.070 |
| GO:0042277 | peptide binding | 28 | 0.192 | GO:0044238 | primary metabolic process | 83 | 0.510 | GO:0030427 | site of polarized growth | 4 | 0.056 |
| GO:0022892 | substrate-specific transporter activity | 22 | 0.151 | GO:0042221 | response to chemical stimulus | 69 | 0.424 | GO:0042995 | cell projection | 3 | 0.042 |
| GO:0019208 | phosphatase regulator activity | 12 | 0.082 | GO:0006996 | organelle organization | 47 | 0.289 | GO:0030312 | external encapsulating structure | 2 | 0.028 |
| GO:0003712 | transcription cofactor activity | 12 | 0.082 | GO:0007017 | microtubule-based process | 42 | 0.258 | GO:0031594 | neuromuscular junction | 2 | 0.028 |
| GO:0019207 | kinase regulator activity | 11 | 0.075 | GO:0044281 | small molecule metabolic process | 39 | 0.240 | GO:0045177 | apical part of cell | 2 | 0.028 |
| GO:0008289 | lipid binding | 9 | 0.062 | GO:0051301 | cell division | 37 | 0.227 | GO:0019028 | viral capsid | 1 | 0.014 |
| GO:0005201 | extracellular matrix structural constituent | 8 | 0.055 | GO:0022613 | ribonucleoprotein complex biogenesis | 35 | 0.215 | GO:0031252 | cell leading edge | 1 | 0.014 |
| GO:0050840 | extracellular matrix binding | 6 | 0.041 | GO:0019637 | organophosphate metabolic process | 34 | 0.209 | GO:0044217 | other organism part | 1 | 0.014 |
| GO:0061134 | peptidase regulator activity | 6 | 0.041 | GO:0045184 | establishment of protein localization | 34 | 0.209 | GO:0044297 | cell body | 1 | 0.014 |
| GO:0030246 | carbohydrate binding | 6 | 0.041 | GO:0009628 | response to abiotic stimulus | 23 | 0.141 | GO:0044463 | cell projection part | 1 | 0.014 |
| GO:0016248 | channel inhibitor activity | 5 | 0.034 | GO:0019748 | secondary metabolic process | 21 | 0.129 | TOTAL | 7,154 | ||
| GO:0003702 | RNA polymerase II transcription factor activity | 5 | 0.034 | GO:0009058 | biosynthetic process | 21 | 0.129 | ||||
| GO:0005198 | structural molecule activity | 4 | 0.027 | GO:0007155 | cell adhesion | 18 | 0.111 | ||||
| GO:0016986 | transcription initiation factor activity | 4 | 0.027 | GO:0061024 | membrane organization | 16 | 0.098 | ||||
| GO:0042165 | neurotransmitter binding | 4 | 0.027 | GO:0007275 | multicellular organismal development | 16 | 0.098 | ||||
| GO:0003682 | chromatin binding | 3 | 0.021 | GO:0016192 | vesicle-mediated transport | 12 | 0.074 | ||||
| GO:0008430 | selenium binding | 3 | 0.021 | GO:0043062 | extracellular structure organization | 10 | 0.061 | ||||
| GO:0030234 | enzyme regulator activity | 2 | 0.014 | GO:0034330 | cell junction organization | 9 | 0.055 | ||||
| GO:0030528 | transcription regulator activity | 2 | 0.014 | GO:0048609 | reproductive process in a multicellular organism | 9 | 0.055 | ||||
| GO:0009055 | electron carrier activity | 2 | 0.014 | GO:0007154 | cell communication | 8 | 0.049 | ||||
| GO:0017056 | structural constituent of nuclear pore | 2 | 0.014 | GO:0003008 | system process | 7 | 0.043 | ||||
| GO:0017080 | sodium channel regulator activity | 2 | 0.014 | GO:0016049 | cell growth | 7 | 0.043 | ||||
| GO:0001871 | pattern binding | 2 | 0.014 | GO:0016458 | gene silencing | 6 | 0.037 | ||||
| GO:0019239 | deaminase activity | 2 | 0.014 | GO:0008219 | cell death | 5 | 0.031 | ||||
| GO:0043021 | ribonucleoprotein binding | 2 | 0.014 | GO:0033036 | macromolecule localization | 4 | 0.025 | ||||
| GO:0008538 | proteasome activator activity | 2 | 0.014 | GO:0048610 | reproductive cellular process | 4 | 0.025 | ||||
| GO:0030337 | DNA polymerase processivity factor activity | 1 | 0.007 | GO:0051236 | establishment of RNA localization | 4 | 0.025 | ||||
| GO:0031406 | carboxylic acid binding | 1 | 0.007 | GO:0071684 | organism emergence from protective structure | 4 | 0.025 | ||||
| GO:0042562 | hormone binding | 1 | 0.007 | GO:0006955 | immune response | 4 | 0.025 | ||||
| GO:0046906 | tetrapyrrole binding | 1 | 0.007 | GO:0007049 | cell cycle | 4 | 0.025 | ||||
| GO:0051540 | metal cluster binding | 1 | 0.007 | GO:0009405 | pathogenesis | 4 | 0.025 | ||||
| TOTAL | 14,570 | GO:0009607 | response to biotic stimulus | 4 | 0.025 | ||||||
| GO:0009791 | post-embryonic development | 4 | 0.025 | ||||||||
| GO:0048646 | anatomical structure formation involved in morphogenesis | 4 | 0.025 | ||||||||
| GO:0009790 | embryonic development | 3 | 0.018 | ||||||||
| GO:0015976 | carbon utilization | 2 | 0.012 | ||||||||
| GO:0032506 | cytokinetic process | 2 | 0.012 | ||||||||
| GO:0045103 | intermediate filament-based process | 2 | 0.012 | ||||||||
| GO:0070882 | cellular cell wall organization or biogenesis | 2 | 0.012 | ||||||||
| GO:0006413 | translational initiation | 2 | 0.012 | ||||||||
| GO:0009566 | fertilization | 2 | 0.012 | ||||||||
| GO:0001906 | cell killing | 1 | 0.006 | ||||||||
| GO:0043473 | pigmentation | 1 | 0.006 | ||||||||
| GO:0009987 | cellular process | 1 | 0.006 | ||||||||
| GO:0022402 | cell cycle process | 1 | 0.006 | ||||||||
| GO:0022411 | cellular component disassembly | 1 | 0.006 | ||||||||
| GO:0022415 | viral reproductive process | 1 | 0.006 | ||||||||
| GO:0023061 | signal release | 1 | 0.006 | ||||||||
| GO:0030030 | cell projection organization | 1 | 0.006 | ||||||||
| GO:0051606 | detection of stimulus | 1 | 0.006 | ||||||||
| GO:0000746 | conjugation | 1 | 0.006 | ||||||||
| GO:0007163 | establishment or maintenance of cell polarity | 1 | 0.006 | ||||||||
| GO:0009653 | anatomical structure morphogenesis | 1 | 0.006 | ||||||||
| TOTAL | 16,268 | ||||||||||
The most probable GO code in the three ontology categories--molecular function, biological process and cellular component-- for each sequence in the 90e data set was selected. For simplicity, only level one and two codes are shown here in order.
Among the most abundant GO annotations at the biological process level, leaving aside metabolism-related features, 'response to stress' was found for 1,070 sequences (6.58%). This finding was expected because the original biological sample was a mixture of intact and regenerating planarians, both normal and irradiated. 'Regulation of biological process' was in the same range, with 1,012 sequences (6.22%). At the GO molecular function level, 'binding' was the most common annotation, although where possible a more specific annotion was provided by drilling down to the 2nd level child annotations on the GO graph. It is interesting to find, among others, 3 'selenium binding' activities, since it has been reported that selenium may play an important role in cancer prevention, immune system function, male fertility, cardiovascular and muscle disorders, and prevention and control of the ageing process [23]. Finding selenium-binding proteins would be evidence of the presence of selenoproteins, which are thought to be responsible for most of the biomedical effects of selenium across eukaryota [24]. When looking at the cellular component level and discarding many of the 'intracellular organelles' due to their co-occurrence with 'nucleotide binding', there are a notably large number of 'protein complexes', 2,918 sequences (40.79%). With 819 sequences (11.45%), another important term on this level is 'vesicle', which correlates with secretory functions, apoptosis, and autophagy.
To prove the usefulness of the Smed454 dataset, we performed several searches on specific groups and gene families for which only scant data has been reported to date in planarians. Planarians are mainly known for their remarkable regenerative capabilities, which depend upon the presence of stem cells named neoblasts. Because of the unique properties of these cells, some studies have used a microarray-based strategy to detect neoblast-specific genes [25,26]. In our Smed454 dataset we were able to identify, in addition to known neoblast markers such as piwis, histones, bruli, vasa or tudor, several other genes annotated as involved in cell cycle or DNA damage and repair (Additional File 6). Within these gene set we find many cyclins and cell cycle division-related genes but also genes related to replication and chromosome maintenance. Finally, genes related to stress response and DNA damage were also identified, probably owing to the use of irradiated animals in the generation of the Smed454 dataset. In addition to these neoblast-related genes we were able to identify large collections of much less well-characterized families in planarians, such as neurotransmitter, peptide and hormone receptors, homeobox domain-containing genes, and genes related to eye function in other animals.
Prediction of planarian transmembrane proteins
Transmembrane (TM) proteins regulate a number of biological processes ranging from catalytic processes in intracellular and extracellular transport to cell-to-cell communication. TM proteins have become particularly interesting as many of them are key initiators of signal transduction pathways, and they can be easily manipulated by small molecule- or antibody-based drugs. To identify putative TM proteins from the planarian transcriptome, we mined the 454 dataset for putative TM protein-encoding messages (see Methods). Considering only the proteins that at least two application predicted would contain one or more transmembrane domains, resulted in a list of 8,597 predicted transmembrane proteins (see Figure 7a), which represents 15,3% of the complete protein database. Protein-BLAST searches were then used to align sequences to each other, and redundant sequences were removed from the predicted transmembrane set. The resulting database contained 4,663 sequences. Functional categorization using the UFO web-server [27] allowed us to assign PFAM protein families to 1,474 of the sequences and gene ontology classifications to 2,464. The top ten PFAM domains (~33% of all assignments) included, for example, the classifications for 'major facilitator superfamily' (a ubiquitous transporter family), '7 transmembrane receptor (rhodopsin family)' and 'ion transport protein' (see Figure 7b). The top ten gene ontology profiles (~49% of all assignments) included 'membrane' (cellular component), 'transport', and 'G-protein coupled receptor protein signalling pathway' (both biological processes, see Figure 7c). The enrichment of our database with proteins that have a predicted function in transport and receptor signalling supports the reliability of our approach. A complete list of the 4,663 predicted transmembrane proteins, the number of predicted transmembrane domains, predicted topology, and functional categorizations (PFAM and GO) are shown in Additional File 7.
Figure 7.
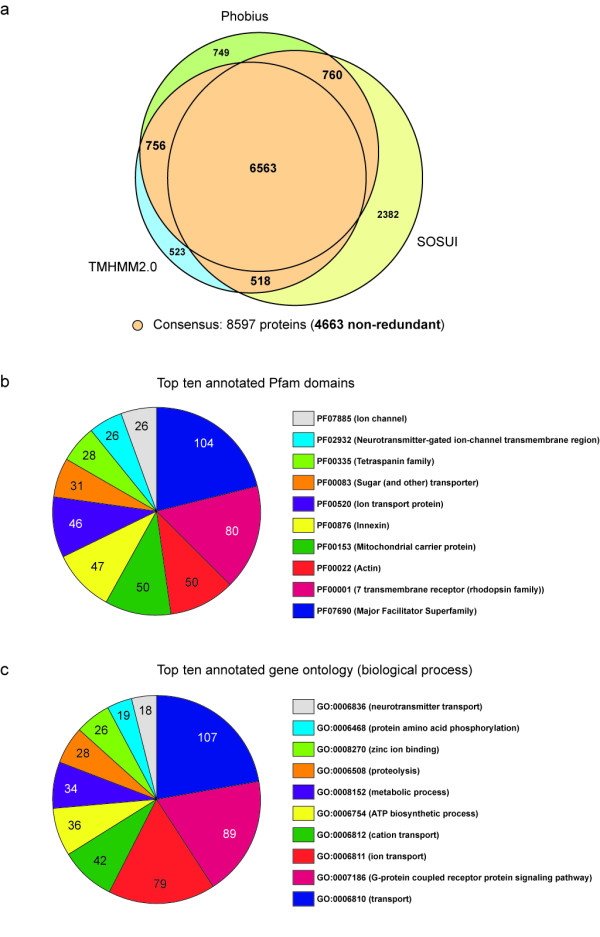
Prediction of planarian transmembrane proteins and functional annotations. A) Venn-diagram showing the overlap between predictions of transmembrane proteins generated by the Phobius, TMHMM2.0 and SOSUI programs for a set of 56,362 protein sequences translated from planarian ESTs. Only proteins predicted to contain one or more transmembrane domains by at least two programs (colored orange, 8,597 proteins, of which 4,663 are non-redundant) were considered for further analysis. B) Top ten PFAM domains and C) gene ontologies (biological process) for the 4,663 non-redundant transmembrane-proteins predicted. The figures indicate the number of proteins contained in a given annotation group.
Neurotransmitter and hormone receptors in Schmidtea mediterranea
Despite our growing knowledge about how planarian neoblasts are regulated at the molecular level [9,25,26,28-31], we are still far from characterizing the complete repertoire of factors that control neoblast biology. Receptors for neurotransmitters, peptides and hormones are among the candidates for a role in the regulation of neoblast proliferation, differentiation and migration. In planarians, some of the data suggest that molecules such as dopamine [32,33], serotonin [34], substance P [35], somatostatin [36] and FMRFamide [37] can accelerate or delay the regeneration rate, probably by regulating neoblast proliferation and/or differentiation. A model has been proposed in which neoblasts express receptors for some of these factors, which in turn regulate the fate of these cells [35]. We found 288 contigs and singletons in the annotated Smed454 dataset with significant homology to neurotransmitter and hormone receptors (Table 3 and Additional File 8), providing a list of potentially interesting candidates.
Table 3.
List of neurotransmitter, peptide and hormone receptor sequence candidates.
| ID | BLASTX HIT | ACCESSION NUMBER | E-VALUE |
|---|---|---|---|
| 90_1623 | adiponectin receptor (Schistosoma mansoni) | XP_002577010.1 | 2,00E-103 |
| 90_11706 | allatostatin receptor, putative (Ixodes scapularis) | XP_002414997.1 | 2,00E-18 |
| 90_9653 | amine GPCR (Schistosoma mansoni) | XP_002576533.1 | 8,00E-25 |
| P02IKPED | atrial natriuretic peptide receptor (Aedes aegypti) | XP_001652228.1 | 7,00E-28 |
| P02HWID8 | beta adrenergic receptor (Aedes aegypti) | XP_001651714.1 | 2,00E-18 |
| 90_17484 | similar to bombesin-like peptide receptor (Ornithorhynchus anatinus) | XP_001514235.1 | 2,00E-12 |
| 90_19322 | C1A receptor, putative (Ixodes scapularis) | XP_002405845.1 | 4,00E-04 |
| 90_4815 | calcitonin receptor, isoform CRA_d (Rattus norvegicus) | AAA65964.1 | 3,00E-50 |
| 90_20672 | cardioexcitatory receptor (Lymnaea stagnalis) | AAB92258.1 | 6,00E-11 |
| 90_6224 | class b secretin-like g-protein coupled receptor GPRmth5 (Pediculus humanus) | XP_002427184.1 | 2,00E-10 |
| P02IZJB4 | similar to putative diuretic hormone receptor II (Nasonia vitripennis) | XP_001606711.1 | 4,00E-22 |
| 90_7506 | type I dopamine receptor (Panulirus interruptus) | ABB87183.1 | 8,00E-69 |
| 90_6802 | dopamine receptor type D2 (Apis mellifera) | NP_001011567.1 | 2,00E-27 |
| 90_8536 | dro/myosuppressin receptor (Schistosoma mansoni) | XP_002570000.1 | 7,00E-26 |
| 90_9052 | FMRFamide receptor (Culex quinquefasciatus) | XP_001849293.1 | 2,00E-17 |
| P02GUXTP | similar to galanin receptor type I (Danio rerio) | XP_690480.1 | 3,00E-06 |
| 90_6830 | glutamate receptor kainate (Schistosoma mansoni) | XP_002576035.1 | 3,00E-70 |
| P02GLFYW | glutamate receptor NMDA (Schistosoma mansoni) | XP_002572261.1 | 3,00E-21 |
| 90_15092 | glutamate receptor, ionotropic, AMPA 1b (Danio rerio) | NP_991293.1 | 5,00E-78 |
| 90_13524 | metabotropic glutamate receptor (Schistosoma mansoni) | XP_002572726.1 | 1,00E-12 |
| 90_18656 | gonadotropin-releasing hormone receptor type I (Capra hircus) | ABL76162.1 | 7,00E-04 |
| 90_4098 | growth hormone secretagogue receptor (Schistosoma mansoni) | XP_002569813.1 | 7,00E-36 |
| 90_976 | growth hormone-inducible transmembrane protein (Osmerus mordax) | AC008873.1 | 2,00E-51 |
| 90_6465 | putative insulin receptor (Echinococcus multilocularis) | CAD30260.1 | 6,00E-61 |
| 90_7253 | lung seven transmembrane receptor (Culex quinquefasciatus) | XP_001868443.1 | 1,00E-68 |
| 90_17047 | metabotropic GABA-B receptor subtype, putative (Ixodes scapularis) | XP_002406087.1 | 4,00E-21 |
| 90_6512 | natriuretic peptide receptor (Xenopus laevis) | NP_001083703.1 | 4,00E-158 |
| 90_12800 | muscarinic acetylcholine (GAR) receptor (Schistosoma mansoni) | XP_002575679.1 | 2,00E-39 |
| 90_1507 | Nicotinic acetylcholine receptor alpha 1 subunit (Aplysia californica) | AF467898_1 | 3,00E-44 |
| 90_223 | neuroendocrine protein 7b2 (Schistosoma mansoni) | XP_002578500.1 | 6,00E-25 |
| 90_6302 | similar to neuromedin U receptor 2 (Strongylocentrotus purpuratus) | XP_001200425.1 | 4,00E-27 |
| 90_29452 | neuropeptide FF receptor 2 isoform 3 (Homo sapiens) | NP_001138228.1 | 2,00E-09 |
| 90_6772 | neuropeptide F-like receptor (Schistosoma mansoni) | XP_002573542.1 | 1,00E-28 |
| 90_5995 | neuropeptide Y receptor Y7 (Oncorhynchus mykiss) | ABB54774.1 | 9,00E-18 |
| 90_25975 | octopamine receptor (Aplysia californica) | AAF37686.1 | 1,00E-25 |
| 90_8498 | odorant receptor (Tetraodon nigroviridis) | CAG08888.1 | 6,00E-05 |
| 90_5999 | similar to olfactory receptor 355 (Bos taurus) | XP_610381.4 | 1,00E-05 |
| 90_2541 | P2Y purinergic receptor (Meleagris gallopavo) | AAA18784.1 | 2,00E-04 |
| 90_8537 | P2X receptor subunit (Schistosoma mansoni) | XP_002580774.1 | 1,00E-72 |
| 90_19040 | pituitary adenylate cyclase activating polypeptide receptor (Oncorhynchus mykiss) | NP_001118113.1 | 1,00E-08 |
| 90_28219 | parathyroid hormone 2 receptor (Danio rerio) | AAI62580.1 | 3,00E-11 |
| 90_6836 | peptide (allatostatin)-like receptor (Schistosoma mansoni) | XP_002572656.1 | 2,00E-66 |
| 90_7984 | peptide (allatostatin/somatostatin)-like receptor (Schistosoma mansoni) | XP_002575539.1 | 2,00E-32 |
| 90_10769 | progesterone receptor membrane component 1 (Danio rerio) | NP_001007393.1 | 7,00E-04 |
| 90_5450 | progestin receptor membrane component 1 (Oryzias latines) | BAE47967.1 | 2,00E-28 |
| P02GZGVI | prolactin releasing hormone receptor (Homo sapiens) | BAG36078.1 | 2,00E-06 |
| P02I1U9K | pyrokinin-like receptor (Dermacentor variabilis) | ACC99623.1 | 2,00E-11 |
| 90_10680 | Rhodopsin-like GPCR superfamily, domain-containing protein (Schistosoma japonicum) | CAX73015.1 | 6,00E-37 |
| 90_2955 | rhodopsin-like orphan GPCR (Schistosoma mansoni) | XP_002579928.1 | 2,00E-42 |
| 90_27829 | ryanodine receptor 44F (Schistosoma japonicum) | CAX69439.1 | 8,00E-16 |
| 90_14326 | serotonin receptor-like planarian receptor 1 (Dugesia japonica) | BAA22404.1 | 3,00E-54 |
| 90_15981 | serotonin receptor 7 (Dugesia japonica) | BAI44327.1 | 2,00E-14 |
| 90_11349 | sex peptide receptor (Tribolium castaneum) | NP_001106940.1 | 5,00E-25 |
| P02HBR62 | SIFamide receptor (Apis mellifera) | NP_001106756.1 | 9,00E-10 |
| 90_19415 | parathyroid hormone-related peptide receptor precursor (Tribolium castaneum) | XP_969953.1 | 7,00E-20 |
| P02FKOY5 | parathyroid hormone receptor 2, isoform CRA_c (Mus musculus) | EDL00229.1 | 3,00E-07 |
| 90_1140 | somatostatin receptor (Culex quinquefasciatus) | XP_001859671.1 | 7,00E-43 |
| P02JZNDR | tachykinin receptor 1 (Mus musculus) | NP_033339.2 | 2,00E-06 |
| P02FL51R | thyroid hormone receptor (Schistosoma mansoni) | XP_002573733.1 | 2,00E-23 |
| P02FHDMB | thyroid stimulating hormone receptor precursor (Canis lupus familiares) | NP_001003285.1 | 5,00E-04 |
| 90_3545 | thyrotropin-releasing hormone receptor 1 (Catostomus commersonii) | AAG31763.1 | 2,00E-51 |
| 90_26294 | tyramine receptor (Bombyx mori) | BAD11157.1 | 1,00E-11 |
Homeobox-containing sequences in Schmidtea mediterranea
Since the first homeobox-containing genes were characterized in planarians [38], a large number of Hox and ParaHox genes that could be accommodated into the classical series of paralogous groups from Plhox1 to Plohox-9 and Xlox to cad/Cdx [39,40] have been described. Some of them show a differentially axial nested expression; while others are ubiquitously expressed [41-43]. Most of this work has been done in the planarians Girardia tigrina and Dugesia japonica. Recently, the first expression of an S. mediterranea Hox gene has been reported [44]. We identified 50 contigs and singletons with significant sequence similarity to homeobox gene sequences in the annotated Smed545 dataset (Table 4), including Hox genes and homeobox-containing genes, some already characterized in other planarian species.
Table 4.
Complete list of homeobox-containing gene sequence candidates.
| ID | BLASTX HIT | ACCESSION NUMBER | E-VALUE |
|---|---|---|---|
| F6AJIXP02J3PG4 | arrowhead [Schistosoma mansoni] | XP_002575389 | 6,00E-09 |
| 90_9219 | barh homeobox protein [Schistosoma mansoni] | XP_002571667 | 9,00E-26 |
| F6AJIXP02J2YIH | brain-specific homeobox [Tribolium castaneum] | EFA05724 | 5,00E-05 |
| 90_23337 | cut, isoform C [Drosophila melanogaster] | NP_001138174 | 3,00E-18 |
| F6AJIXP02HF7ZO | cut, isoform C [Drosophila melanogaster] | NP_001138174 | 2,00E-10 |
| 90_8368 | Cut-like homeobox 1 [Mus musculus] | AAH14289 | 2,00E-23 |
| 90_3019 | distalless, Dlx-1 [Platynereis dumerilii] | CAJ38799 | 8,00E-07 |
| 90_14605 | DjotxB [Dugesia japonica] | BAF80446 | 4,00E-65 |
| F6AJIXP02FICZL | Eye absent protein [Dugesia japonica] | CAD89530 | 1,00E-74 |
| F6AJIXP02IV6Y0 | gsx family homeobox protein [Schistosoma mansoni] | XP_002574396 | 3,00E-12 |
| 90_24312 | H6-like-homeobox [Drosophila melanogaster] | NP_732244 | 2,00E-15 |
| 90_8293 | homeobox protein distal-less dlx [Schistosoma mansoni] | XP_002574393 | 4,00E-07 |
| F6AJIXP02JJ1QK | Homeobox protein DTH-2 [Girardia tigrina] | Q00401 | 3,00E-40 |
| 90_8753 | homeobox prox 1 [Danio rerio] | NP_956564 | 5,00E-19 |
| 90_12057 | homeodomain protein Tlx [Capitella teleta] | ACH89436 | 1,00E-23 |
| 90_8083 | Hox class homeodomain protein AbdBa Hox protein [Schmidtea mediterranea]. | ABW79872 | 1,00E-26 |
| 90_7618 | Hox class homeodomain protein DjAbd-Ba [Dugesia japonica] | BAB41079 | 2,00E-16 |
| 90_6369 | Hox class homeodomain protein DjAbd-Bb [Dugesia japonica] | BAB41078 | 1,00E-108 |
| F6AJIXP02ILMDY | Hox class homeodomain protein DjAbd-Bb [Dugesia japonica] | BAB41078 | 3,00E-33 |
| F6AJIXP02HN15J_2 | Hypothetical protein CBG18604 [Caenorhabditis briggsae] | XP_002638395 | 7,00E-05 |
| 90_28860 | ladybird homeobox corepressor 1-like protein [Mus musculus | NP_001103213 XP_001479028 | 8,00E-33 |
| 90_6629 | lim domain binding protein [Schistosoma mansoni] | XP_002576324 | 6,00E-05 |
| F6AJIXP02GEYYP | lim domain homeobox 3/4 transcription factor [Saccoglossus kowalevskii) | NP_001158395 | 4,00E-23 |
| 90_10783 | lim homeobox protein [Schistosoma mansoni] | XP_002579046 | 2,00E-13 |
| 90_11027 | lim homeobox protein [Schistosoma mansoni] | XP_002579046 | 1,00E-26 |
| 90_10828 | LIM homeobox transcription factor 1 alpha [Mus musculus] | EDL39177 | 2,00E-14 |
| 90_13775 | LIM motif-containing protein kinase 1 [Schistosoma japonicum] | CAX72746 | 2,00E-11 |
| 90_9432 | LIM-homeodomain protein AmphiLim1/5 [Branchiostoma floridae] | ABD59002 | 5,00E-05 |
| 90_8762 | LIM-homeodomain transcription factor islet [Branchiostoma floridae | AAF34717 | 2,00E-15 |
| 90_6339 | Nk1 protein [Platynereis dumerilii] | CAJ38797 | 1,00E-11 |
| F6AJIXP02G077U | paired-like homeobox 2a [Danio rerio] | NP_996953 | 5,00E-16 |
| 90_6703 | phtf [Drosophila melanogaster] | NP_610232 | 2,00E-55 |
| 90_25126 | PLOX2-Dj [Dugesia japonica] | BAA77402 | 2,00E-42 |
| 90_21567 | PLOX4-Dj [Dugesia japonica] | BAA77404 | 2,00E-21 |
| 90_23010 | PLOX5-Dj [Dugesia japonica] | BAA77405 | 6,00E-22 |
| F6AJIXP02IVOTI | PLOX5-Dj [Dugesia japonica] | BAA77405 | 1,00E-17 |
| 90_21710 | pre-B-cell leukemia transcription factor 1 2 3 4 (pbx) [Schistosoma mansoni) | XP_002572195 | 2,00E-27 |
| 90_3405 | PREDICTED: similar to UBX domain protein 4 [Hydra magnipapillata] | XP_002162754 | 2,00E-06 |
| F6AJIXP02HI24E | PREP homeodomain-like protein [Schmidtea mediterranea] | ADB54565 | 2,00E-47 |
| F6AJIXP02JSRJD | PREP homeodomain-like protein [Schmidtea mediterranea] | ADB54565 | 1,00E-32 |
| F6AJIXP02GVFDM | prospero-like protein [Schistosoma mansoni] | XP_002578694 | 1,00E-21 |
| F6AJIXP02IUJ5Q | prospero-like protein [Schistosoma mansoni] | XP_002578694 | 4,00E-25 |
| F6AJIXP02HZIDG | short stature homeobox protein 2 isoform c [Homo sapiens | NP_001157150 | 5,00E-08 |
| 90_7545 | SIX homeobox 2 [Gallus gallus] | NP_001038160 | 7,00E-36 |
| F6AJIXP02HBGHT | SJCHGC06100 protein [Schistosoma japonicum] | AAW24487 | 6,00E-11 |
| 90_3395 | UBX domain containing 8, isoform CRA_d [Mus musculus] | EDL41153 | 3,00E-14 |
| 90_1176 | UBX domain-containing protein 4 [Mus musculus] | NP_080666 | 1,00E-06 |
| 90_2625 | ubx6(yeast)-related [Schistosoma mansoni] | XP_002576054 | 2,00E-16 |
| 90_24438 | visual system homeobox protein [Tribolium castaneum] | CAX64460 | 9,00E-23 |
| F6AJIXP02G5JJX_1 | Zn finger homeodomain 2 [Tribolium castaneum] | EFA01350 | 1,00E-05 |
Eye genes in Schmidtea mediterranea
The structural simplicity of the planarian eye in conjunction with the regenerative abilities of these organisms provides a unique system for dissecting the genetic mechanisms that allow a simple visual structure to be built [45,46]. Despite great morphological differences, there is evidence that the early morphogenesis of animal eyes requires the regulatory activity of Pax6, Sine oculis (Six), Eyes absent (Eya) and Dachshund (Dach), a gene network known as the retinal determination gene network (RDGN) [47-50]. Most of the genetic elements of the RDGN have been characterized in planarians [51-54]. In addition, the following planarian genes have been identified as being involved in eye regeneration: Djeye53, Dj1020HH [55]; Smed-netR, Smed-netrin2 [56]; Gt/Smed/Dj ops [46,57]; Djsnap-25 [58]; and Smednos [59]. In order to characterize new S. mediterranea eye network genes, we analyzed the Smed454 annotated dataset and found a collection of genes, ranging from transcription factors to eye-realizator genes, which have been implicated in eye development in other systems. These are good candidates for expanding our knowledge about the genetic network responsible for planarian eye regeneration (Table 5 and Additional File 9).
Table 5.
List of eye-related gene sequence candidates.
| ID | BLASTX HIT | ACCESSION Nr. | E-VALUE |
|---|---|---|---|
| 90_7233 | abl interactor 2 [Schistosoma japonicum] | CAX69750.1 | 6.00E-019 |
| 90_4001 | adaptor-related protein complex [Schistosoma mansoni] | XP_002574891.1 | 3.00E-072 |
| 90_30923 | arginine/serine-rich splicing factor [Schistosoma mansoni] | XP_002574990.1 | 2.00E-026 |
| 90_482 | ATPase protein [Schistosoma japonicum] | AAW26203.1 | 3.00E-049 |
| 90_3152 | beta-catenin-like protein 2 [Schmidtea mediterranea] | ABW79874.1 | 0 |
| 90_12909 | BMP [Schmidtea mediterranea] | ABV04322.1 | 3.00E-090 |
| 90_120 | cat eye syndrome protein [Schistosoma japonicum] | AAX27345.2 | 4.00E-035 |
| P02FKNEB | CaTaLase family member (ctl-2) [Caenorhabditis elegans] | NP_001022473.1 | 1.00E-029 |
| 90_205 | Chaperonin Containing TCP-1 family member (cct-3) [Caenorhabditis elegans] | NP_494218.2 | 1.00E-090 |
| C90_6158 | disks large homolog 1 isoform 1 [Homo sapiens] | NP_001091894.1 | 2.00E-027 |
| P02GJNCV | extradenticle 1 protein [Schistosoma japonicum] | AAW24487.1 | 3.00E-013 |
| P02JKJ4Z_2 | eye53 [Dugesia japonica] | BAD20650.1 | 6.00E-016 |
| 90_8483 | eyes absent protein [Dugesia japonica] | CAD89531.1 | 2.00E-064 |
| 90_651f | ascin protein [Schistosoma japonicum] | XP_002574990.1 | 5.00E-045 |
| 90_14368 | heat shock protein 70 [Lumbricus terrestris] | ACB77918.1 | 4.00E-038 |
| 90_9533 | Heparan sulfate 6-O-sulfotransferase 2 [Danio rerio] | AAH45453.1 | 1.00E-042 |
| 90_6564 | histone-lysine n-methyltransferase suv9 [Schistosoma mansoni] | XP_002574171.1 | 3.00E-061 |
| 90_15456 | homeodomain protein NK4 [Platynereis dumerilii] | ABQ10640.1 | 8.00E-023 |
| 90_12892 | homeotic protein six3-alpha [Mus musculus] | S74256 | 1.00E-082 |
| 90_325 | importin-7 [Culex quinquefasciatus] | XP_001843364.1 | 2.00E-147 |
| 90_4360 | intraflagellar transport 57 homolog [Xenopus (Silurana) tropicalis] | NP_001016561.1 | 1.00E-044 |
| 90_11027 | lim homeobox protein [Schistosoma mansoni] | XP_002579046.1 | 5.00E-027 |
| 90_8432 | lozenge [Schistosoma mansoni] | XP_002580418.1 | 8.00E-032 |
| 90_8924 | Male ABnormal family member (mab-21) [Caenorhabditis elegans] | NP_497940.2 | 1.00E-046 |
| P02HSHWR | mothers against decapentaplegic homolog 4 [Mus musculus] | NP_032566.2 | 5.00E-018 |
| 90_5640 | muscleblind-like protein [Schistosoma mansoni] | XP_002575346.1 | 3.00E-025 |
| P02F0EF6 | neurogenic differentiation [Platynereis dumerilii] | CAQ57533.1 | 2.00E-012 |
| P02GMLJM | nuclear transcription factor X-box binding 1 (nfx1) [Schistosoma bovis] | XP_002577564.1 | 5.00E-014 |
| 90_828 | phenylalanine hydroxylase [Caenorhabditis elegans] | AAD31643.1 | 2.00E-145 |
| 90_2925 | protein [Schistosoma japonicum] | AAW24487.1 | 4.00E-126 |
| 90_7228 | protein kinase [Schistosoma mansoni] | XP_002576342.1 | 2.00E-077 |
| 90_2256 | Protein pob [Schistosoma japonicum] | CAX75988.1 | 4.00E-089 |
| 90_4436 | Rab-protein 6 [Drosophila melanogaster] | NP_477172.1 | 8.00E-085 |
| P02GENUT_1 | retinaldehyde dehydrogenase 1 [Eleutherodactylus coqui] | ACE74542.1 | 8.00E-008 |
| 90_11988 | runt protein [Branchiostoma lanceolatum] | AAN08565.1 | 4.00E-017 |
| P02FN7BT | Septin-7 (CDC10 protein homolog) [Schistosoma japonicum] | CAX83064.1 | 3.00E-012 |
| 90_3747 | serine/threonine protein kinase [Schistosoma mansoni] | XP_002580180.1 | 9.00E-094 |
| P02FICZL | six1-2 protein [Dugesia japonica] | CAD89530.1 | 8.00E-86 |
| P02IZDJZ_1 | SRY-related HMG box B protein [Platynereis dumerilii] | CAY12631.1 | 3.00E-028 |
| P02HE4J6 | strabismus protein CBR-VANG-1 [Platynereis dumerilii] | CAJ26300.1 | 1.00E-006 |
| 90_9483 | tetratricopeptide repeat protein 10 tpr10 [Schistosoma mansoni] | XP_002573898.1 | 4.00E-048 |
| 90_16088 | tyrosine kinase [Schistosoma mansoni] | XP_002576978.1 | 2.00E-031 |
| 90_11388 | ubiquitin conjugating enzyme E2 [Schistosoma mansoni] | XP_002578016.1 | 3.00E-053 |
| 90_1263 | vacuolar ATP synthase proteolipid subunit 1 2 3 [Schistosoma japonicum] | XP_002571892.1 | 9.00E-049 |
| 90_12567 | vermilion [Drosophila ananassae] | XP_001963597.1 | 2.00E-012 |
| 90_5500 | white pigment protein [Drosophila melanogaster] | CAA26716.2 | 2.00E-020 |
| 90_13309 | YY1 transcription factor [Schistosoma japonicum] | CAX73893.1 | 5.00E-049 |
| 90_10118 | zinc finger protein 42 homolog [Homo sapiens] | NP_777560.2 | 6.00E-031 |
| 90_9460 | 14-3-3 zeta isoform [Schistosoma bovis] | AAT39382.1 | 2.00E-023 |
| P02ILIK3 | 52-kD bracketing protein [Drosophila melanogaster] | CAA44483.1 | 1.00E-016 |
Conclusions
The inherent complexity of the planarian genome and methodological difficulties initially prevented the complete genome assembly of S. mediterranea. High-throughput sequencing technologies are now well established and help molecular biologists to unravel the molecular components of organisms. We present a 454 sequencing dataset that can be used to decipher the transcriptome of the planarian S. mediterranea, an organism that has great potential for the study of regeneration processes.
We obtained more than half a million sequencing reads and assembled them into different datasets using a number of different similarity thresholds. The complete dataset has been made publicly available via web [21]. About 50,000 contigs in one of those sets (90e) were mapped against the most up-to-date genome scaffolds and to the set of known proteins from NCBI NR. Interestingly, we found a large number of transcribed sequences not covered by the genome sequence (more than 3 Mbp). The novel 454 contigs will allow us to extend current genomic sequences and connect up to 8,000 pairs of genome scaffolds. Furthermore, a preliminary analysis of the planarian splice sites was made on a collection of 454 contigs mapped univocally to the genome. Annotation of the sequences yielded a number of gene candidates in different functional categories that will be useful for further experimental studies. However, many of the novel contigs have no similarity to known proteins and will require further validation if we want to understand the transcriptional inventory of the planarian at a functional level. We also provided a preliminary gene annotation for S. mediterranea, focusing our rankings on four different gene families; these serve as applied examples of the usefulness of this new sequence resource.
Methods
Animals and RNA isolation
Schmidtea mediterranea from the BCN-10 clonal line were used. Animals were starved one week prior to experiments and irradiated at a lethal dose of 100Gy. Total RNA was isolated from a mixed sample of planarians that contained non-irradiated intact and regenerating planarians (1, 3, 5 and 7 days of regeneration) as well as irradiated intact and regenerating animals (1, 3, 5 and 7 days of regeneration). RNA was extracted with TRIzol® (Invitrogen) following the manufacturer's instructions.
cDNA library construction and 454 sequencing
First, 5 μg of total RNA was used to construct a cDNA library. RNA quality was assessed in a Bioanalyzer 2100 (Agilent-Bonsai Technologies). 5 μg of full-length double-stranded cDNA was then processed by the standard Genome Sequencer library-preparation method using the 454 DNA Library Preparation Kit (Titanium chemistry) to generate single-stranded DNA ready for emulsion PCR (emPCR™). The cDNA library was then nebulized according to the fragmentation process used in the standard Genome Sequencer shotgun library preparation procedure. The cDNA library was sequenced according to GS FLX technology (454/Roche). Reads were assembled by MIRA[60] version 3 using enhanced 454 parameters.
Mapping to genomic and functional annotation
BLAT[61] was used with default parameters to map the Smed454 90e dataset on the S. mediterranea draft genome assembly v3.1 [14] since the 454 sequences should be very similar to the corresponding genomic sequences, except for the lack of introns. Perl scripts were developed to classify all HSPs into the categories shown in Figure 3. 90e contigs having two or more collinear HSPs covering more than 100bp of the contig, and for which HSPs had more than 90% identity to the genomic contigs and length of the HSP larger than 50 bp, were chosen as 1-to-1 matches to genome. Once the sequences of the 90e/genomic contig pairs were retrieved, exonerate[62] was used to refine the alignments over the splice sites (using as parameters model = est2genome and bestn = 10). Perl scripts were used to retrieve the splice sites coordinates from exonerate output, as well as the sequences from genomic contigs. After clipping the donor and acceptor splice sites for each intron, nucleotide frequencies were computed and the corresponding position weight matrices for U2/U12 sites were drawn as pictograms using compi[19]. Known S. mediterranea genes were compared with contigs from 90e using BLASTN[63] with the following cut-offs: e-value = 0.001, identity score > 80%, HSP length > 50 bp.
GO functional annotation was computed on the BLASTX[63] results of the three assembly datasets (90, 98, and 90e) against all proteins from NCBI NR. BLASTX parameters were set to e-value = 10e-25 and maximum number of descriptions and alignments to report = 250, which produced around 26 million HSPs for each set. After that, only HSPs with a minimum length of 80 bp and a similarity score of at least 80% were considered. GO annotation was performed on those HSPs using the e-value selection criteria and supporting sequences described for Blast2GO[64]. Further Perl scripts were used to summarize the data shown in Table 2 and Additional File 3.
RT-PCR
In order to validate the expression of a random subset of novel 454 transcripts, RT-PCRs were performed on planarian cDNA generated with Superscript III (Invitrogen) following the manufacturer's instructions. Additional File 3 includes a list of the contigs validated and the primers used for each of them.
Prediction of transmembrane proteins from ESTs
A total of 53,867 assembled ESTs (90e database) and 2,495 additional mRNAs were translated into all six reading frames using the 'transeq' program from the EMBOSS package [65]. The longest open reading frame for each EST/mRNA was then extracted and used as a protein database (containing 56,362 protein sequences overall) for the prediction of membrane-spanning proteins. We followed an approach described by Almen et al. [66] basing our analysis on consensus predictions of alpha-helices and using three applications: Phobius[67], TMHMM2.0[68], and SOSUI[69]. Phobius and TMHMM2.0 both use hidden Markov models based on different training sets to predict membrane topology. SOSUI evaluates proteins for their hydrophobic and amphiphilic properties to make its predictions. The use of all three programs should improve prediction accuracy. We first ran Phobius, which can predict both transmembrane helices and signal peptides. Signal peptide sequences are similar to transmembrane segments owing to their hydrophobic nature [70]. To avoid false positive predictions, we excluded signal peptides before running TMHMM2.0 and SOSUI.
Abbreviations
bp: base pairs (nucleotide length unit); EST: Expressed Sequence Tag; GC%: percent of guanine+cytosine sequence content; HSP: High-scoring Segment Pair; GO: Gene Ontology; WUSL: Washington University in St Louis; TM: transmembrane; RDGN: retinal determination gene network; Gy: gray.
Authors' contributions
JFA performed the computational analyses on the assemblies, the GO characterization, the mapping into the genome and the analysis of spliced sites, and prepared all the corresponding figures and tables. GRE analyzed the coverage of known annotated genes and generated the corresponding table. TH performed the sequence analysis of planarian transmembrane proteins, generated the corresponding figure and table and designed primers for RT-PCRs. GRE, SF, FC and KB designed the primers and performed the RT-PCRs. FC and SF analyzed the annotated data to characterize the neurotransmitter, peptide and hormone receptors and prepared the corresponding tables. ES and BC analyzed the annotated data to characterize homeobox-containing and eye-related genes and prepared the corresponding tables. JFA, KB, FC and ES conceived of the study, participated in its design and coordination, and helped draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
GO annotation for 90e contigs not mapping onto the WUSL 3.1 genome assembly. 8,831 90e contigs were not found in the genome. 3,480 had a BLASTX hit to a sequence of NCBI NRprot; yet only 2,401 had a hit to a protein functionally annotated in the GO database. This file contains the description of the best HSP for 71 of those annotated contigs, after filtering out as described above. (Header: CONTIG ID = Smed454 sequence identifier, E-VALUE = BLASTX HSP E-value, ALN_SCORE = HSP alignment score, IDENTITIES = number of identical amino acids, POSITIVES = number of similar amino acids, SEQUENCE ID = Protein sequence identifier, ACCESSION NUMBER = Protein sequence full accession number, SEQUENCE DESCRIPTION = Full protein GenBank description).
Splice sites for a subset of Smed454 sequences mapped onto the Schmidtea mediterranea genome. (Header: GID = Genomic contig IDentifier from WUSLv3.1 genome assembly--including the start and end nucleotide coordinates for the complete match--, CIG=90e contig IDentifier, INTNUM = Intron number within the 90e contig, EXO = splice signals found by exonerate, ORI = sequence orientation--here -1 means that the match was found on the reverse strand of the genomic contig--, CEXO = corrected splice site signals after reverse complementing the genomic sequence when required, ILEN = Intron length in bp, IORI = Intron start--relative to the match coordinates--, IEND = Intron end--relative to the match coordinates--, STRAND, SSSEQ = Splice sites sequences--where a point separates three nucleotides from the 5' and 3' exons, and the three dots in the middle denote intron sequence not shown for clarity--).
List of 90e transcripts validated by RT-PCR. (Header: # = Number, CONTIG=90e contig ID, PRIMER_FORWARD = 5' to 3' sequence of the forward primer used, REVERSE_FORWARD = 5' to 3' sequence of the reverse primer used, AMPLICON SIZE = Size amplified in bp, SET = refers to the subset of origin of the 90e contig: no hit genome, hit genome, - blast (no BLASTX hit), +blast (BLASTX hit)).
Smed454 sequences matching known Schmidtea mediterranea genes. (Header: ACCESSION NUMBER = Known gene sequence identifier as target, NAME = Description for that sequence, LENGTH = Nucleotide length for that sequence, A&T CONTENT = Sequence composition, 454 90e CONTIG/SINGLETON = Smed454 sequence identifier as query, LENGTH = Nucleotide sequence length for this sequence, ALIGNMENT LENGTH = HSP length, START = Start nucleotide of alignment on target, END = Final nucleotide of alignment on target, IDENTITY = Identity score, BITSCORE = Alignment bit score, E-VALUE = HSP BLAST e-value, HIT LENGTH = Un-gapped length of the alignment on the target, %COVERAGE = Sum of co-linear HSPs on target coordinates divided by the total length of the target, #SEQs = Number of co-linear HSPs considered, avg%COV = The coverage divided by the number of co-linear HSPs).
Gene Ontology for all three Smed454 sets: 90, 98 and 90e. Level one and two GO codes are shown in order to simplify the listings. Although there are small changes in GO frequencies, annotation is consistent throughout all three sets. (Header: GO = Gene Ontology unique identifier, Count = Number of sequences with a given GO annotation, Freq% = Frequencies for every GO annotation. The total shown does not include the un-annotated and over-represented features, that is, the first two rows on each table).
List of cell cycle, cell division, DNA repair or DNA damage candidates. Short list of candidates annotated as genes involved in cell cycle, cell division, DNA repair or DNA damage. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).
Summary report for the consensus set of 4,663 predicted transmembrane proteins including functional annotations. (Header: Sequence_ID = Protein sequence identifier, Sequence_AA = Amino acid sequence, Length[aa] = Length of amino acid sequence, Phobius_TM = Phobius prediction of number of transmembrane domains, Phobius_SP = Phobius prediction of signal peptide, Phobius_Top = Phobius prediction of membrane topology, TMHMM_TM = TMHMM2.0 prediction of number of transmembrane domains, TMHMM_Top = TMHMMv2.0 prediction of membrane topology, SOSUI_TM = SOSUI prediction of number of transmembrane domains, SOSUI_Top = SOSUI prediction of membrane topology, UFO_PFAM = UFO annotation of Pfam protein families, UFO_GO = UFO annotation of gene ontologies).
List of neurotransmitter, peptide and hormone receptor sequence candidates. Complete complement of Smed454 dataset contigs and singletons showing homology to neurotransmitter and hormone receptors, totalling 287 sequences. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).
List of eye-related gene sequence candidates. Complete complement of Smed454 dataset contigs and singletons showing homology to eye-related genes, totalling 95 sequences. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).
Contributor Information
Josep F Abril, Email: jabril@ub.edu.
Francesc Cebrià, Email: fcebrias@ub.edu.
Gustavo Rodríguez-Esteban, Email: gustavorodriguez@ub.edu.
Thomas Horn, Email: t.horn@dkfz-heidelberg.de.
Susanna Fraguas, Email: susannafraguas@ub.edu.
Beatriz Calvo, Email: beatrix.9629@gmx.de.
Kerstin Bartscherer, Email: kerstin.bartscherer@mpi-muenster.mpg.de.
Emili Saló, Email: esalo@ub.edu.
Acknowledgements
Genomic sequence data was produced by the Washington University Genome Sequencing Center in St. Louis. DNA library construction, 454 pyrosequencing and the contig browser were generated by Skuldtech (Montpellier, France). We thank Ann King for language revision and editorial advice, and Franziska Konert for technical assistance. SF and GRE are supported by FPI fellowships, and BC is supported by an FPU fellowship (both MICINN, Spain). TH is supported by a fellowship of the Studienstiftung des deutschen Volkes. FC is a Ramón y Cajal researcher (MICINN, Spain). This work was funded by grants BFU2008-01544 (MICINN) to ES, BFU2008-00710 (MICINN) to FC, BFU2009-09102(MICINN) to JFA, and 2009SGR1018 (AGAUR) to ES and FC. Finally, K.B. was funded by a FRONTIER grant from the University of Heidelberg DFG Excellence Initiative.
References
- Agata K, Tanaka T, Kobayashi C, Kato K, Saitoh Y. Intercalary regeneration in planarians. Dev Dyn. 2003;226(2):308–316. doi: 10.1002/dvdy.10249. [DOI] [PubMed] [Google Scholar]
- Newmark PA, Sánchez Alvarado A. Not your father's planarian: a classic model enters the era of functional genomics. Nat Rev Genet. 2002;3(3):210–219. doi: 10.1038/nrg759. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Sánchez-Alvarado A. Fundamentals of planarian regeneration. Annu Rev Cell Dev Biol. 2004;20:725–757. doi: 10.1146/annurev.cellbio.20.010403.095114. [DOI] [PubMed] [Google Scholar]
- Saló E. The power of regeneration and the stem-cell kingdom: freshwater planarians (Platyhelminthes) Bioessays. 2006;28(5):546–559. doi: 10.1002/bies.20416. [DOI] [PubMed] [Google Scholar]
- Baguñà J, Saló E, Auladell C. Regeneration and pattern formation in planarians. III. Evidence that neoblasts are totipotent stem cells and the source of blastema cells. Development. 1989;107:77–86. [Google Scholar]
- Newmark PA, Sánchez-Alvarado A. Bromodeoxyuridine specifically labels the regenerative stem cells of planarians. Dev Biol. 2000;220(2):142–153. doi: 10.1006/dbio.2000.9645. [DOI] [PubMed] [Google Scholar]
- Cebrià F. Regenerating the central nervous system: how easy for planarians! Dev Genes Evol. 2007;217(11-12):733–748. doi: 10.1007/s00427-007-0188-6. [DOI] [PubMed] [Google Scholar]
- Scimone ML, Meisel J, Reddien PW. The Mi-2-like Smed-CHD4 gene is required for stem cell differentiation in the planarian Schmidtea mediterranea. Development. 2010;137(8):1231–1241. doi: 10.1242/dev.042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Taboada E, Moritz S, Zeuschner D, Stehling M, Scholer HR, Saló E, Gentile L. Smed-SmB, a member of the LSm protein superfamily, is essential for chromatoid body organization and planarian stem cell proliferation. Development. 2010;137(7):1055–1065. doi: 10.1242/dev.042564. [DOI] [PubMed] [Google Scholar]
- Forsthoefel DJ, Newmark PA. Emerging patterns in planarian regeneration. Curr Opin Genet Dev. 2009;19(4):412–420. doi: 10.1016/j.gde.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Alvarado A, Newmark PA, Robb SM, Juste R. The Schmidtea mediterranea database as a molecular resource for studying platyhelminthes, stem cells and regeneration. Development. 2002;129(24):5659–5665. doi: 10.1242/dev.00167. [DOI] [PubMed] [Google Scholar]
- Zayas RM, Hernandez A, Habermann B, Wang Y, Stary JM, Newmark PA. The planarian Schmidtea mediterranea as a model for epigenetic germ cell specification: analysis of ESTs from the hermaphroditic strain. Proc Natl Acad Sci USA. 2005;102(51):18491–18496. doi: 10.1073/pnas.0509507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius JU, Wagner L, Schuler GD. In: The NCBI Handbook. Information NCfB. Bethesda (MD), editor. 2003. UniGene: a unified view of the transcriptome. [Google Scholar]
- Schmidtea mediterranea genome sequencing project. http://genome.wustl.edu/genomes/view/schmidtea_mediterranea/
- Proposal for the Sequencing of a New Target Genome: White Paper for a Planarian Genome Project. http://genome.wustl.edu/images/uploads/Whitepaper/Schmidtea_mediterranea_WP.pdf
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sánchez-Alvarado A, Yandell M. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb SM, Ross E, Sánchez-Alvarado A. SmedGD: the Schmidtea mediterranea genome database. Nucleic Acids Res. 2008. pp. D599–606. [DOI] [PMC free article] [PubMed]
- Emrich SJ, Barbazuk WB, Li L, Schnable PS. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007;17(1):69–73. doi: 10.1101/gr.5145806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abril JF, Castelo R, Guigo R. Comparison of splice sites in mammals and chicken. Genome Res. 2005;15(1):111–119. doi: 10.1101/gr.3108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009. pp. D26–31. [DOI] [PMC free article] [PubMed]
- Smed454 datasets web site. http://planarian.bio.ub.es/datasets/454/
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield DL. Selenium: Its Molecular Biology and Role in Human Health. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- Castellano S, Novoselov SV, Kryukov GV, Lescure A, Blanco E, Krol A, Gladyshev VN, Guigo R. Reconsidering the evolution of eukaryotic selenoproteins: a novel nonmammalian family with scattered phylogenetic distribution. EMBO Rep. 2004;5(1):71–77. doi: 10.1038/sj.embor.7400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhoffer GT, Kang H, Sánchez-Alvarado A. Molecular analysis of stem cells and their descendants during cell turnover and regeneration in the planarian Schmidtea mediterranea. Cell Stem Cell. 2008;3(3):327–339. doi: 10.1016/j.stem.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L, Salvetti A, Marincola FM, Lena A, Deri P, Mannini L, Batistoni R, Wang E, Gremigni V. Deciphering the molecular machinery of stem cells: a look at the neoblast gene expression profile. Genome Biol. 2007;8(4):R62. doi: 10.1186/gb-2007-8-4-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinicke P. UFO: a web server for ultra-fast functional profiling of whole genome protein sequences. BMC Genomics. 2009;10:409. doi: 10.1186/1471-2164-10-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonuccelli L, Rossi L, Lena A, Scarcelli V, Rainaldi G, Evangelista M, Iacopetti P, Gremigni V, Salvetti A. An RbAp48-like gene regulates adult stem cells in planarians. J Cell Sci. 2010;123(Pt 5):690–698. doi: 10.1242/jcs.053900. [DOI] [PubMed] [Google Scholar]
- Guo T, Peters AH, Newmark PA. A Bruno-like gene is required for stem cell maintenance in planarians. Dev Cell. 2006;11(2):159–169. doi: 10.1016/j.devcel.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Pearson BJ, Sánchez-Alvarado A. A planarian p53 homolog regulates proliferation and self-renewal in adult stem cell lineages. Development. 2010;137(2):213–221. doi: 10.1242/dev.044297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Oviedo NJ, Jennings JR, Jenkin JC, Sánchez-Alvarado A. SmedWI-2 is a PIWI-like protein that regulates planarian stem cells. Science. 2005;310(5752):1327–1330. doi: 10.1126/science.1116110. [DOI] [PubMed] [Google Scholar]
- Franquinet R. [The role of serotonin and catecholamines in the regeneration of the Planaria Polycelis tenvis] J Embryol Exp Morphol. 1979;51:85–95. [PubMed] [Google Scholar]
- Franquinet R, Martelly I. Effects of serotonin and catecholamines on RNA synthesis in planarians; in vitro and in vivo studies. Cell Differ. 1981;10(4):201–209. doi: 10.1016/0045-6039(81)90002-6. [DOI] [PubMed] [Google Scholar]
- Franquinet R, Le Moigne A, Hanoune J. The adenylate cyclase system of planaria Polycelis tenuis: activation by serotonin and guanine nucleotides. Biochim Biophys Acta. 1978;539(1):88–97. doi: 10.1016/0304-4165(78)90124-1. [DOI] [PubMed] [Google Scholar]
- Baguñà J, Saló E, Romero R. Effects of activators and antagonists of the neuropeptides substance P and substance K on cell proliferation in planarians. Int J Dev Biol. 1989;33(2):261–266. [PubMed] [Google Scholar]
- Bautz A, Schilt J. Somatostatin-like peptide and regeneration capacities in planarians. Gen Comp Endocrinol. 1986;64(2):267–272. doi: 10.1016/0016-6480(86)90013-4. [DOI] [PubMed] [Google Scholar]
- Kreshchenko ND. Functions of flatworm neuropeptides NPF, GYIRF and FMRF in course of pharyngeal regeneration of anterior body fragments of planarian, Girardia tigrina. Acta Biol Hung. 2008;59(Suppl):199–207. doi: 10.1556/ABiol.59.2008.Suppl.29. [DOI] [PubMed] [Google Scholar]
- García-Fernández J, Baguñà J, Saló E. Planarian homeobox genes: cloning, sequence analysis, and expression. Proc Natl Acad Sci USA. 1991;88(16):7338–7342. doi: 10.1073/pnas.88.16.7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saló E, Baguñà J. Regeneration in planarians and other worms: New findings, new tools, and new perspectives. J Exp Zool. 2002;292(6):528–539. doi: 10.1002/jez.90001. [DOI] [PubMed] [Google Scholar]
- Saló E, Tauler J, Jiménez E, Bayascas JR, González J, García-Fernàndez J, Baguñà J. Hox and parahox genes in flatworms: characterization and expression. Amer Zool. 2001;41:652–663. [Google Scholar]
- Bayascas JR, Castillo E, Saló E. Platyhelminthes have a hox code differentially activated during regeneration, with genes closely related to those of spiralian protostomes. Dev Genes Evol. 1998;208(8):467–473. doi: 10.1007/s004270050204. [DOI] [PubMed] [Google Scholar]
- Nogi T, Watanabe K. Position-specific and non-colinear expression of the planarian posterior (Abdominal-B-like) gene. Dev Growth Differ. 2001;43(2):177–184. doi: 10.1046/j.1440-169X.2001.00564.x. [DOI] [PubMed] [Google Scholar]
- Orii H, Kato K, Umesono Y, Sakurai T, Agata K, Watanabe K. The planarian HOM/HOX homeobox genes (Plox) expressed along the anteroposterior axis. Dev Biol. 1999;210(2):456–468. doi: 10.1006/dbio.1999.9275. [DOI] [PubMed] [Google Scholar]
- Iglesias M, Gómez-Skarmeta JL, Saló E, Adell T. Silencing of Smed-betacatenin1 generates radial-like hypercephalized planarians. Development. 2008;135(7):1215–1221. doi: 10.1242/dev.020289. [DOI] [PubMed] [Google Scholar]
- Saló E, Batistoni R. Animal models in eye research. Tsonis PA: Academic Press; 2008. The planarian eye: a simple and plastic system with great regenerative capacity; pp. 15–26. [Google Scholar]
- Saló E, Pineda D, Marsal M, González J, Gremigni V, Batistoni R. Genetic network of the eye in Platyhelminthes: expression and functional analysis of some players during planarian regeneration. Gene. 2002;287:67–74. doi: 10.1016/s0378-1119(01)00863-0. [DOI] [PubMed] [Google Scholar]
- Gehring WJ. New perspectives on eye development and the evolution of eyes and photoreceptors. J Hered. 2005;96(3):171–184. doi: 10.1093/jhered/esi027. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Ikeo K. Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet. 1999;15(9):371–377. doi: 10.1016/S0168-9525(99)01776-X. [DOI] [PubMed] [Google Scholar]
- Silver SJ, Rebay I. Signaling circuitries in development: insights from the retinal determination gene network. Development. 2005;132(1):3–13. doi: 10.1242/dev.01539. [DOI] [PubMed] [Google Scholar]
- Treisman JE. A conserved blueprint for the eye? Bioessays. 1999;21(10):843–850. doi: 10.1002/(SICI)1521-1878(199910)21:10<843::AID-BIES6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Mannini L, Rossi L, Deri P, Gremigni V, Salvetti A, Saló E, Batistoni R. Djeyes absent (Djeya) controls prototypic planarian eye regeneration by cooperating with the transcription factor Djsix-1. Dev Biol. 2004;269(2):346–359. doi: 10.1016/j.ydbio.2004.01.042. [DOI] [PubMed] [Google Scholar]
- Pineda D, González J, Callaerts P, Ikeo K, Gehring WJ, Saló E. Searching for the prototypic eye genetic network: Sine oculis is essential for eye regeneration in planarians. Proc Natl Acad Sci USA. 2000;97(9):4525–4529. doi: 10.1073/pnas.97.9.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda D, González J, Marsal M, Saló E. Evolutionary conservation of the initial eye genetic pathway in planarians. Belg J Zool. 2001;131:77–82. [Google Scholar]
- Pineda D, Rossi L, Batistoni R, Salvetti A, Marsal M, Gremigni V, Falleni A, González-Linares J, Deri P, Saló E. The genetic network of prototypic planarian eye regeneration is Pax6 independent. Development. 2002;129(6):1423–1434. doi: 10.1242/dev.129.6.1423. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kumamoto H, Okamoto K, Umesono Y, Sakai M, Sánchez-Alvarado A, Agata K. Morphological and functional recovery of the planarian photosensing system during head regeneration. Zoolog Sci. 2004;21(3):275–283. doi: 10.2108/zsj.21.275. [DOI] [PubMed] [Google Scholar]
- Cebrià F, Newmark PA. Planarian homologs of netrin and netrin receptor are required for proper regeneration of the central nervous system and the maintenance of nervous system architecture. Development. 2005;132(16):3691–3703. doi: 10.1242/dev.01941. [DOI] [PubMed] [Google Scholar]
- Sánchez Alvarado A, Newmark PA. Double-stranded RNA specifically disrupts gene expression during planarian regeneration. Proc Natl Acad Sci USA. 1999;96(9):5049–5054. doi: 10.1073/pnas.96.9.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Pulvers JN, Inoue T, Tarui H, Sakamoto H, Agata K, Umesono Y. Regeneration-dependent conditional gene knockdown (Readyknock) in planarian: Demonstration of requirement for Djsnap-25 expression in the brain for negative phototactic behavior. Dev Growth Differ. 2007;49(5):383–394. doi: 10.1111/j.1440-169X.2007.00936.x. [DOI] [PubMed] [Google Scholar]
- Handberg-Thorsager M, Saló E. The planarian nanos-like gene Smednos is expressed in germline and eye precursor cells during development and regeneration. Dev Genes Evol. 2007;217(5):403–411. doi: 10.1007/s00427-007-0146-3. [DOI] [PubMed] [Google Scholar]
- Chevreux B. MIRA: An automated genome and EST assembler. Duisburg: Heidelberg; 2005. [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- exonerate: A generic tool for sequence alignment. http://www.ebi.ac.uk/~guy/exonerate/
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talón M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Almen MS, Nordstrom KJ, Fredriksson R, Schioth HB. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009;7:50. doi: 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic Acids Res. 2007. pp. W429–432. [DOI] [PMC free article] [PubMed]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14(4):378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- Ahram M, Litou ZI, Fang R, Al-Tawallbeh G. Estimation of membrane proteins in the human proteome. In Silico Biol. 2006;6(5):379–386. [PubMed] [Google Scholar]
- Hintze JL, Nelson RD. Violin plots: a box plot-density trace synergism. The American Statistician. 1998;52(2):181–184. doi: 10.2307/2685478. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GO annotation for 90e contigs not mapping onto the WUSL 3.1 genome assembly. 8,831 90e contigs were not found in the genome. 3,480 had a BLASTX hit to a sequence of NCBI NRprot; yet only 2,401 had a hit to a protein functionally annotated in the GO database. This file contains the description of the best HSP for 71 of those annotated contigs, after filtering out as described above. (Header: CONTIG ID = Smed454 sequence identifier, E-VALUE = BLASTX HSP E-value, ALN_SCORE = HSP alignment score, IDENTITIES = number of identical amino acids, POSITIVES = number of similar amino acids, SEQUENCE ID = Protein sequence identifier, ACCESSION NUMBER = Protein sequence full accession number, SEQUENCE DESCRIPTION = Full protein GenBank description).
Splice sites for a subset of Smed454 sequences mapped onto the Schmidtea mediterranea genome. (Header: GID = Genomic contig IDentifier from WUSLv3.1 genome assembly--including the start and end nucleotide coordinates for the complete match--, CIG=90e contig IDentifier, INTNUM = Intron number within the 90e contig, EXO = splice signals found by exonerate, ORI = sequence orientation--here -1 means that the match was found on the reverse strand of the genomic contig--, CEXO = corrected splice site signals after reverse complementing the genomic sequence when required, ILEN = Intron length in bp, IORI = Intron start--relative to the match coordinates--, IEND = Intron end--relative to the match coordinates--, STRAND, SSSEQ = Splice sites sequences--where a point separates three nucleotides from the 5' and 3' exons, and the three dots in the middle denote intron sequence not shown for clarity--).
List of 90e transcripts validated by RT-PCR. (Header: # = Number, CONTIG=90e contig ID, PRIMER_FORWARD = 5' to 3' sequence of the forward primer used, REVERSE_FORWARD = 5' to 3' sequence of the reverse primer used, AMPLICON SIZE = Size amplified in bp, SET = refers to the subset of origin of the 90e contig: no hit genome, hit genome, - blast (no BLASTX hit), +blast (BLASTX hit)).
Smed454 sequences matching known Schmidtea mediterranea genes. (Header: ACCESSION NUMBER = Known gene sequence identifier as target, NAME = Description for that sequence, LENGTH = Nucleotide length for that sequence, A&T CONTENT = Sequence composition, 454 90e CONTIG/SINGLETON = Smed454 sequence identifier as query, LENGTH = Nucleotide sequence length for this sequence, ALIGNMENT LENGTH = HSP length, START = Start nucleotide of alignment on target, END = Final nucleotide of alignment on target, IDENTITY = Identity score, BITSCORE = Alignment bit score, E-VALUE = HSP BLAST e-value, HIT LENGTH = Un-gapped length of the alignment on the target, %COVERAGE = Sum of co-linear HSPs on target coordinates divided by the total length of the target, #SEQs = Number of co-linear HSPs considered, avg%COV = The coverage divided by the number of co-linear HSPs).
Gene Ontology for all three Smed454 sets: 90, 98 and 90e. Level one and two GO codes are shown in order to simplify the listings. Although there are small changes in GO frequencies, annotation is consistent throughout all three sets. (Header: GO = Gene Ontology unique identifier, Count = Number of sequences with a given GO annotation, Freq% = Frequencies for every GO annotation. The total shown does not include the un-annotated and over-represented features, that is, the first two rows on each table).
List of cell cycle, cell division, DNA repair or DNA damage candidates. Short list of candidates annotated as genes involved in cell cycle, cell division, DNA repair or DNA damage. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).
Summary report for the consensus set of 4,663 predicted transmembrane proteins including functional annotations. (Header: Sequence_ID = Protein sequence identifier, Sequence_AA = Amino acid sequence, Length[aa] = Length of amino acid sequence, Phobius_TM = Phobius prediction of number of transmembrane domains, Phobius_SP = Phobius prediction of signal peptide, Phobius_Top = Phobius prediction of membrane topology, TMHMM_TM = TMHMM2.0 prediction of number of transmembrane domains, TMHMM_Top = TMHMMv2.0 prediction of membrane topology, SOSUI_TM = SOSUI prediction of number of transmembrane domains, SOSUI_Top = SOSUI prediction of membrane topology, UFO_PFAM = UFO annotation of Pfam protein families, UFO_GO = UFO annotation of gene ontologies).
List of neurotransmitter, peptide and hormone receptor sequence candidates. Complete complement of Smed454 dataset contigs and singletons showing homology to neurotransmitter and hormone receptors, totalling 287 sequences. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).
List of eye-related gene sequence candidates. Complete complement of Smed454 dataset contigs and singletons showing homology to eye-related genes, totalling 95 sequences. (Header: ID = Smed454 sequence identifier, BLASTX HIT = Description of the best sequence hit, ACCESSION NUMBER = Sequence identifier of the best sequence hit, E-VALUE = BLASTX e-value for that sequence hit).