

Abstract
Purpose
NRAS and BRAF mutations are common in cutaneous melanomas, although rarely detected mutually in the same tumor. Distinct clinical correlates of these mutations have not been described, despite in vitro data suggesting enhanced oncogenic effects. This study was designed to test the hypothesis that primary human cutaneous melanomas harboring mutations in NRAS or BRAF display a more aggressive clinical phenotype than tumors wild type at both loci.
Experimental Design
Microdissection of 223 primary melanomas was carried out, followed by determination of the NRAS and BRAF mutational status. Genotypic findings were correlated with features known to influence tumor behavior, including age, gender, Breslow depth, Clark level, mitotic rate, the presence of ulceration, and AJCC staging.
Results
Breslow depth and Clark level varied significantly among the genotypes, with NRAS mutants showing the deepest levels and wild type tumors the least depth. Ulceration also differed significantly among the genotypes, with BRAF mutants demonstrating the highest rate. Additionally, tumors with mutated NRAS were more likely to be located on the extremities. Patients whose tumors carried either mutation presented with more advanced AJCC stages compared to patients with wild type tumors, and specifically, were more likely to have Stage III disease at diagnosis. Overall survival did not differ among the three groups.
Conclusions
Distinct clinical phenotypes exist for melanomas bearing NRAS and BRAF mutations, whether considered together or separately, and are associated with features known to predict aggressive tumor behavior. The impact of these mutations is most evident at earlier stages of disease progression.
Keywords: melanoma, NRAS, BRAF, mutation, phenotype
Introduction
Cutaneous melanomas have long been known to harbor activating mutations of the NRAS gene in 15–20% of primary tumors (1). Although definitive clinical correlates of mutated NRAS have not been established in the melanoma patient population, at least one study has suggested aggressive behavior of this melanoma subset (2). The most common site of mutation is codon 61 in exon 2, although exon 1 mutations are occasionally found (1–3). The codon 61 mutations are heterogeneous, with C181A (Q61K) and A182G (Q61R) found most frequently. There is in vitro evidence to suggest that codon 61 mutations result in prolongation of the GTP-bound state of N-Ras, leading to enhanced signaling through growth-promoting pathways such as the mitogen-activated protein kinase (MAPK) pathway (4).
In this context, considerable excitement was generated some years ago when activating mutations of BRAF, which encodes the kinase immediately downstream of N-Ras, were reported in 60% of cultured melanoma cell lines and 50% of primary human tumors (5, 6). These mutations for the most part consist of T1799A (V600E) in exon 15 and are rarely found in melanomas bearing NRAS mutations. Contrary to expectations, clinical studies have been largely unsuccessful in demonstrating a clear phenotype associated with mutated BRAF (7–10). Adding to the debate surrounding the clinical relevance of BRAF mutations in primary melanoma is the observation that these mutations occur in benign nevi at a similar or greater frequency (11).
In an attempt to further clarify the impact of mutated NRAS and BRAF on clinical melanoma behavior, we determined the mutational status of 223 primary tumors and correlated these findings with pathologic and clinical data. Here, we report that NRAS- and BRAF-mutated tumors are more invasive than tumors wild type at both loci. Furthermore, patients with tumors carrying either mutation are more likely to present with stage III disease.
Materials and Methods
Study population
The study was approved by The University of Texas M. D. Anderson Cancer Center (MDACC) Institutional Review Board and conducted according to the Health Insurance Portability and Accountability Act (HIPAA) guidelines. A written informed consent was obtained from all subjects. Tumor samples consisted of consecutive entries into the MDACC Melanoma Informatics, Tissue Resource, and Pathology Core (Melanoma Tumor Bank) as part of a larger MDACC Melanoma Specialized Program of Research Interest (SPORE) project.
DNA extraction and amplification
For each primary tumor, three 5 micron-thick, formalin-fixed, paraffin-embedded tissue sections were prepared for laser capture microdissection (LCM) as previously described (12). Microdissection of tumor cells was performed by a board-certified pathologist (VRG) using the PixCell II Laser Capture Microdissection System (Arcturus, Mountain View, Ca). Dissected cells were incubated for 72 hours at 37°C in 50–100 ul of lysis buffer (10 mM Tris-HCl pH 8.0, 1% Tween-20, 1 mM EDTA, and 0.04% proteinase K). After a 5-minute high-speed centrifugation, the sample was heated to 95°C for 8 minutes to inactivate the proteinase K. PCR was performed using the GeneAmp Gold PCR Reagent Kit (Applied Biosystems, Foster City, CA). Primers and conditions for NRAS exon 2 and BRAF exon 15 amplification (Sigma Genosys, The Woodlands, TX) are listed below (5, 13):
-
NRAS exon 2:Forward: 5’-CCCCTTACCCTCCACAC-3’
Reverse: 5’-AGGTTAATATCCGCAAATGAC-3
95 °C 45 s, 55 °C 45 s, 72°C 60 s; 40 cycles; magnesium chloride 1.5 mM
-
BRAF exon 15:Forward: 5’-TCATAATGCTTGCTCTGATAGGA-3’
Reverse: 5’-GGCCAAAAATTTAATCAGTGGA-3’
94 °C 30 s, 57 °C 60 s, 72 °C 60 s; 40 cycles; magnesium chloride 1.5 mM
PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Sequencing of the PCR products in both directions was performed by the MDACC DNA Core Facility using an ABI Prism 3100 DNA Genetic Analyzer (Applied Biosystems) using Big Dye v.3.1 dye terminator chemistry (Applied Biosystems). Chromatogram results were interpreted independently by two reviewers (JAE and VRG). All mutations were identified on both strands.
Statistical analysis
Associations with mutation types were evaluated using the three genotypes (mutated NRAS, mutated BRAF, and both wild type), as well as mutational status collapsed into two categories (either mutation vs. no mutation). Fisher’s exact test was used to examine associations between mutation status and the following factors: patient gender, Breslow thickness ≥ 1 mm, Clark level, ulceration, and AJCC staging. The associations between genotype and ordinal variables (patient age, Breslow depth, and mitotic rate) were determined using the Kruskal-Wallis test. Overall survival was computed from the date of pathologic diagnosis until the date of death. Patients alive at the end of the study period were censored at the date of last follow-up. Survival curves were constructed using the Kaplan Meier method and the log-rank test was used to evaluate equality across strata. Associations between independent variables and survival were further investigated using Cox proportional hazards regression models. All p-values were two-sided and p-values < 0.05 were considered statistically significant. Analyses were conducted using SAS for Windows (release 9.1, SAS Institute, Cary, North Carolina, USA).
Results
Patient population
A total of 297 primary melanomas were microdissected, representing consecutive entries into the MDACC Melanoma Tumor Bank. Of this initial set, sequences for both BRAF exon 15 and NRAS exon 2 were successfully determined in 223 cases. The remaining 74 samples were excluded from analysis for the following reasons: one or both exons could not be adequately amplified or sequenced (39 cases); the material remaining in the tissue block did not show invasive melanoma (21 cases); the tumor was too small for microdissection (14 cases).
Based on the results of PCR and sequencing, each of the 223 cases was assigned one of the following genotypic designations: mutated NRAS (MN), 31 patients (13.9%); mutated BRAF (MB), 109 patients (48.9%); wild type at both loci (WW), 80 patients (35.9%); and mutated at both loci (MM), 3 patients, (1.3 %). For analytical purposes, the three MM cases were examined separately, such that the final analysis included 220 subjects. The study population consisted of 118 males and 102 females, with a median age of 49 years (range 18–77 years). The distribution of histologic types included superficial spreading 70%, nodular 19%, lentigo maligna 4%, and unclassified 7%. The rates of specific mutations are shown in Table 1.
Table 1.
Summary of BRAF and NRAS mutations
| DNA mutation | AA Mutation | N (%) | |
|---|---|---|---|
| BRAF (N = 109) | T1799A | V600E | 93 (85.3) |
| GT1798AA | V600K | 9 (8.3) | |
| A1801G | K601E | 2 (1.8) | |
| TG1799AA | V600E | 2 (1.8) | |
| T1790A | L597Q | 1 (0.9) | |
| T1785A | F595L | 1 (0.9) | |
| T1799A, C1834G | V600E, Q612E | 1 (0.9) | |
| NRAS (N = 31) | C181A | Q61K | 15 (48.4) |
| A182G | Q61R | 11 (35.5) | |
| A182T | Q61L | 3 (9.7) | |
| AA182TG | Q61L | 1 (3.2) | |
| C181A, A183G | Q61K | 1 (3.2) |
AA, amino acid
Association of mutational status with tumor histopathology
To test the hypothesis that mutation-bearing tumors display a more aggressive clinical phenotype, various relevant patient characteristics and histologic features were compared among the three genotypes. These included age, gender, Breslow depth, Clark level, mitotic rate, and the presence of ulceration (Table 2). Results show that the median Breslow depth varied significantly among the three genotypes, with MN tumors showing the deepest invasion (1.40 mm) and WW the least invasion (0.93 mm), while MB tumors were intermediate (1.28 mm) (p = 0.006). The same pattern was seen if Breslow depth was examined at a cutoff of 1 mm (p = 0.021). Clark levels followed suit, with mutated and WW tumors showing the most and least aggressive patterns, respectively (p < 0.001). Ulceration also differed significantly among the genotypes, with MB tumors demonstrating the highest rate, whereas rates for MN and WW were considerably lower and similar to each other (p = 0.045). The three genotypes did not differ in terms of gender, age, or mitotic index.
Table 2.
Association of mutational genotype with histopathologic findings
| MN | MB | WW | p | |
|---|---|---|---|---|
| N | 31 | 109 | 80 | |
| Median age in yrs (range) | 51 (19–75) | 48 (19–77) | 49.5 (18–73) | n.s. |
| Gender, percent female | 48 | 45 | 48 | n.s. |
| Median Breslow in mm (range) | 1.40 (0.49–16.00) | 1.28 (0.22–11.00) | 0.93 (0.11–10.00) | 0.006 |
| Breslow ≤ 1 mm (%) | 9 (29.0) | 45 (41.7) | 45 (56.3) | 0.021 |
| Clark level (%) 2 | 1 (3.2) | 12 (11.0) | 21 (26.3) | |
| 3 | 15 (48.4) | 42 (38.5) | 38 (47.5) | |
| 4 | 13 (41.9) | 55 (50.5) | 21 (26.3) | |
| 5 | 2 (6.5) | 0 (0.0) | 0 (0.0) | < 0.001 |
| Median mitotic figures/mm2 (range) | 1 (0–20) | 2 (0–18) | 1 (0–43) | n.s. |
| Ulceration present (%) | 3 (9.7) | 24 (22.4) | 8 (10.1) | 0.045 |
n.s., not significant
Association of mutational status with patient staging and outcomes
All patients were assigned a pathologic stage according to 2009 American Joint Committee on Cancer (AJCC) criteria (14). As shown in Table 3, there was a trend for patients with either mutation to present with a higher stage than those with wild type tumors (p = 0.104). If the MN and MB categories were collapsed, these findings reached significance (p = 0.027). In particular, 52 subjects presented with Stage III disease and patients in this subset were more likely to have mutated tumors (p = 0.031 for MN or MB vs. WW). As further seen in Table 3, the majority of Stage IIIC tumors, i.e. those with larger volume nodal disease, carried one or the other mutation (findings not significant). In spite of this, there was no difference in survival among the stage III patients when stratified according to the presence or absence of mutation (Fig. 1). Furthermore, with a median follow-up of 39 months, overall survival did not differ between the three mutational groups as a whole or stratified by stage at presentation (data not shown).
Table 3.
Stage at presentation by genotype, with Stage III sub-categories
| AJCC Stage | Stage III sub-category | |||||
|---|---|---|---|---|---|---|
| I | II | III | IIIA | IIIB | IIIC | |
| MN (%) | 18 (58.1) | 5 (16.1) | 8 (25.8) | 4 (50.0) | 2 (25.0) | 2 (25.0) |
| MB (%) | 57 (52.3) | 20 (18.4) | 32 (29.4) | 17 (53.1) | 4 (12.5) | 11 (34.4) |
| WW (%) | 57 (71.3) | 11 (13.8) | 12 (15.0) | 7 (58.3) | 4 (33.3) | 1 (8.3) |
Figure 1.
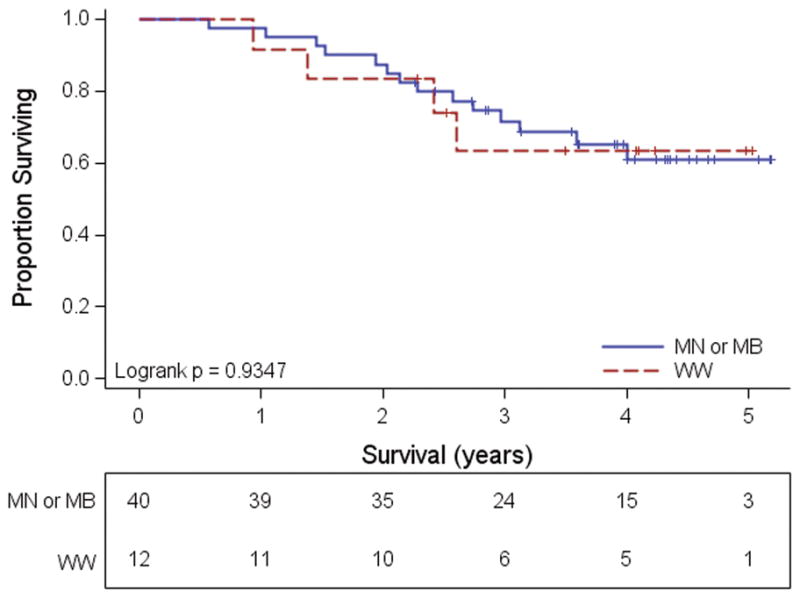
Survival of patients presenting with Stage III disease according to the presence or absence of mutation. Patients with tumors carrying either NRAS or BRAF mutations are combined into a single category.
Tumor localization by mutational status
It has been noted previously that NRAS mutated tumors display a propensity for developing on the upper extremities (10). A similar finding was seen in our patient population, with 90% of MN tumors located on either extremity and only 6% on the trunk (Fig. 2). In contrast, MB tumors were localized to the extremities in 41% of cases, with a similar proportion (46%) found on the trunk. The site distribution of WW tumors was similar to that of MB tumors. Differences in location among the three genotypes were statistically significant (p < 0.0001).
Figure 2.

Influence of genotype on tumor localization. Anatomic location of tumors for each genotype is shown in A. Conversely B shows the distribution of genotypes for each general locale. “Extremity” (Ext) includes hand, arm, shoulder, foot, and leg. “Trunk” (Tr) includes back, chest, abdomen, and buttocks. Scalp, face, ears, and neck are included in “Head and Neck” (HN).
Primary tumors with uncommon mutational status
Three tumors carried mutations in both BRAF exon 15 and NRAS exon 2, designated MM (Table 4). The ages of these three patients (58, 58, and 57 years) were somewhat greater than the median age of 49 years for the other groups. Also interesting was the presence of tandem nucleotide mutations in BRAF exon 15 in two of the three tumors. Otherwise, the MM tumors were not remarkable for any distinct features, and fell across the spectrum of the other three genotypes. One MM patient was found to have regional nodal metastasis on presentation and underwent a successful lymphadenectomy. All three patients remain alive and disease-free at 40, 44, and 49 months from diagnosis.
Table 4.
Uncommon genotypes
| NRAS | BRAF | Age (years) | Gender | Breslow ( mm ) | Clark | Mitotic Index (figures/mm2) | Ulceration | |
|---|---|---|---|---|---|---|---|---|
| Double mutant | A182G | TG1799AA | 58 | M | 0.85 | 3 | 1 | no |
| C181A | T1799A | 58 | M | 1.46 | 4 | 1 | no | |
| A182G | GT1798AA | 57 | F | 1.2 | 4 | 3 | yes | |
| Only mutated DNA | C181A | Wild type | 49 | F | 3.9 | 5 | 5 | no |
| A182G | Wild type | 51 | M | 2.2 | 4 | 2 | no | |
| Wild type | T1799A | 35 | F | 3.1 | 3 | 3 | no | |
| Wild type | GT1798AA | 77 | M | 3.8 | 4 | 9 | no | |
| Wild type | T1799A | 57 | M | 5 | 4 | 16 | yes |
Another uncommon mutational finding was the exclusive presence of mutated DNA, whether NRAS or BRAF, as indicated by a solitary mutation peak on the sequencing chromatogram. This can occur as a result of identical mutations on both strands or a mutation on one strand and deletion of that stretch of DNA on the other. This finding was observed in the tumors of five patients, two with mutated NRAS and three with mutated BRAF (Table 4). Notably, all five tumors displayed aggressive features with a median Breslow depth of 3.8 mm and a median mitotic rate of 5 figures/mm2. Three of these patients developed regional metastases, but all remain alive and disease-free with a median follow-up of 49.5 months.
Discussion
Since the recognition of the high frequency of BRAF mutations in cutaneous melanoma, this anomaly has become a favored target for drug design. However, justification for such directed therapy has largely been based on in vitro studies indicating that the BRAF V600E mutation is activating and drives the MAPK pathway. The data that we now present provide clinical justification for the therapeutic targeting of mutated B-Raf, as well as mutated N-Ras, in the treatment of melanoma. Our findings demonstrate a correlation of NRAS and/or BRAF mutations with other factors well-known to negatively influence prognosis, such as invasion and ulceration, and are consistent with our previous report that NRAS and BRAF mutations are acquired as melanoma cells progress from the radial to the vertical growth phase (12). In keeping with these observations, patients with mutated tumors are more likely to present with regional metastases. It is interesting to note, however, that in our study population to date, survival does not differ between Stage III patients whose primary tumors do or do not carry mutations, even though the mutated tumors tended to produce larger volume nodal disease.
A similar attempt to establish clinical correlates of NRAS and BRAF mutations in melanoma has been published by Edlundh-Rose et al. (10). This group examined 294 tumors, the majority of which were metastases, and reported the characteristics of the primary tumors from which they were derived. Although this approach selected for tumors that eventually metastasized and assumed genotypic agreement between primaries and metastases, many of their findings were consistent with ours, particularly the invasiveness of (presumed) NRAS-mutated primary tumors and lack of survival differences based on genotype. Taken together, these data suggest that the effects of NRAS and BRAF mutations may be limited to early disease stages and that other factors are more influential after regional metastases have occurred. It is also possible that the mutation pattern in our study population differs between the primary and metastatic tumors, as we have not sequenced the exons in question in metastases from these patients. However, the literature would suggest that the mutational status is maintained throughout the various stages of disease progression (15).
The high frequency of ulceration in BRAF mutated tumors is notable, reaching a rate of 22% compared to around 10% in NRAS-mutated and wild type tumors. Because BRAF and NRAS are components of the same signaling pathway, it is difficult to reconcile the differences in ulceration rates on the basis of downstream MAPK effectors. Implicit here is that the mechanism of melanoma ulceration, while poorly understood, may be linked to factors unique to BRAF mutation and may ultimately shed light on BRAF-specific molecular processes.
The difference in tumor localization based on genotype is compelling, and here, NRAS mutants distinguish themselves from the two other genotypes with their propensity for the extremities. BRAF mutated tumors have been reported to arise less frequently in skin with chronic sun exposure and actinic damage (16). Although less well documented, melanomas with NRAS mutations are reported as more common on skin with continuous sun exposure (3, 10). Our data generally support these previous reports in terms of sites more likely to be sun-exposed (extremities) or protected (trunk). To further address the suggested association of mutations and sun exposure, we are presently examining the tumor sections for solar elastosis, an accepted marker of chronic sun damage. Alternatively, other differences in the external environment or even differences that occur in melanocytes as they migrate to and develop in central vs. peripheral regions of the body may influence the risk of one or another mutation.
In conclusion, our data provide convincing evidence for distinct clinical phenotypes of melanomas bearing NRAS and BRAF mutations, whether considered together or separately, and largely point to an inferior patient outcome. Conversely, patients with tumors wild type at both loci might be expected to exhibit a less aggressive form of this disease. Although melanoma has reliable histopathologic predictors of tumor behavior, genotypic data may assist in decision-making for patients with borderline cases, particularly those at risk for complications of sentinel node biopsy or general anesthesia, or patients generally reluctant to undergo invasive procedures. Our ultimate goal is to strengthen and expand these correlative findings as we continue to follow this patient cohort.
Statement of Translational Relevance.
In this study, we present evidence for aggressive behavior of cutaneous melanomas bearing the common mutations of NRAS or BRAF when compared to tumors wild type at both loci. The translational significance of these findings are twofold. First, our data provide clinical support for drug development targeting the mutated forms of these important oncogenic signaling molecules. Second, with the rising frequency of NRAS and BRAF mutational analysis in the clinical setting, our findings support a more conservative approach to wild type primary tumors, particularly in the case of patients at risk for complications of sentinel node biopsy or general anesthesia, or patients generally reluctant to undergo invasive procedures.
Acknowledgments
The authors would like to thank the members of the MDACC Melanoma Informatics, Tissue Resource, and Pathology Core for carefully processing and providing the materials used in this study. This work was supported by NIH P50 CA093459 and NIH CA16672 (DNA Analysis Facility). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
- 1.Albino AP, Nanus DM, Mentle IR, et al. Analysis of ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene. 1989;4:1363–74. [PubMed] [Google Scholar]
- 2.Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP. RAS mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102:285–90. doi: 10.1111/1523-1747.ep12371783. [DOI] [PubMed] [Google Scholar]
- 3.van’t Veer LJ, Burgering BMT, Versteeg R, et al. N–ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol. 1989;9:3114–6. doi: 10.1128/mcb.9.7.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polakis P, McCormick F. Structural requirements for the interaction of p21ras with GAP, exchange factors, and its biological effector target. J Biol Chem. 1993;13:9157–60. [PubMed] [Google Scholar]
- 5.Davies H, Bignell GR, Cox C, et al. Mutation of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 6.Goydos JS, Mann B, Kim HJ, et al. Detection of B-RAF and N-RAS mutations in human melanoma. J Am Coll Surg. 2005;200:362–70. doi: 10.1016/j.jamcollsurg.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 7.Dong J, Phelps RG, Qiao R, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–5. [PubMed] [Google Scholar]
- 8.Shinozaki M, Fujimoto A, Morton DL, Hoon DSB. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004;10:1753–7. doi: 10.1158/1078-0432.ccr-1169-3. [DOI] [PubMed] [Google Scholar]
- 9.Akslen LA, Angelini S, Straume O, et al. BRAF and NRAS mutations are frequent in nodular melanoma but are not associated with tumor cell proliferation or patient survival. J Invest Dermatol. 2005;125:312–7. doi: 10.1111/j.0022-202X.2005.23788.x. [DOI] [PubMed] [Google Scholar]
- 10.Edlundh-Rose E, Egyhazi S, Omholt K, et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006;16:471–8. doi: 10.1097/01.cmr.0000232300.22032.86. [DOI] [PubMed] [Google Scholar]
- 11.Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 12.Greene VR, Johnson MM, Grimm EA, et al. Frequencies of NRAS and BRAF mutations increase from the radial to the vertical growth phase in cutaneous melanoma. J Invest Dermatol. 2009;129:1483–8. doi: 10.1038/jid.2008.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar R, Angelini S, Hemminki K. Activating BRAF and N-Ras mutations in sporadic primary melanomas: an inverse association with allelic loss on chromosome 9. Oncogene. 2003;22:9217–24. doi: 10.1038/sj.onc.1206909. [DOI] [PubMed] [Google Scholar]
- 14.Balch CM, Gershenwald JE, Soong S, et al. Final Version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;36:6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483–8. [PubMed] [Google Scholar]
- 16.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct set of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]