

Abstract
Epigenetic modifications work with genetic mechanisms to determine transcriptional activity and, while somatically heritable they are also reversible, making them good therapeutic candidates. Epigenetic changes can precede disease pathology and thus are diagnostic indicators for risk, and can act as prognostic indicators for disease progression. Histone deacetylase inhibitors and DNA methylation inhibitors have been FDA approved for several years and are clinically successful. More recently, histone methylation and microRNAs have also gained attention as potential therapeutic targets. The presence of multiple epigenetic aberrations within a diseased tissue and the abilities of cells to develop resistance suggest that combination therapies may be most beneficial. This review will focus on recent examples of using epigenetic modifications to evaluate disease risk, progression and clinical response and will describe the latest clinical advances in epigenetic therapies concentrating on treatments which combine epigenetic therapeutics with each other and with cytotoxic agents to increase clinical response.
Epigenetics is defined as heritable changes in gene expression that do not result from an alteration in the DNA sequence itself. DNA methylation, histone variants and modifications, and nucleosome positioning work together to determine the epigenetic landscape of a cell. DNA methylation occurs when a methyl group is added to the 5′ position of the cytosine ring of CpG dinucleotides. Histones can be covalently modified by the addition of a variety of modifications (methyl, acetyl, phospho, ubiqityl, or sumo groups) and whether the modification has a facilitory or inhibitory effect on transcription depends on which residue is modified and the type of modification. Nucleosomes consist of DNA wrapped around a core of 2 copies of each H2A, H2B, H3 and H4 histone proteins, thus integrating DNA methylation and histone modifications. Variants of core histone proteins, such as H3.3 and H2A.Z, also occur at specific genomic loci to alter the stability of nucleosome occupancy. The localization of nucleosomes within genomic regulatory regions plays an important role in creating permissible or refractory environments for transcription. These different aspects of epigenetic regulation work in concert to determine the epigenetic state of a cell and hence, its transcription profile.
Epigenetic Disease Mechanisms
Epigenetic aberrations have been well established in cancer1, 2 and occur in several other diseases including diabetes3, lupus4, asthma5 and a variety of neurological disorders2, 6, 7,8 (Table 1 and references within). In cancer, there is a global loss of DNA methylation (hypomethylation), particularly in gene bodies and intergenic regions, including repetitive elements, leading to genomic instability. This global hypomethylation is accompanied by increased de novo methylation (hypermethylation) of many promoters of tumor suppressor and other genes that are contained within CpG islands, resulting in permanent gene silencing (Figure 1). In addition to changes in DNA methylation, there is a global loss of H4K16 acetylation and H4K20 tri-methylation, as well as increased expression of BMI1, a component of the polycomb repressive complex 1 (PRC1), and EZH2, a component of PRC2, which act to inhibit gene expression1, 9. Interestingly, recent evidence has demonstrated that genes that are targets of the PRC in embryonic stem cells are more likely than others to become methylated in cancer, potentially linking different epigenetic silencing mechanisms10-12.
Table 1.
Selected Examples of Known Epigenetic Alterations Associated with Diseases
| Epigenetic Aberration | Enzyme Responsible | Disease | Epigenetic Alteration | Comments | Ref |
|---|---|---|---|---|---|
| DNA Methylation | DNMT1, DNMT3A, DNMT3B and DNMT3L | Rett Syndrome | Inability to “read” DNA methylation | Mutation in MeCP2 | 6, 7, 8 |
| Diabetes | Hypermethylation of PPARGC1A gene promoter | 3 | |||
| Cancer | Global Hypomethylation, hypermethylation of some CpG island promoters, including CIMP | 1, 2, 6 | |||
| Systemic Lupus Erythematosus | Hypomethylation of CpG islands at specific promoter regions | Decreased DNMT1 and DNMT3B expression | 4 | ||
| ICF Syndrome | Hypomethylation at specific sites | DNMT3B Mutation | 6, 7, 8 | ||
| ATR-X Syndrome | Hypomethylation of specific repeat and satellite sequences | ATRX Mutation | 6, 7 | ||
| Histone Acetylation | Histone Acetyltransferases (HATs) and HDACs | Rubinstein-Taybi Syndrome | Hypoacetylation | Mutation in CBP, a known HAT | 6, 7, 8 |
| Diabetes | Hyperacetylation at inflammatory genes promoters | 3 | |||
| Asthma | Hyperacetylation | Increased HAT activity and decreased HDAC activity | 5 | ||
| Cancer | H4K16 acetylation loss | Hypomethylation of DNA repetitive sequences | 1 | ||
| Histone Methylation | Histone Methyltransferases (HMTs) and Histone Demethylates (HDMs) | Cancer | H4K20 tri-methylation loss | Hypomethylation of DNA repetitive sequences | 1 |
| Sotos Syndrome | Decreased H4K20 and H3K36 tri-methylation | Loss of function of NSD1, a histone methyltransferase | 98 | ||
| Huntington's Disease (HD) | Increased H3K9 tri-methylation and possibly increased H3K27 tri-methylation | Increased HMT (ESET) expression and enhanced PRC2 activity | 7 | ||
| miRNA Expression | N/A | Cancer | Decreased miR-101 | Increased EZH2, H3K27 trimethylation | 61, 74 |
| Decreased miR-143 | Increased DNMT3A | 75 | |||
| Decreased miR-29 | Increased DNMT3A and 3B | 76 | |||
| Increased miR-21 | Decreased PTEN | 83 | |||
| Increased miR-155 | Lower survival rates | 82 |
Figure 1. Epigenetic Aberrations of CpG Island Promoters in Cancer and the Epigenetic Therapies That Target Them.

Tumor suppressor genes (e.g. FBXO32, MLH1 & RUNX3) are expressed in normal cells and become silenced in cancer cells. This can occur by PRC reprogramming (e.g. FBXO32), where the polycomb group protein EZH2 catalyses the methylation of H3K27 or by 5mC reprogramming (e.g. MLH1, RUNX3) due to de-novo DNA methylation by DNMT3A and DNMT3B. Polycomb mediated repression can be targeted by inhibitors of PRC2, like DZNep and re-expression of these genes can be enhanced by HDAC and LSD1 inhibitors allowing acetylation of H3/4 and methylation of H3K4, respectively. Polycomb mediated repression can also be reversed by inducing miR-101 expression, which inhibits the expression and function of EZH2. 5mC reprogramming can be reversed, mainly by DNMT inhibitors, but also by re-expression of miR-143 and miR-29, two miRNAs that target de-novo DNMTs. LSD1 inhibitors may also reactivate tumor suppressor genes by inhibiting DNMT1 stabilization leading to loss of DNA methylation maintenance. Genes, which are polycomb repressed in normal cells (e.g. PAX7), can undergo epigenetic switching by gaining DNA methylation, thus losing their plasticity during transformation. Currently it is not known whether the treatment of cancer cells with DNMTi alone will reverse epigenetic switching to restore the polycomb repressed state or whether it will re-activate this set of genes. Cancer-Testis Antigens (CTAs, e.g. NY-ESO-1) can become silenced by DNA methylation in cancer. Treatment with DNMT inhibitors can induce CTA expression, allowing the immune system to recognize and kill the cancer cells. Black arrows represent epigenetic alterations during transformation and gray arrows represent the reversion of this alteration by epigenetic therapy.
Epigenetic modifications can be used to stratify disease13 and predict clinical outcome14, 15. H3 acetylation and H3K9 di-methylation can discriminate between prostate cancer (PCA) and non-malignant prostate tissue and H3K4 tri-methylation can be a significant predictor of PSA recurrence16. EZH2 expression is an independent prognostic marker that correlates with the aggressiveness of prostate, breast and endometrial cancers17. Expression of the DNA repair gene O(6)-methylguanine-DNA methyltransferase (MGMT) antagonizes chemotherapy and radiation treatment18 thus, silencing of MGMT by hypermethylation correlates with positive treatment response. Furthermore, epigenetic alterations can also precede tumor formation and thus are potential diagnostic indicators of disease risk19. For example, H. pylori bacterial infection is associated with DNA hypermethylation of specific genes, which are often methylated in cancer20. Thus, reversal of epigenetic alterations that occur as a result of an acute illness may prevent the progression to a more chronic disease state.
With the growing development of technologies to analyze the epigenome, a new field: pharmaco-epigenomics - is emerging, where epigenetic profiles can be used to identify molecular pathways for cancer drug sensitivity21 and be used in determining the best therapeutic approach. In non-small-cell lung cancer, an unmethylated IGFBP3 promoter is indicative of responsiveness to cisplatin based chemotherapy22. A polymorphism in the CYP2C19*2 variant of cytochrome P450 requires higher valproic acid (VPA) doses to achieve target plasma concentration levels23. Furthermore, monitoring epigenetic changes can be used to measure treatment efficacy and disease progression. Methylation of PITX2 can be used to predict outcome of early breast cancer patients following adjuvant tamoxifen therapy24. Patients with p16 hypermethylation had lower bladder cancer recurrence rates after IL-2 treatment compared to patients with no p16 hypermethylation after IL-2 treatment25. Since epigenetic mechanisms determine which genes and hence signaling pathways can be activated, the presence of distinct modifications on specific genes and subsets of genes can aid at several steps in determining and monitoring optimal therapeutic approaches.
Epigenetic modifications are somatically heritable yet also reversible, making them good candidates for potential drug treatments. Multiple epigenetic abnormalities are typically present in diseased tissue leading to an altered epigenetic landscape. Cancer cells may become ‘addicted’ to the aberrantly developed epigenetic landscape and, consequently, be more sensitive than normal cells to epigenetic therapy, in a process similar to an inverted oncogene addiction. One example of oncogene addiction is MET, a tyrosine kinase that acts as a receptor for Hepatocyte Growth Factor (HGF) and functions in normal cells to control tissue homeostasis26. MET can be aberrantly activated in cancer, by ligand dependent mechanisms or by overexpression26. Interestingly, while MET plays roles in both normal and cancer cells, the latter are more sensitive to MET inhibition due to increase reliance on MET signaling26. Thus, cancer cells become dependent (and consequently addicted) to increased activity of a few highly important oncogenes. It is possible that cancer cells undergo a parallel process by which they become dependent (and consequently addicted) to aberrant silencing/inactivation of a few highly important tumor suppressor genes. Since it is well known that several tumor suppressor genes are silenced in cancer by epigenetic mechanisms1 it is possible that cancer cells become addicted to their aberrant epigenetic landscape and, consequently, more sensitive to the epigenetic therapy than normal cells.
DNA Methylation
In cancer there is global hypomethylation accompanied by hypermethylation of a subset of gene promoters contained within CpG islands leading to gene silencing (Figure 1)1. This hypermethylation has recently been described to also extend past the boundaries of CpG islands into “DNA shores” 27. DNA methyltransferases 3A/B are responsible for de novo DNA methylation patterns, which are then copied to daughter cells during S-phase by DNMT1. DNA methylation inhibitors have been well characterized and tested in clinical trials28. 5- Azacytidine (5-Aza-CR; Vidaza) is a nucleoside analog that incorporates into RNA and DNA and is FDA approved to treat high risk myelodysplastic syndromes (MDS) patients and successful clinical results have recently been reported (Table 2)29. 5-Aza-2-deoxycytidine (5-Aza-CdR; Decitabine) is the deoxy derivative of 5-Aza-CR and is incorporated only into DNA. At low doses both azanucleosides act by sequestering DNMT enzymes after incorporation into DNA thus leading to global demethylation as cells divide, while at higher doses they induce cytotoxicity. Potential new drugs that can be used in the clinic include zebularine, a cytidine analog that acts similarly to 5-Aza-CR, but has lower toxicity, increased stability and specificity30 and S110, a decitabine derivative that has increased stability and activity, which has shown promise in pre-clinical studies (Figure 2)31. In addition to inhibiting DNA methyltransferase activity, azanucleosides also act through unspecific mechanisms, which likely contribute to their clinical effectiveness. Promoter DNA methylation can be used to molecularly classify21, 32,33 predict progression of cancer34, 35 and direct therapeutic approach36,37. For example, DNA methylation of specific promoters may identify a sub-population of colorectal cancer that is responsive to 5-fluorouracil36. Furthermore, reversing MLH1 silencing by DNA methylation inhibitors restores sensitivity to cisplatin38 suggesting that inclusion of DNA methylation inhibitors may increase the effectiveness of conventional chemotherapy. Successful conventional chemotherapy depends on activation of pro-apoptotic genes that respond to cytotoxic agents leading to cell death. DNA methylation of these pro-apoptotic genes can block cell death from occurring leading to chemotherapeutic resistance, thus reactivation of epigenetically silenced apoptotic genes can increase efficacy of chemotherapy. For example, APAF1 is silenced in metastatic melanoma cells and treatment with 5-Aza-CdR restores expression and chemosensivity37. Conversely, methylation induced silencing of DNA repair genes can be detrimental by leading to microsatellite instability39 or beneficial by preventing the repair of genes targeted by chemotherapy causing cells to undergo apoptosis rather than repair40. Methylation induced silencing of cancer-testis antigens, such as NY-ESO-1, can protect cancer cells from being recognized by T cells. Treating cancer cells with demethylating agents can induce the expression of these antigens, allowing a reaction by engineered lymphocytes41 suggesting that epigenetic therapy can be combined with immunotherapy for better results (Figure 1).
Table 2.
Selected Clinical Trials of Epigenetic Therapy in Cancer
| Epigenetic Target | Agent | Phase of Study | Disease | Findings | n | Reference |
|---|---|---|---|---|---|---|
| DNMT inhibitor alone | ||||||
| DNMTs | 5-Aza-CR | II/III | MDS and AML | Complete remission in 10-17% and hematological improvement in 23-36% | 309 | 99 |
| III | MDS | Better overall survival (24.5 vs. 15 months) than conventional care | 358 | 29 | ||
| 5-Aza-CdR | II | MDS and CMML | Anti-MDS and anti-CMML activities with a safe toxicity profile. 34% of patients achieved complete response and 73% had an objective response | 95 | 100 | |
| HDACi Alone | ||||||
| HDAC | Phenylbutyrate | I | MDS and AML | Well tolerated. No patients achieved complete or partial remission, although 4 achieved hematological improvement. | 27 | 101 |
| Vorinostat (SAHA) | I | Relapsed or refractory AML, CLL, MDS, ALL, CML | Seven out 31 AML patients showed hematological improvement, including 2 complete response and 2 complete response with incomplete blood count recovery | 41 | 102 | |
| Advanced solid and hematologic malignancies | One complete response (diffuse large B-cell lymphoma), three partial responses (cutaneous T-cell lymphoma) | 73 | 103 | |||
| Combination therapy | ||||||
| DNMTs and HDAC | 5-Aza-CR and VPA | I | Advanced solid cancers | The combination is safe; 25% of the patients showed stable disease (median, 6 months) | 55 | 95 |
| 5-Aza-CR and Phenylbutyrate | I | Refractory solid tumors | The combination is safe. No clinical benefit | 27 | 97 | |
| HDAC | Vorinostat (SAHA) and Doxorubicin | I | Solid tumors | Two out of 24 partial responses (breast and prostate cancer) and 2 stable disease for more than 8 months (melanoma) | 32 | 104 |
| Vorinostat (SAHA) plus Carboplatin and Paclitaxel | II | Advanced non-small-cell lung cancer | Better response ratio (34% vs. 12.5%), progression-free survival (6 months vs. 4.1) and overall survival (13 months vs. 9.7) than placebo plus Carboplatin and Paclitaxel | 94 | 93 | |
Figure 2. Chemical structure of selected compounds that target epigenetic modifications.
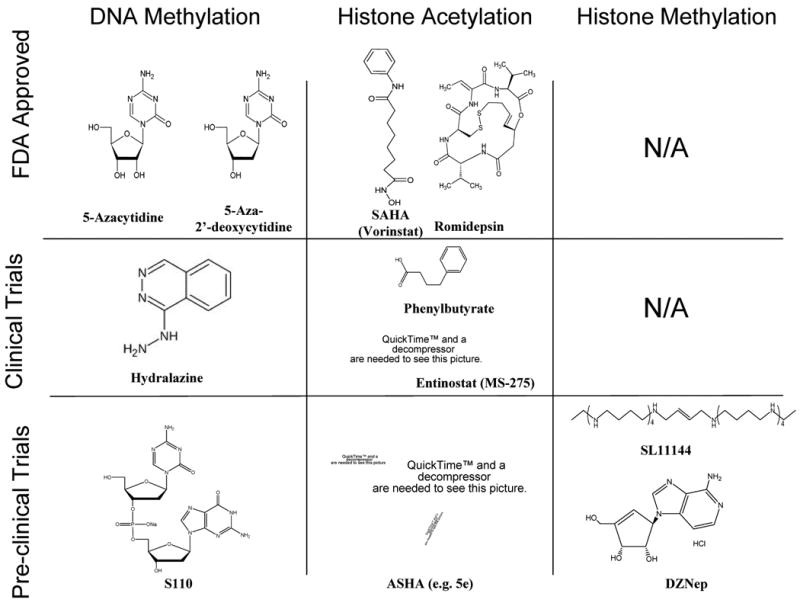
At the present time, several molecules, which target epigenetic alterations in pathological states are at different stages of drug development. The nucleoside analogs 5-Azacytidine and 5-Aza-2′-deoxycytidine are FDA approved to treat high-risk myelodysplastic syndromes (MDS) patients and successful clinical results have been reported. The drug Hydralazine is currently being investigated in clinical trials as a putative demethylating agent against solid tumors and S110, a dinucleotide containing 5-aza-CdR, has been shown, in vitro, to demethylate DNA with a decreased deamination by cytidine deaminase. Targeting histone acetylation has also been a successful example of epigenetic therapy. Several histone deacetylase inhibitors are FDA approved such as the hydroxamic acid-based compound SAHA and the depsipeptide, Romidepsin, while others are currently in clinical trials for cancer (Phenylbutyrate and Entinostat) and neurologic diseases (Entinostat) and new molecules targeting specific HDACs are under pre-clinical investigations (e.g. PCI-34051, which targets HDAC8). More recently, significant effort is underway to find new molecules able to target histone methylation. To our knowledge there are no drugs targeting histone methylation which are FDA approved or in clinical trials. However, pre-clinical trials suggest anti-tumor activity of the oligoamine analogue SL-11144, a LSD1 inhibitor and the S-adenosylhomocysteine hydrolase inhibitor 3-Deazaneplanocin A (DZNep), a drug that depletes cellular levels of PRC2 components.
Despite the clinical successes achieved with DNA methylation inhibitors there is still room for improvement. Currently available DNA methylation inhibitors block DNA methylation at the enzymatic level leading to global DNA methylation inhibition. This is therapeutically beneficial as tumor suppressor genes are hypermethylated in cancer, however global hypomethylation may lead to activation of oncogenes and/or increased genomic instability. DNA hypomethylation can activate promoters within repetitive elements, for example LINE-1 hypomethylation can activate an alternative transcript of the MET oncogene in bladder cancer42, suggesting a risk of global DNA demethylation agents. Developing DNA methylation inhibitors that targeted specific genes or groups of genes would overcome this limitation. Furthermore, DNA methylation inhibitors act during S-phase of the cell cycle, thus they preferentially affect rapidly growing cells. This is advantageous when treating rapidly dividing cancer cells but may be less clinically useful in treating diseases that are not characterized by rapid cell cycling. Furthermore, upon azanucleoside withdrawal the DNA methylation levels return to pre-treatment levels6 demonstrating a continual need for DNA methyltransferase inhibition. Thus, while DNA methylation inhibitors are clinically successful, their lack of specificity, cell cycle dependency and need for continual administration leave room for the development of better therapies.
Histone Modifications
While DNA methylation is considered to be a very stable epigenetic modification, histone modifications are comparably more labile with their levels being maintained by a balance between histone modifying enzymes, which add or remove specific modifications. Aberrant histone modification levels result from an imbalance in these modifying enzymes in diseased tissue, thus correcting the increased or decreased level of a particular enzyme will restore the natural balance in the affected cells. In cancer, both histone methyltransferase and histone demethylase enzymes are misregulated and there is a high expression level of histone deacetylases (HDAC) and a global reduction in histone acetylation levels1, 9, 43, 44, 45. HDAC inhibitors have long been studied in the clinical setting as potential therapies (Figure 2) and recent clinical trials have been extensively reviewed elsewhere (Table 2)46. Histone deacetylase inhibitors (HDACi) can affect acetylation of non-histone proteins, as well as histones, potentially leading to more global effects46. Furthermore, non-isoform selective HDACi only target approximately 10% of all acetylation sites 47, thus more work is necessary to understand the underlying basis for target specification of global and isoform specific HDACi. Currently, significant effort is underway to find new molecules that are able to selectively inhibit specific HDACs48, 49 and thus avoid the side effects that occur with a global HDACi50. To date, specific inhibitors of HDAC6 (class II) and HDAC8 (class I) have been developed48, 49. The development of specific HDACi combined with a better understanding of the pathophysiology of diseases associated with alterations in HDACs will allow more rational therapy and, potentially, reduce side effects. For example, the HDAC inhibitor PCI-34051, which is derived from a low molecular weight hydroxamic acid scaffold, was recently shown to selectively inhibit HDAC8 and induce apoptosis specifically in T-cell lymphomas and not other tumor or normal cells, showing HDC8 plays an important role in the pathophysiology of this disease and suggesting the therapy with HDAC8 specific inhibitor may lead to less side effects49. While the identification of additional specific HDAC inhibitors will increase specificity and the possibility of personalized treatments, it may also potentially limit the likelihood of success in combinatorial therapy.
Histone methyltransferase and demethylase enzymes are relatively more specific than HDACs in that they target a limited number of residues51 however, like HDACs, lysine and arginine methyltransferase enzymes methylate non-histone proteins as well as histone proteins52, 53. A great deal of effort is underway to find drugs able to revert specific histone methylation marks or to target histone methyltransferase or histone demethylases. In this regard, a new class of oligoamine analogs was recently found that act as potent inhibitors of Lysine specific demethylase 1 (LSD1) (Figure 2). LSD1 targets the activating H3K4 mono and di-methylation mark but can also target the repressive H3K9me2 mark when complexed with the androgen receptor43, 54. Treatment of colon cancer cells with LSD1 inhibitors (e.g. SL11144) resulted in increased H3K4 methylation, decreased H3K9me2, and re-expression of the SFRP2 gene55, demonstrating context specificity of LSD1 and its inhibitors. LSD1 inhibition in neuroblastoma, resulted in decreased proliferation in vitro and reduced xenograft growth56. Interestingly, LSD1 can also demethylate DNMT1 resulting in destabilization and loss of global DNA methylation maintenance57. The ability of LSD1 to affect both histone and DNA methylation, make it a promising target for epigenetic therapy.
The repression mediated by the H3K27 tri-methylation mark, occurs through the actions of two multi-subunit complexes, PRC1 and PRC2, H3K27me3 is deposited by EZH2 and then recognized and bound by PRC1, which can further recruit additional proteins to establish a repressed chromatin configuration1. Gene promoters which are marked by PRC2 (i.e. polycomb target genes) in embryonic stem cells have recently been shown to be far more likely than other genes to become methylated in cancer10-12. Similarly, polycomb targets in normal prostate cells also become methylated in prostate cancer58. Thus alterations in chromatin structure do not always coincide with changes in gene expression, rather DNA methylation replacement of polycomb repressive marks acts to “lock in” an inactive chromatin state through a process called “epigenetic switching”58. The mechanism underlying the predisposition of polycomb targets for DNA methylation is not fully understood, however some links have recently been uncovered. CBX7, a component of the PRC1 complex, can directly interact with DNMT1 and 3B at polycomb target genes59.
While there is significant promise for drugs that target histone methylation enzymes, more work is necessary to determine their specificities and stability and there are currently no such drugs undergoing clinical trials. To date, the S-adenosylhomocysteine hydrolase inhibitor, 3-deazaneplanocin A (DZNep) has the most promising pre-clinical results of drugs, which target histone methylating enzymes (Figure 2). DZNep depletes cellular levels of PRC2 components (EZH2, EED and SUZ12) and consequently reduces H3K27me3 levels and induces apoptosis in breast cancer but not normal cells60. The effect of DZNep is similar to those observed when EZH2 is depleted by RNAi, suggesting that this drug may be more effective in cancers like prostate and breast, which rely on abnormally high EZH2 expression levels61. On the other hand, a subsequent study showed that DZNep also decreased H4K20me3 demonstrating that DZNep lacks specificity and acts more as a global histone methylation inhibitor, suggesting the need for further development of histone methylation inhibitors62.
EZH2 activity can also be regulated by signaling cascades. For example, AKT phosphorylates EZH2 at serine 21, suppressing its methyltransferase activity and, consequently, reducing H3K27me363. H3K27me3 levels can be restored using the PI3K/AKT inhibitor LY294002, opening a new therapeutic opportunity to repair epigenetic alterations by targeting upstream signaling pathways. Furthermore, in prostate cancer, the oncogenic ETS transcription factor ERG can bind to the EZH2 promoter and induce over-expression. Thus, the pharmacological disruption of ERG activity could reduce EZH2 over-expression observed in cancer64. EZH2 is a particularly important example because it is frequently over-expressed and aberrantly targeted to genes in cancer58, a process termed PRC reprogramming (Figure 1).
G9a and GLP (G9a like protein) are histone methyl-transferases that catalyze H3K9 di-methylation and are often over-expressed in tumors65. Knockdown of G9a in prostate cancer cells demonstrates a critical role for this protein in regulating centrosome duplication and chromatin structure, suggesting that G9a is important to perpetuate the malignant phenotype and, thus, a putative target in cancer therapy66. As a result there is significant interest in developing G9a/GLP inhibitors and thus far the most efficient inhibitor is BIX-01294 (adiazepin-quinazolin-amine derivative), which is able to transiently reduce bulk H3K9me2 levels in several cell lines67. BIX-01294 binds to the SET domain of GLP in the same groove where the target lysine (H3K9) binds, preventing the binding of the peptide substrate and, consequently, the deposition of methylation marks at H3K968.
Several other histone methyltransferases and demethylases have also been associated with diseases and, thus, could be potential targets for epigenetic therapy. For instance, MMSET, a H4K20 methyltransferase, is overexpressed in myeloma cell lines and required for cell viability69. SMYD3, an H3K4 methyltransferase, is also highly expressed in cancer and seems to play a role in carcinogenesis as a coactivator of ERalpha70. GASC1, an H3K9 and H3K36 demethylase, is often amplified in cancer and its inhibition results in decreases cell proliferation71.
Despite a lack of specificity, targeting histone modifications has been clinically successful. Hopefully the development of therapeutics that target specific histone modifying enzymes will retain or increase their therapeutic success while decreasing side effects due to the lack of specificity. On the other hand targeting individual histone modifying enzymes may decrease clinical efficacy due to compensation by other histone modifying enzymes potentially leading to resistance. Designing personalized cocktails of inhibitors based on an individual's need may help overcome the potential problems of compensation and resistance.
microRNAs
miRNAs are able to induce heritable changes in gene expression without altering DNA sequence, and thus contribute to the epigenetic landscape. In addition miRNAs can both regulate, and be regulated by, additional epigenetic mechanisms. Expression of miRNAs is misregulated in several diseases including cancer72 and neurodegeneration73. For example, miR-101 targets EZH2 for degradation and is down-regulated in several types of cancer, resulting in increased EZH2 expression and consequently H3K27me3 levels and decreased tumor suppressor gene expression61, 74. Re-expression of miR-101 leads to reduced H3K27me3 and inhibits colony formation and cancer cell proliferation61, 74. Expression of miR-143 in colorectal cancer cells75 and the miR-29 family in lung cancer cells76 reduced DNMT3A and DNMT3A and B levels, respectively, and resulted in decreased cell growth and colony formation. Treatment of cells with 5-Aza-CdR and 4-phenylbutyric acid resulted in miR-127 activation, which in turn downregulated the BCL6 oncogene in bladder cancer cells77. In fact, treatment with 5-Aza-CdR alone is sufficient to reactivate miR148a, miR34b/c and miR-9, a group of miRNAs with metastasis suppression activity78. In addition to inducing aberrantly repressed miRNAs using epigenetic drugs, replacement gene therapy may also be useful in re-establishing miRNA expression. Viral vectors generated by cloning individual or groups of human miRNAs has been successful in pre-clinical assays using a mouse model of hepatocellular carcinoma in which miR-26a expression by an adeno-associated virus (AAV) resulted in apoptosis and inhibition of cancer cell proliferation in the absence of toxicity79. microRNA gene therapy has an advantage, over the conventional RNAi, in that it is unlikely to generate a strong type I interferon response because double-strand RNA is not introduced to the cell and furthermore, the AAV vector is less immunogenic than previous generations of viral vectors used for gene transfer80.
Abnormally high expression of miRNAs can be targeted using recently developed locked-nucleic-acid (LNA)-modified phosphorothioate oligonucleotide technology. Locked-nucleic-acid-modified oligonucleotides contain an extra bridge in their chemical composition leading to enhanced stability compared to their unmodified counterparts. miRNAs can be generated using these LNA modified phosphorothioate oligonucleotides to create LNA-antimiRs, which can be delivered systemically. In pre-clinical assays with primates, intravenous injections of LNA-antimiR complementary to the 5′ end of miR-122 antagonized liver specific expression of this miRNA without toxicity81. Based on these promising results, phase 1 trials are currently underway. LNA-antimiRs may be used to target aberrantly expressed miRNAs in other diseases, such as cancer. For example, miR-155 is up-regulated in lung adenocarcinoma compared to non-cancerous lung tissue and patients with higher miR-155 expression had lower survival rates than patients with lower miR-155 expression, suggesting that miR-155 is a promising target for LNA-antimiR therapy (Table1)82. Several other miRNAs are up-regulated in cancer and could, theoretically, be used as LNA-antimiR targets. For example, miR-21, is up-regulated in several types of cancer (lung, breast, colon, gastric and prostate carcinomas, endocrine pancreatic tumours, glioblastomas and cholangiocarcinomas) and targets the tumor suppressor PTEN (Table 1)83. Thus, miRNAs can alter the epigenetic machinery and also be regulated by epigenetic alterations, creating a highly controlled feedback mechanism, making it a suitable target for epigenetic therapy and possibly an epigenetic drug itself.
One unique advantage of targeting miRNAs is the ability of one miRNA to regulate several target genes and multiple cellular process. In that way, if the level of one or a few miRNAs has changed in a pathological state, several different pathways could be consequently altered. Rather than trying to identify and directly target the proteins in multiple pathways it would be more effective to restore the physiological level and functions of the misregulated miRNA(s). This clinical potential highlights the importance of better understanding miRNA profiles in healthy and diseased tissues in order to develop better therapeutic strategies. Furthermore, multiple miRNAs that target different steps of an over-active pathway can be combined to increase efficacy and allow for customization of therapies to individual patients. While the unique composition of miRNA based therapy provides many benefits, additional research is necessary to determine the best method of delivery and increase miRNA stability to ensure efficacy.
Combined Epigenetic Therapies
The presence of multiple epigenetic aberrations in a single tissue, the ability to develop resistance and the discovery that common sets of genes are regulated by distinct epigenetic mechanisms at different biological stages demonstrates the need and highlights the potential success of combinatorial therapy. Combining epigenetic therapies to increase efficacy has been done for over 25 years84, 85 and has demonstrated both additive and synergistic effects depending on the targets6, 44. Extensive work has evaluated the clinical benefits of combined DNMT and HDAC inhibition and comprehensive reviews have been published elsewhere1, 39, 46, 86, 87. A recent phase II multicenter study examining the combination of 5-Aza-CR and the HDAC inhibitor VPA in patients with higher risk MDS found that therapeutic levels of VPA may increase 5-Aza-CR efficacy23. Sequential administration of DNMT and HDAC inhibitors demonstrated clinical efficacy in patients with hematologic malignancies46, 86. However, other studies found no correlation between baseline methylation levels or methylation reversal and positive clinical outcome in MDS and AML patients following 5-Aza-CR and entinostat treatment88. The mechanism behind the clinical efficacy of combined DNMT and HDAC remains controversial and additional studies investigating potential genetic or epigenetic determinants of responsiveness will be helpful. In addition to inducing apoptosis in cancer cells, another therapeutic approach is to induce differentiation of cancer cells. To this end, 5-Aza-CR and VPA treatment followed by all trans-retinoic acid in patients with acute myeloid leukemia or high-risk myelodysplastic syndrome resulted in global hypomethylation and histone acetylation and clinical response in nearly half of treated patients89.
While targeting histone de-methylation enzymes is still in its infancy, early pre-clinical studies alone (described above) and combined with other epigenetic therapies are promising. SFRP2 (a negative regulator of Wnt signaling) expression was re-established in vitro and in vivo in a human colon cancer model following LSD1 and DNMT inhibition resulting significant growth inhibition of established tumors55. Interestingly, in addition to histone residues, LSD1 can also demethylate DNMT1 providing the ability to target both histone methylation and DNA methylation using a single compound. Co-treatment with the HDAC inhibitor, Panobinostat, further enhanced DZNep's reduction of EZH2 levels leading to increased p16, p21, p27 and FBX032 expression and apoptosis in cultured AML cells and mouse models90. Thus, while clinical trials targeting histone methylation and demethylation enzymes are not currently underway it is an area of active investigation with significant promise.
Epigenetic and Cytotoxic Therapies
Conventional chemotherapy can rapidly induce cell death in cancer cells. However, resistance to standard chemotherapy can be established through epigenetic and DNA repair mechanisms22. As a result, epigenetic therapeutics can be combined with more conventional therapies to induce responsiveness or overcome resistance to cytotoxic treatments. Preconditioning with epigenetic drugs could reverse the epigenetic alteration(s) that conferred resistance restoring chemotherapeutic sensitivity. For example, 5-Aza-CR treatment can reverse DNA methylation, thereby overcoming the gene silencing, which led to chemotherapeutic resistance91. On the other hand, methylation induced silencing of DNA repair genes (i.e. MGMT) correlates with a positive clinical response to chemotherapy. Thus, whether the combination of DNA methylation inhibitors and chemotherapy will be successful may depend on the epigenetic profile of an individual tumor. Clinical findings in patients with previously untreated non-small cell lung cancer treated with the HDAC inhibitor, vorinostat, in combination with carboplatin and paclitaxil were sufficiently promising to warrant a phase 2 study which also showed promising results (Table 2)92, 93.
Conclusions/Perspective
Epigenetic therapy has been established as a successful treatment approach for a variety of malignancies. Inhibition of DNMTs and HDACs are two FDA approved cancer treatments. While the mechanism(s) behind the therapeutic benefit of DNMT and HDAC inhibition are not fully understood, ongoing and future studies that combine genomic sequencing and expression data may provide the keys to understanding the mechanism(s) underlying responsiveness. Furthermore, histones can be phosphorylated, ubiquitylated and sumoylated in addition to methylated and acetylated, however these modifications have been less well studied in the context of disease and may offer additional therapeutic targets.
One important challenge in epigenetic therapy is defining which genes are the drivers (i.e. group of genes necessary to be epigenetically silenced for disease to occur) and the passengers (i.e. group of genes that are epigenetically silenced due to aberrant activity of the epigenetic machinery, but are not necessary for disease to occur). Recent advances in high throughout technologies like genome-wide sequencing, combined with RNA profiling, chromatin immunoprecipitation (ChIP) or bisulfite conversion, have led to the generation of large amounts of data that can be integrated to form a comprehensive understanding of the epigenetic alterations that are common and specific to various disease states. Assimilating these large datasets will likely assist in identifying epigenetic alterations that are causational and those that are merely correlative. Thus, in the future it may be possible for patients to be screened, using high throughout technologies, and classified by epigenetic alterations of the driver genes allowing the use of personalized targeted therapies.
Currently, epigenetic therapies are successfully used in the clinic to treat hematological malignancies, however little success has been achieved in treating solid cancers (Table 2)94-97. Initial clinical trials used treatment regimens later found to be less than optimal and resulted in few cases of positive clinical response. Administering more recently developed dosing and treatment schedules in addition to subtyping tumor types based on molecular signatures may increase the efficacy of epigenetic therapy for solid tumors. Furthermore, solid tumors are heterogeneous cell populations, often consisting of cells at a variety of stages of differentiation, thus determining which of these cells harbor epigenetic alterations and ensuring that therapeutic agents maintain stability and are able to penetrate the cellular mass and reach affected cells and will increase the likelihood of clinical success.
The recognition of epigenetics as a significant contributor to normal development and disease opens an expanding new avenue for drug discovery and therapeutics. Epigenetic therapies can be combined with conventional therapies to develop personalized treatments, used to convert unresponsive tumors and may allow for lower dosing which can limit side effects of treatment improving quality of life and treatment compliance.
References
- 1.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fouse SD, Costello JF. Epigenetics of neurological cancers. Future Oncol. 2009;5:1615–1629. doi: 10.2217/fon.09.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol. 2010;299:F14–25. doi: 10.1152/ajprenal.00200.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, Berdasco M, Fraga MF, O'Hanlon TP, Rider LG, Jacinto FV, Lopez-Longo FJ, Dopazo J, Forn M, Peinado MA, Carreno L, Sawalha AH, Harley JB, Siebert R, Esteller M, Miller FW, Ballestar E. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2009;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. Copd. 2005;2:445–455. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]
- 6.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 7.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 8.Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 11.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I, Cedar H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 12.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, Berman DM, Jenuwein T, Pruitt K, Sharkis SJ, Watkins DN, Herman JG, Baylin SB. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 15.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, Valk PJ, Lowenberg B, Delwel R, Melnick A. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellinger J, Kahl P, von der Gathen J, Rogenhofer S, Heukamp LC, Gutgemann I, Walter B, Hofstadter F, Buttner R, Muller SC, Bastian PJ, von Ruecker A. Global levels of histone modifications predict prostate cancer recurrence. Prostate. 2010;70:61–69. doi: 10.1002/pros.21038. [DOI] [PubMed] [Google Scholar]
- 17.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 18.Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, Hegi ME. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 19.Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2009;101:36–45. doi: 10.1111/j.1349-7006.2009.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo T, Oka T, Sato H, Shinnou Y, Washio K, Takano M, Morito T, Takata K, Ohara N, Ouchida M, Shimizu K, Yoshino T. Accumulation of aberrant CpG hypermethylation by Helicobacter pylori infection promotes development and progression of gastric MALT lymphoma. Int J Oncol. 2009;35:547–557. doi: 10.3892/ijo_00000366. [DOI] [PubMed] [Google Scholar]
- 21.Shen L, Kondo Y, Ahmed S, Boumber Y, Konishi K, Guo Y, Chen X, Vilaythong JN, Issa JP. Drug sensitivity prediction by CpG island methylation profile in the NCI-60 cancer cell line panel. Cancer Res. 2007;67:11335–11343. doi: 10.1158/0008-5472.CAN-07-1502. [DOI] [PubMed] [Google Scholar]
- 22.Ibanez de Caceres I, Cortes-Sempere M, Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, Manguan-Garcia C, Cejas P, Lopez-Rios F, Paz-Ares L, de Castrocarpeno J, Nistal M, Belda-Iniesta C, Perona R. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene. 2009;29:1681–1690. doi: 10.1038/onc.2009.454. [DOI] [PubMed] [Google Scholar]
- 23.Voso MT, Santini V, Finelli C, Musto P, Pogliani E, Angelucci E, Fioritoni G, Alimena G, Maurillo L, Cortelezzi A, Buccisano F, Gobbi M, Borin L, Di Tucci A, Zini G, Petti MC, Martinelli G, Fabiani E, Fazi P, Vignetti M, Piciocchi A, Liso V, Amadori S, Leone G. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin Cancer Res. 2009;15:5002–5007. doi: 10.1158/1078-0432.CCR-09-0494. [DOI] [PubMed] [Google Scholar]
- 24.Martens JW, Margossian AL, Schmitt M, Foekens J, Harbeck N. DNA methylation as a biomarker in breast cancer. Future Oncol. 2009;5:1245–1256. doi: 10.2217/fon.09.89. [DOI] [PubMed] [Google Scholar]
- 25.Jarmalaite S, Andrekute R, Scesnaite A, Suziedelis K, Husgafvel-Pursiainen K, Jankevicius F. Promoter hypermethylation in tumour suppressor genes and response to interleukin-2 treatment in bladder cancer: a pilot study. J Cancer Res Clin Oncol. 2009;136:847–854. doi: 10.1007/s00432-009-0725-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 27.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoo CB, Cheng JC, Jones PA. Zebularine: a new drug for epigenetic therapy. Biochem Soc Trans. 2004;32:910–912. doi: 10.1042/BST0320910. [DOI] [PubMed] [Google Scholar]
- 31.Yoo CB, Jeong S, Egger G, Liang G, Phiasivongsa P, Tang C, Redkar S, Jones PA. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–6408. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 32.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U S A. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanemura A, Terando AM, Sim MS, van Hoesel AQ, de Maat MF, Morton DL, Hoon DS. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res. 2009;15:1801–1807. doi: 10.1158/1078-0432.CCR-08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 37.Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 38.Strathdee G, MacKean MJ, Illand M, Brown R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999;18:2335–2341. doi: 10.1038/sj.onc.1202540. [DOI] [PubMed] [Google Scholar]
- 39.Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69:1911–1934. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 40.Fukushima T, Takeshima H, Kataoka H. Anti-glioma therapy with temozolomide and status of the DNA-repair gene MGMT. Anticancer Res. 2009;29:4845–4854. [PubMed] [Google Scholar]
- 41.Wargo JA, Robbins PF, Li Y, Zhao Y, El-Gamil M, Caragacianu D, Zheng Z, Hong JA, Downey S, Schrump DS, Rosenberg SA, Morgan RA. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol Immunother. 2009;58:383–394. doi: 10.1007/s00262-008-0562-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 44.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 45.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 46.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 47.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 48.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia. 2008;22:1026–1034. doi: 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- 50.Bruserud O, Stapnes C, Ersvaer E, Gjertsen BT, Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr Pharm Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- 51.Lall S. Primers on chromatin. Nat Struct Mol Biol. 2007;14:1110–1115. doi: 10.1038/nsmb1107-1110. [DOI] [PubMed] [Google Scholar]
- 52.Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins. Curr Opin Genet Dev. 2008;18:152–158. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 53.Lee YH, Stallcup MR. Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol Endocrinol. 2009;23:425–433. doi: 10.1210/me.2008-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, Metzger E, Schule R, Eggert A, Buettner R, Kirfel J. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- 58.Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–12984. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohammad HP, Cai Y, McGarvey KM, Easwaran H, Van Neste L, Ohm JE, O'Hagan HM, Baylin SB. Polycomb CBX7 promotes initiation of heritable repression of genes frequently silenced with cancer-specific DNA hypermethylation. Cancer Res. 2009;69:6322–6330. doi: 10.1158/0008-5472.CAN-09-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, Kim JH, Han B, Tan P, Kumar-Sinha C, Lonigro RJ, Palanisamy N, Maher CA, Chinnaiyan AM. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–310. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 64.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, Gong Y, Cheng H, Laxman B, Vellaichamy A, Shankar S, Li Y, Dhanasekaran SM, Morey R, Barrette T, Lonigro RJ, Tomlins SA, Varambally S, Qin ZS, Chinnaiyan AM. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17:443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang J, Dorsey J, Chuikov S, Perez-Burgos L, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285:9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JP. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 68.Chang Y, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, Snyder JP, Bedford MT, Cheng X. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marango J, Shimoyama M, Nishio H, Meyer JA, Min DJ, Sirulnik A, Martinez-Martinez Y, Chesi M, Bergsagel PL, Zhou MM, Waxman S, Leibovitch BA, Walsh MJ, Licht JD. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim H, Heo K, Kim JH, Kim K, Choi J, An W. Requirement of histone methyltransferase SMYD3 for estrogen receptor-mediated transcription. J Biol Chem. 2009;284:19867–19877. doi: 10.1074/jbc.M109.021485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 72.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eacker SM, Dawson TM, Dawson VL. Understanding microRNAs in neurodegeneration. Nat Rev Neurosci. 2009;10:837–841. doi: 10.1038/nrn2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Friedman JM, Liang G, Liu CC, Wolff EM, Tsai YC, Ye W, Zhou X, Jones PA. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res. 2009;69:2623–2629. doi: 10.1158/0008-5472.CAN-08-3114. [DOI] [PubMed] [Google Scholar]
- 75.Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li JJ, Rocken C, Ebert MP, Kwok TT, Sung JJ. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer. 2009;101:699–706. doi: 10.1038/sj.bjc.6605195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 78.Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM, Esteller M. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008;105:13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCaffrey AP, Fawcett P, Nakai H, McCaffrey RL, Ehrhardt A, Pham TT, Pandey K, Xu H, Feuss S, Storm TA, Kay MA. The host response to adenovirus, helper-dependent adenovirus, and adeno-associated virus in mouse liver. Mol Ther. 2008;16:931–941. doi: 10.1038/mt.2008.37. [DOI] [PubMed] [Google Scholar]
- 81.Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 83.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 84.Jahangeer S, Elliott RM, Henneberry RC. beta-Adrenergic receptor induction in HeLa cells: synergistic effect of 5-azacytidine and butyrate. Biochem Biophys Res Commun. 1982;108:1434–1440. doi: 10.1016/s0006-291x(82)80067-3. [DOI] [PubMed] [Google Scholar]
- 85.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 86.Kuendgen A, Lubbert M. Current status of epigenetic treatment in myelodysplastic syndromes. Ann Hematol. 2008;87:601–611. doi: 10.1007/s00277-008-0477-9. [DOI] [PubMed] [Google Scholar]
- 87.Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23–36. doi: 10.1038/nrc2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, Licht J, McConnell MJ, Nasrallah C, Kim MK, Zhang W, Sun Y, Murgo A, Espinoza-Delgado I, Oteiza K, Owoeye I, Silverman LR, Gore SD, Carraway HE. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114:2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, Cortes J, Wierda WG, Ouzounian S, Quezada A, Pierce S, Estey EH, Issa JP, Kantarjian HM, Garcia-Manero G. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 90.Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, Balusu R, Koul S, Atadja P, Marquez VE, Bhalla KN. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Crea F, Giovannetti E, Cortesi F, Mey V, Nannizzi S, Gallegos Ruiz MI, Ricciardi S, Del Tacca M, Peters GJ, Danesi R. Epigenetic mechanisms of irinotecan sensitivity in colorectal cancer cell lines. Mol Cancer Ther. 2009;8:1964–1973. doi: 10.1158/1535-7163.MCT-09-0027. [DOI] [PubMed] [Google Scholar]
- 92.Ramalingam SS, Parise RA, Ramanathan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ, Belani CP. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007;13:3605–3610. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 93.Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, Thomas S, Espinoza-Delgado I, Vokes EE, Gandara DR, Belani CP. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rasheed W, Bishton M, Johnstone RW, Prince HM. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther. 2008;8:413–432. doi: 10.1586/14737140.8.3.413. [DOI] [PubMed] [Google Scholar]
- 95.Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva Lde P, Yang H, Alexander S, Wolff J, Kurzrock R. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. 2008;14:6296–6301. doi: 10.1158/1078-0432.CCR-08-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Balch C, Fang F, Matei DE, Huang TH, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009;150:4003–4011. doi: 10.1210/en.2009-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin J, Gilbert J, Rudek MA, Zwiebel JA, Gore S, Jiemjit A, Zhao M, Baker SD, Ambinder RF, Herman JG, Donehower RC, Carducci MA. A phase I dose-finding study of 5-azacytidine in combination with sodium phenylbutyrate in patients with refractory solid tumors. Clin Cancer Res. 2009;15:6241–6249. doi: 10.1158/1078-0432.CCR-09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berdasco M, Ropero S, Setien F, Fraga MF, Lapunzina P, Losson R, Alaminos M, Cheung NK, Rahman N, Esteller M. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci U S A. 2009;106:21830–21835. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silverman LR, McKenzie DR, Peterson BL, Holland JF, Backstrom JT, Beach CL, Larson RA. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 100.Kantarjian HM, O'Brien S, Shan J, Aribi A, Garcia-Manero G, Jabbour E, Ravandi F, Cortes J, Davisson J, Issa JP. Update of the decitabine experience in higher risk myelodysplastic syndrome and analysis of prognostic factors associated with outcome. Cancer. 2007;109:265–273. doi: 10.1002/cncr.22376. [DOI] [PubMed] [Google Scholar]
- 101.Gore SD, Weng LJ, Zhai S, Figg WD, Donehower RC, Dover GJ, Grever M, Griffin CA, Grochow LB, Rowinsky EK, Zabalena Y, Hawkins AL, Burks K, Miller CB. Impact of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin Cancer Res. 2001;7:2330–2339. [PubMed] [Google Scholar]
- 102.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 103.Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Munster PN, Marchion D, Thomas S, Egorin M, Minton S, Springett G, Lee JH, Simon G, Chiappori A, Sullivan D, Daud A. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer. 2009;101:1044–1050. doi: 10.1038/sj.bjc.6605293. [DOI] [PMC free article] [PubMed] [Google Scholar]