

Abstract
Recently, we reported that membrane androgen receptors (mARs) are expressed in colon tumors triggering strong apoptotic responses. In the present study, we analyzed mAR-induced downstream effectors controlling cell survival and migration of Caco2 colon cancer cells. We show that long-term activation of mAR downregulated the activity of PI-3K and Akt and induced de-phosphorylation/activation of the proapoptotic Bad (p-Bad). Moreover, treatment of APCMin/+ mice, which spontaneously develop intestinal tumors, with mAR-activating testosterone conjugates reduced the tumor incidence by 80% and significantly decreased the expression of p-Akt and p-Bad levels in tumor tissue. Furthermore, mAR activation strongly inhibited Caco2 cell migration. In accordance with these findings, vinculin, a protein controlling cell adhesion and actin reorganization, was effectively phosphorylated upon mAR activation. Phosphorylation inhibitors genistein and PP2 inhibited actin reorganization and restored motility. Moreover, silencing vinculin by appropriate siRNA’s, or blocking actin reorganization by cytochalasin B, restored the migration potential. From these results we conclude that mAR activation inhibits the prosurvival signals Akt/Bad in vitro and in vivo and blocks migration of colon cancer cells via regulation of vinculin signaling and actin reorganization, supporting the powerful tumoristatic effect of those receptors.
INTRODUCTION
Recent studies established the expression of functional membrane androgen receptors (mARs) inducing rapid nongenomic androgen actions in tumors, including prostate (1,2), breast (3,4) and colon (5) as well as in other cell types such as macrophages and T cells (6,7), C6 (8) and vascular smooth muscle cells (9). The exact molecular identity of mAR remains unknown. It has been reported that signals emanating from this receptor, such as intracellular calcium or inositol 1,4,5-trisphosphate, are sensitive to pertussis-toxin inhibition (10,11), indicating that mAR may be a specific G-protein coupled receptor (GPCR) or a receptor in close association with a GPCR. Moreover, stimulation of mARs by membrane-impermeable testosterone conjugates triggered specific early signaling pathways and induced proapoptotic responses that could not be blocked by antiandrogens (1,5,12,13,14), implying that mAR effects are most likely different from those manifested upon activation of the intracellular androgen receptors (iARs).
The mAR-dependent nongenomic signaling was recently characterized in detail in prostate and breast cancer cells (15), and key prosurvival and proapoptotic gene products were identified that regulate the apoptotic response induced by mAR activation (16). According to these reports it was postulated that mAR might be a significant novel target for cancer treatment. Most recently, we reported that mARs (but not membrane-bound iARs) are expressed in colon tumors (5). Activation of mARs by testosterone-albumin conjugates triggered strong apoptotic responses and considerably reduced the colonic tumor incidence following chemical cancerogenesis in Balb/c mice (5). Moreover, mARs were predominantly expressed in colon tumor cells, whereas very low or even undetectable mARs were expressed in normal tissues (5). Although the proapoptotic responses were clearly dependent on mAR stimulation, downstream events regulating the expression and/or function of key prosurvival mediators in colon tumors remained undefined. In addition, the profound antitumorigenic mAR action has not been functionally correlated with changes in other key mechanisms such as cell motility and invasiveness.
The main goal of this work was to address the role of mAR activation toward major characteristics of tumor cells, namely cell survival and cell migration. Because mAR activation has been shown to promote strong apoptotic regression (2,5), we analyzed the functionality of the prosurvival PI-3K/Akt pathway. In addition, because this signaling pathway also plays a major role in the invasive potential of tumor cells, we further studied the migration potential upon mAR activation. Our findings indicate that prosurvival signaling prevails in colon tumor cells but is strongly downregulated upon mAR stimulation in vitro and in colon tumor tissues isolated from APCMin/+ mice following treatment with mAR agonists. Furthermore, mAR activation blocked migration and invasiveness of colon tumor cells, mainly recruiting the adhesion-and actin cytoskeleton–regulator vinculin. These results provide novel mechanistic insights into the regulation of the proapoptotic and antimigratory mAR effects in colon tumors.
MATERIALS AND METHODS
Cell Cultures and Wound Healing Assay
The Caco2 human colon cancer cell line and IEC06 nontransformed intestinal cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and were studied between passages 60 and 70. On the basis of previous titration experiments (5), we used a 10−7 mol/L testosterone–human serum albumin (HSA) concentration for mAR stimulation. For the wound healing assay, confluent cell cultures were scraped with a pipette tip across a 24-well plate. Following wounding, culture medium was replaced with fresh medium, and cells were exposed to 10−7 mol/L testosterone-HSA (Sigma, St. Louis, MO, USA) in the presence or absence of 10−6 mol/L cy-tochalasin B (Sigma) for the indicated time. It should be noted that the method for the wound healing assay requires that confluent cells are used, to provide the necessary cell layer to create wounding by scraping. On the other hand, subconfluent cells were used in Matrigel and Transwell assays (see below) to ensure correct evaluation of cell motility and invasiveness.
Matrigel and Transwell Assays
Matrigel assays were performed by using BD BioCoat™ BD Matrigel™ Invasion Chambers (BD Bioscience, San Jose, CA, USA). Matrigel was placed in each insert with 8.0-μm pore size in a 24-well plate. The chamber was allowed to polymerize at 37°C for 1 h. The inserts were then washed with serum-free DMEM, and 100 μL of complete cell culture medium with 1 × 105 cells was then seeded onto the insert. Five hundred microliters of complete cell culture medium with 10−7 mol/L testosterone-HSA or testosterone–bovine serum albumin (BSA) (Sigma) in the presence or absence of 10−6 mol/L cytochalasin B (Sigma) was added into the well below the insert. In control experiments, cells were preincubated with 10−7 mol/L testosterone-albumin conjugates (TAC) for 2 h. Then, TAC was washed out with complete cell culture medium and 500 μL were added into the well below the insert. After a 24-h incubation, the insert was wiped with a wet cotton swab. The lower surface was gently rinsed with phosphate-buffered saline (PBS), the cells were fixed and stained with 4,6-diamidino-2-phenylindole (DAPI) for 10 min, rinsed again with sterile water and allowed to dry. After the membranes were removed from the inserts, they were mounted with ProLong Gold antifade reagent (Invitrogen, Paisley, UK). To determine the total number of migrating cells, the slices were viewed and imaged under the microscope, and the number of cells/field in 10 random fields was counted. Experiments were performed in triplicates.
The Transwell assay was performed using Transwell inserts (BD Bioscience). The inserts were then washed with serum-free DMEM, and 100 μL of complete cell culture medium with 1 × 105 cells were seeded onto the insert. We then added 500 μL of complete cell culture medium with 10−7 mol/L testosterone-HSA or testosterone-BSA (Sigma) in the presence or absence of 10−6 mol/L cytochalasin B (Sigma), 10−6 mol/L anastrozole (Sigma), 10−6 mol/L flutamide (Sigma), 5 × 10−6 mol/L PP2 (Sigma) or 5 × 10−6 mol/L genistein (Sigma) into the well underside of the insert for 24 h at 37°C and 5% CO2. In control experiments, cells were preincubated with 10−7 mol/L TAC for 2 h. Then, TAC was washed out with complete cell culture medium and 500 μL was added into the well below the insert. After 24 h incubation, cells were fixed, stained with DAPI for 10 min and microscopically analyzed as described above.
Immunoprecipitation and Western Blotting
Cells were incubated with 10−7 mol/L testosterone-HSA, free testosterone or free HSA for the indicated time, washed twice with ice-cold PBS and suspended in 500 μL ice-cold lysis buffer (50 mmol/L Tris/HCl, 1% TritonX-100 pH 7.4, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 0.15% NaCl, 1 mmol/L EDTA, 1 mmol/L sodium orthovanadate) containing pro-tease inhibitor cocktail (Sigma). The protein concentration was determined using the Bradford assay (Bio-Rad, Munich, Germany). Sixty micrograms of protein were solubilized in sample buffer at 95ºC for 5 min and resolved by 10% SDS–polyacrylamide gel electrophoresis. For immunoblotting, proteins were electrotransferred onto a polyvinylidene difluoride membrane and blocked with 5% nonfat milk in Tris-buffered saline–0.10% Tween 20 at room temperature for 1 h. Then, the membrane was incubated with phospho-Akt (Thr308), phospho-PI-3K p85 (Tyr458)/p55 (Tyr199) (1:1000; Cell Signaling, Danvers, MA, USA), or phospho-Bad (Ser136) (1:100, Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight. After washing (in PBS with 0.05% Tween [PBST]) and subsequent blocking, the blot was incubated with secondary antirabbit antibody (1:2000; Cell Signaling) or antimouse antibody (1:5000, GE Healthcare, Piscataway, NJ, USA) for 1 h at room temperature. After washing, antibody binding was detected with the enhanced chemiluminescence detection reagent (Amersham, Freiburg, Germany). For controls the blots were stripped in stripping buffer (Carl Roth, Karlsruhe, Germany) at 56°C for 30 min. After washing with PBST, blots were blocked with Tris-buffered saline with Tween + 5% milk for 1 h at room temperature. Then, they were incubated with anti-Akt or anti-Bad (1:100; Santa Cruz Biotechnology) antibodies at 4°C overnight. After washing with PBST and incubation with antirabbit antibody (1:2000; Cell Signaling), antibody binding was detected and quantified with Quantity One Software (Bio-Rad).
For immunoprecipitation, equal amounts of protein (500 μg) in the presence or absence of 50 μmol/L genistein were subjected to immunoprecipitation with a monoclonal antiphosphotyrosine antibody (7 μg/500 μg total protein, Santa Cruz Biotechnology). After incubation with the antibody for 1 h at 4°C, 50 μL of the homogeneous protein A suspension was added into the mixture and incubated overnight at 4°C on a rocking platform. After three washing steps, samples were resuspended in SDS sample buffer, subjected to SDS electrophoresis and transferred to nitrocellulose membrane. Proteins were incubated with vinculin monoclonal antibody (1:100; Santa Cruz Biotechnology) followed by the appropriate antimouse antibody (1:5000; GE Healthcare). Detection of protein bands was performed with an enhanced chemiluminescence kit. Bands were quantified with Quantity One Software (Bio-Rad).
Immunofluorescence Analysis and Confocal Laser Scanning Microscopy
For testosterone-HSA–fluorescein isothiocyanate (FITC) staining, 5-μm–thick frozen tissue sections from the adenomatous polyposis coli (APC) mouse tumors were fixed with 4% paraformaldehyde (PFA) for 15 min and incubated with 5% BSA/1 × PBS/0.3% Triton for 1 h at room temperature. After two washes with PBS with 1.5% fetal bovine serum, specimens were exposed to testosterone-HSA-FITC (10−7 mol/L; Sigma) for 1 h at room temperature. Nuclei were stained with DRAQ-5 dye (1:1000; Biostatus, Leicestershire, UK) for 10 min at room temperature.
To quantify the expression of phosphorylated Akt and Bad, 5-μm–thick frozen tissue sections from the APC mice colon tumors (at least three tumors of similar size from each animal) were fixed with 4% PFA for 15 min at room temperature. After washing twice with PBS the slides were incubated with 5% normal goat serum/1 × PBS/0.3% Triton for 1 h at room temperature. Then, the specimens were exposed overnight at 4°C to phospho-Akt (Thr308) (1:800; Cell Signaling) or phospho-Bad (Ser136) (1:100, Santa Cruz Biotechnology). The slides were rinsed three times with PBS and incubated for 1.5 h at room temperature with secondary FITC goat antirabbit antibody (1:500; Invitrogen). After three washing steps the nuclei were stained for 10 min at room temperature with DRAQ-5 dye (1:1000; Biostatus).
To determine the phosphorylation of vinculin, cells were cultured on glass cover slips with testosterone-HSA or control without testosterone-HSA for different time periods, which are indicated in the figure legends. After washing twice with PBS, cells were incubated with 4% PFA for 15 min and then incubated with 5% normal goat serum/1 × PBS/0.3% Triton for 1 h at room temperature. Then, the cells were exposed to antivinculin antibodies (1:400; Gene Tex, Irvine, CA, USA) at 4°C overnight. The cells were rinsed three times with PBS and incubated with secondary FITC goat antirabbit antibody (1:500; Invitrogen) or goat antimouse antibody (1:500; Invitrogen) for 1.5 h at room temperature. For F-actin staining, cells were incubated with rhodamine-phalloidin (1:100; Molecular Probes, Eugene, OR, USA) for 40 min in the dark. After three washing steps the nuclei were stained with DRAQ-5 dye (1:1000; Biostatus) for 10 min at room temperature. All the slides and cover slips were mounted with ProLong Gold antifade reagent (Invitrogen). Images were taken on a Zeiss LSM 5 EXCITER Confocal Laser Scanning Microscope (Carl Zeiss MicroImaging, Jena, Germany) with a water-immersion Plan-Neofluar 40×/1.3 NA DIC. Images were analyzed with the instrument’s software.
Small Interfering RNA Experiments
Caco2 cells were grown in DMEM medium containing 10% fetal calf serum under standard culture conditions (37°C, 5% CO2). We then seeded 4× 104 cells in 24-well plates and cultivated them with fresh culture medium for 8 h. The cells were subsequently transfected with validated small interfering RNA (siRNA) for vinculin (ID# s14764; Ambion, Darmstadt, Germany) or with a negative control siRNA by using an siPORT Amine (Ambion) transfection agent according to the manufacturer’s protocol. The efficiency of silencing was checked by Western blot 72 h after transfection. Upon silencing, 37.6% of the vinculin protein was still detectable in cells treated with siRNA for Vinculin compared with cells treated with a negative control siRNA.
In Vivo Experiments
In vivo animal experiments were carried out in 2- to 6-month-old age-matched mice of both sexes with mutated apc resulting in spontaneous colon tumor development (APCMin/+) obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The animals were housed under controlled environmental conditions (22°–24°C, 50–70% humidity and a 12-h light/dark cycle). Throughout the study the mice had free access to standard pelleted food (C1000; Altromin, Lage, Germany) and tap water. All animal experiments were conducted according to the German law for the care and welfare of animals and were approved by local authorities.
The animals were divided into two groups. Group A (n = 6) received 2× 5 mg/kg subcutaneous TAC injections 3 times per wk for 8 wks. In the control group B (n = 4) similar doses of normal saline were given. At the end of 8 wks all animals were anesthetized with ether and killed. After death, the entire colorectum from the colorectal junction to the anal verge was examined. Then, the colon was opened longitudinally, washed with PBS, and divided into three portions (proximal, middle and distal). Tumors were counted with a dissecting microscope at 3× magnification. After inspection the colon was fixed in a 40% g/L formaldehyde buffer solution (pH 7.4).
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling Assay
The colonic cancer tissues (at least three independent tumors from each animal) were cut into 8-μm frozen sections and subsequently fixed in 4% PFA for 30 min at room temperature. After rinsing with PBS the samples were permeabilized in a solution of 0.1% Triton X-100 in sodium citrate for 2 min. Samples, washed with PBS, were then incubated in the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) reaction mix for 1 h at 37°C, according to the manufacturer’s instructions (Roche, Mannheim, Germany). Nuclei were stained with DRAQ5™ (Biostatus). Sections were analyzed with a confocal laser scanning microscope (Carl Zeiss).
APOPercentage Apoptosis Assay
Caco2 cells were cultured in 96-well plates for the APOPercentage apoptosis assay (Biocolor, Belfast, Ireland). In the presence or absence of 10−6 mol/L anastrozole (Sigma), the cells were stimulated or not with 10−7 mol/L TAC for 24 h in serum-containing medium. Untreated cells cultured in serum-free medium were used as a positive control for the apoptotic response.
Statistical Analysis
Data are provided as means ± SEM, n represents the number of independent experiments. Data were tested for significance by using the unpaired Student t test or ANOVA as appropriate. Differences were considered statistically significant when P values were < 0.05. Statistical analysis was performed with GraphPad InStat version 3.00 for Windows 95 (GraphPad Software, San Diego, CA; USA, www.graphpad.com).
All supplementary materials are available online at www.molmed.org.
RESULTS
mAR Stimulation Inhibits Akt Activity and Induces Bad Dephosphorylation in Caco2 but Not in IEC06 Cells
Activated Akt is a prosurvival factor controlling phosphorylation and activity of various proapoptotic gene products (17,18). Consistent with its well-documented oncogenic role in tumor cells, high basal levels of active, phosphorylated Akt (p-Akt) were detected in Caco2 colon tumor cells (Figure 1A, B). Stimulation of mARs with TAC induced a long-term and profound dephosphorylation of this kinase that became evident 2 h after TAC treatment and continued for at least 12 h (Figure 1A), compared with control untreated p-Akt levels (Figure 1B). Interestingly, in nontransformed IEC06 intestinal cells that do not express mARs (5), p-Akt levels were very low and remained unchanged during TAC treatment (data not shown), indicating mAR specificity for the regulation of this pro-survival factor. Additional control experiments performed by treatments either with testosterone not linked to HSA or HSA not linked to the androgen indicated clearly that long-term p-Akt downregulation was not obvious when Caco2 cells were treated with testosterone (Supplemental Figure 1A). Although this finding may seem contradictory to existing results with testosterone-HSA, it is consistent with a dual role of testosterone in simultaneous binding of mARs (thereby inducing p-Akt downregulation) and iARs (thereby inducing p-Akt upregulation) within the same cell. This assumption is further supported in experiments showing regeneration of the long-term p-Akt downregulation when cells were pre-treated with flutamide to block iAR-testosterone interactions (Supplemental Figure 1B). In addition, p-Akt expression was also not influenced upon treatment of the cells with HSA (Supplemental Figure 1C). Based on the well-documented role of Akt in inactivating the proapoptotic function of Bad via phosphorylation (19,20), we further analyzed the activity of Bad. Figure 1C clearly shows that mAR stimulation by TAC resulted in de-phosphorylation/activation of Bad following kinetics similar to those of Akt, reaching minimum levels of phosphorylated Bad (p-Bad) after 12 h. In line with these findings, the Akt-upstream regulator PI-3K was dephosphorylated upon long-term TAC treatment (Figure 1D), indicating that the prosurvival PI-3K/Akt signaling is downregulated in mAR-activated Caco2 cells.
Figure 1.

Dephosphorylation/inactivation of Akt and Bad and PI-3K in Caco2. Caco2 cells were exposed to 10−7 mol/L testosterone-HSA for the indicated time periods. The ratio of the cellular content of the phosphorylated residues (Thr 308) versus the total isoform of Akt (A, B) and the phosphorylated residues (Ser 136) versus the total isoform of Bad (C) were measured in cell lysates by Western blotting using specific antibodies for each form and were normalized to the corresponding control. Blots show a representative experiment, the numbers below each lane correspond to the mean values ± SE from three independent experiments (*P < 0.05; **P < 0.01) indicating the fold-increase in the phosphorylation level for the indicated time point normalized to the controls. (B) Control experiment showing variation of p-Akt levels over time (representative blot from two distinct experiments). (D) Following cell lysis equal amounts of total lysates were immunoblotted (IB) with a specific antibody against phospho-PI-3K (p85) and total PI-3K. Immunoblots were analyzed by densitometry. The intensity of phospho-PI-3K (p85) was normalized to the intensity of the corresponding total PI-3K band. Blots show a representative experiment, whereas the relative fold-increases are indicated as mean values ± SE from three independent experiments of PI-3K phosphorylation with that of untreated cells taken as one (*P < 0.05; ***P < 0.001).
p-Akt and p-Bad Are Downregulated in Colon Tumor Tissues Treated by Testosterone-HSA
Having established a role of AKT/Bad downstream of mAR activation in colon cancer cells, we sought to determine the possible inactivation of p-Akt/p-Bad in colon tumor specimens isolated from testosterone-HSA–treated mice. To address this, we first assessed the 8-wk incidence of colon tumors spontaneously developed in APCMin/+ mice in the presence or absence of continuous testosterone-HSA treatment. Notably, tumor tissues isolated from these mice express considerable amounts of mARs (Figure 2A). In our experiments, animals were divided in two groups comprising six and four animals. One group (six animals) was treated subcutaneously (3 times/wk, for 8 wks) with 5 mg/kg testosterone-HSA, whereas the other group (four animals) remained untreated. As shown in Figure 2B, testosterone-HSA treatment resulted in a significant reduction of the tumor incidence by 80%. The histological analysis of tumors by TUNEL assay confirmed that apoptotic cells were present in appreciable numbers predominantly in the tumors of animals treated with testosterone-HSA (Figure 2C, left panels), whereas they were significantly less abundant in the nontreated animals (Figure 2C, right panels). Immunohistochemical analysis with either anti–p-Akt (Thr308) or anti–p-Bad (Ser136) antibodies revealed strong expression of p-Akt and p-Bad in colonic tumor tissue from untreated mice and marked downregulation of both phosphorylated proteins in colon tumor tissues from testosterone-HSA–treated animals (Figure 2D). These results clearly prove the antitumor potential of testosterone-albumin conjugates in colon cancer and corroborate previously reported data obtained in a chemically induced colon tumor model in Balb/c mice (5). Furthermore, our results identify Akt/Bad as downstream targets of mAR in colon cancer cells in vitro and in vivo.
Figure 2.

In vivo testosterone-HSA effects on tumor incidence in APCMin/+ mice. APCMin/+ mutant mice of both sexes were used in this study. (A) Confocal laser scanning microscopic analysis of APCMin/+ colon tumor frozen sections stained with testosterone-HSA-FITC or HSA-FITC, showing specific FITC-related fluorescence at the cell membranes. Visualization of the nuclei was evident by DRAQ5™ staining. Magnification 100×. (B) Arithmetic means ± SEM of colonic tumor incidence in APCMin/+ mice. The mice were either treated with 5 mg/kg testosterone-HSA subcutaneously 3 times/wk for 8 wk, (n = 6 animals, black bar) or treated with 5 mg/kg of normal saline subcutaneously 3 times/wk for 8 wk (n = 4 animals, blue bar); ** indicates significant difference between both groups (P < 0.01). (C) After treatment, the APCMin/+ colonic cancer tissue was cut to 8-μm frozen sections, and fragmented DNA was assessed using TUNEL assay according to the manufacturer’s instructions. Representative confocal laser scanning microscopy analyzed samples from at least three independent tumors from each animal; magnification 100×. (D) Representative confocal laser scanning microscopic analysis of TAC-treated and untreated APCMin/+ frozen colon tumor sections prepared from at least three tumors of similar size from each animal stained with (a) anti–phospho-Akt (Thr308) and (b) anti–phospho-Bad (Ser136) antibodies. Antirabbit FITC was used as secondary antibody and DRAQ5™ for nuclei staining; magnification 100×.
mAR Activation Inhibits Cell Motility in Colon Cancer Cells
Inhibition of Akt-dependent pathways has recently been linked to reduced cell motility in colon cancer (21). Thus, we sought to determine whether mAR-stimulation regulates motility and invasiveness of colon cancer cells. To this end, we assessed the migration capacity of Caco2 cells treated or untreated with testosterone-HSA by using the Matrigel, Transwell and wound healing assays, respectively. As shown in Figure 3A, the Matrigel assay revealed that cell invasiveness was reduced by 90% following testosterone-HSA treatment of Caco2 cells. Very similar results were also obtained when cells were treated with testosterone-BSA (data not shown), indicating that both testosterone albumin conjugates produce similar effects in the inhibition of cell motility. In line with this finding, cell motility assessed by the wound healing assay confirmed the inhibition of the migratory capacity of colon cancer cells by mAR stimulation (Figure 3B). The inhibition of the migratory potential of tumor cells by mAR stimulation was further corroborated by the Transwell assay (Figure 3C). Interestingly, in the presence of nontoxic concentrations of cytochalasin B that block the well-described mAR-induced actin reorganization, the inhibition of cell motility was partially restored in all experimental procedures (Figure 3A–C). Finally, to further confirm the androgen specificity of the mAR-induced effects, we analyzed cell motility and the apoptotic response of TAC-treated Caco2 cells in the presence of the aromatase inhibitor anastrozole. As shown in Figure 4, neither the migration potential nor the apoptotic response were influenced by this inhibitor.
Figure 3.

Effect of testosterone-HSA on Caco2 human colon cancer cell migration and adhesion. (A) Cells were cultured with 10−7 mol/L testosterone-HSA in the presence or absence of 10−6 mol/L cytochalasin B on the Matrigel-coated upper compartment of Tran-swell culture chambers, provided with an 8-μm pore-size polycarbonate filter, according to the manufacturer’s instructions. Matrigel was removed by scraping 24 h later, and invaded cells, attached to the lower surface of the filter, were stained with DAPI. The slices were imaged under the microscope and the number of cells in 10 random fields was counted. Bars represent the % of cell invasion in control and treated cells; (n = 3), ***P < 0.001. (B) Wound healing assay of Caco2 cells. Following 24-h culture the confluent monolayer was scratched with a pipette tip to create a cell-free area. Testosterone-HSA 10−7 mol/L was added, and wound closure was documented by microphotography of the same region after 24 h. Bars represent the width of the wound in control and treated cells; *P < 0.05; ***P < 0.001. (C) Cells in the presence or not of 50 μmol/L genistein and PP2 were cultured with 10−7 mol/L testosterone-HSA in the absence or presence of 10−6 mol/L cytochalasin B on the Transwell culture chambers, provided with an 8-μm pore-size polycarbonate filter, according to the manufacturer’s instructions. After 24 h, invaded cells, attached to the lower surface of the filter, were stained with DAPI. The slices were imaged under the microscope, and the number of cells in 10 random fields was counted. Bars represent the % of cell invasion in control and treated cells; (n = 3), ***P < 0.001.
Figure 4.
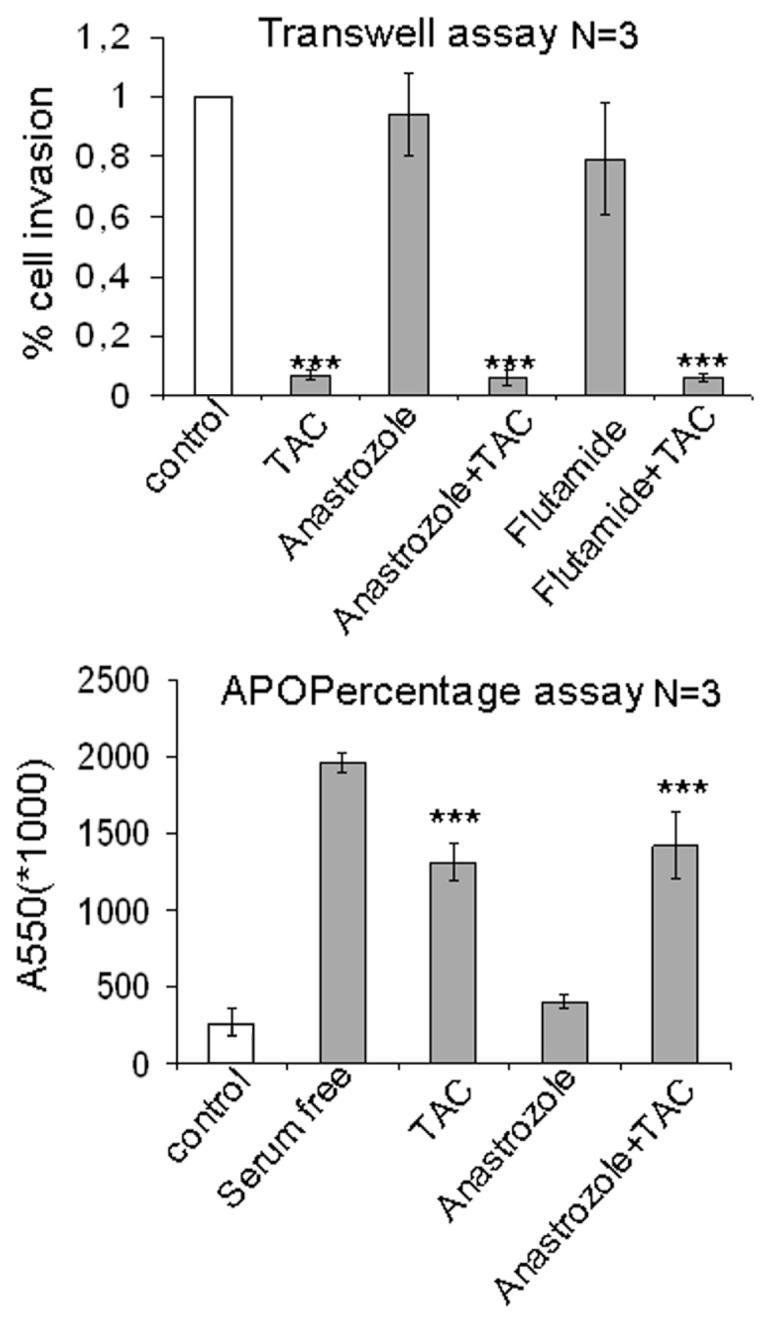
Motility and proapoptotic effects of testosterone-HSA in the absence or presence of inhibitors in Caco2 cells. (A) Cells both in and out of the presence of 10−6 mol/L anastrozole or 10−6 mol/L flu-tamide were cultured with 10−7 mol/L testosterone-HSA on the Transwell culture chambers, provided with an 8-μm pore-size polycarbonate filter, according to the manufacturer’s instructions. After 24 h, invaded cells, attached to the lower surface of the filter, were stained with DAPI. The slices were imaged under the microscope, and the number of cells in 10 random fields was counted. Bars represent the % of cell invasion in control and treated cells; (n = 3), ***P < 0.001. (B) Quantitative APOP-ercentage apoptosis assay of TAC-stimulated Caco2 cells and similar experiments in the presence of anastrozole. Cells were exposed or not exposed to 10−7 mol/L testosterone-HSA for 24 h and proapoptotic responses were assessed by the APOPercentage apoptosis assay. Equally, cells pretreated or not pretreated with 10−6 mol/L anastrozole were exposed to testosterone-HSA for 24 h. Cells serum starved for comparable periods of time served as a positive control for apoptosis. Bars present the mean OD measured at 550 nm; *** P < 0.001, n = 3.
mAR Activation Triggers Vinculin Phosphorylation
The findings presented in Figure 3 suggest that actin restructuring may control cell migration upon mAR activation in colon cancer cells. As tyrosine phosphorylation of specific proteins is linked to actin cytoskeleton dynamics and cellular motility (22,23), we sought to determine whether mAR effects on cell motility were evident in the presence of genistein and PP2, which are widely used tyrosine phosphorylation inhibitors. Figure 3C shows that this was indeed the case, because genistein and PP2 efficiently blocked mAR-induced inhibition of cell migration. Notably, neither inhibitor alone had an effect in this assay (data not shown). We subsequently checked for molecular targets potentially regulating these effects. We focused on vinculin, an adhesion protein that participates in cell-cell adhesions and that has been reported to regulate migration and actin organization (24). As shown in Figure 5A, mAR stimulation with testosterone-HSA disclosed a substantial increase in the phosphorylation of vinculin within 15 min that remained for at least 12 h. Confocal microscopy analysis fully confirmed the Western-blotting data. Indeed, immunostaining of testosterone-HSA–treated Caco2 cell preparations with antivinculin antibodies (Figure 5B) revealed gradually increased vinculin spots observed at the focal adhesions. Triple immunostaining with antivinculin antibodies, rhodamine-phalloidin for F-actin staining and DRAQ-5–dye nuclear staining clearly documented the formation of actin filaments emanating from the vinculin spots of focal adhesions (Figure 5B). Interestingly, in the presence of the tyrosine phosphorylation inhibitor genistein, vinculin phosphorylation was effectively blocked (Figure 6A). Moreover, confocal laser scanning analysis revealed that both the expression of the characteristic vinculin spots and the formation of actin filaments, shown in Figure 5B, disappeared in mAR-stimulated Caco2 cells in the presence of genistein (Figure 6B). These results, coupled with the observed blocking effect of genistein and PP2 in mAR-induced cell motility of Caco2 cells (Figure 3C), provide indirect evidence that vinculin phosphorylation may participate in mAR signaling toward actin reorganization and cell motility regulation.
Figure 5.

Effects of testosterone-HSA on vinculin phosphorylation and on Caco2 cell morphology. (A) Caco2 cells were stimulated with 10−7 mol/L testosterone-HSA for the indicated time periods. Following cell lysis equal amounts of proteins were immunoprecipitated (IP) with an anti-phosphotyrosine (p Tyr) antibody. The tyrosine-phosphorylated as well as equal amounts of total lysates were immunoblotted (IB) with a specific antibody against vinculin. The immunoblots were analyzed by densitometry. The intensity of phosphorylated vinculin bands was normalized to the intensity of the corresponding total vinculin bands. Blots show a representative experiment, whereas the relative fold-increase (mean values ± SE from three independent experiments) in vinculin phosphorylation with that of untreated cells taken as one are indicated. (**P < 0.01). (B) Confocal laser scanning microscopic analysis of vinculin and actin in mAR-activated Caco2 cells. Cells treated or not with 10−7 mol/L testosterone-HSA for different time periods were cultured on cover slips, fixed and stained with mouse antivinculin, anti–mouse-FITC as secondary antibody, DRAQ5™ for nuclei staining and rhodamine-phalloidin for filamentous actin staining; magnification 100×.
Figure 6.

Phosphorylation of vinculin in TAC-treated Caco2 cells in the presence of genistein. (A) Caco2 cells in the presence of genistein were stimulated with 10−7 mol/L testosterone-HSA for the indicated time periods. Following cell lysis equal amounts of proteins were immunoprecipitated (IP) with an antiphosphotyrosine (p-Tyr) antibody. The tyrosine-phosphorylated as well as equal amounts of total lysates were immunoblotted (IB) with a specific antibody against vinculin. The intensity of phosphorylated vinculin bands was normalized to the intensity of the corresponding total vinculin bands. (B) Confocal laser-scanning microscopic analysis of vinculin and actin in mAR-activated Caco2 cells in the presence of genistein. Cells treated with 10−7 mol/L testosterone-HSA for different time periods were cultured in coverslips, fixed and stained with mouse antivinculin, anti–mouse-FITC as secondary antibody, DRAQ5™ for nuclei staining and rhodamine-phalloidin for filamentous actin staining. Magnification 100×.
Vinculin Is Necessary for Actin Reorganization and Migration of mAR Stimulated Caco2 Cells
To provide direct and specific experimental verification for the regulatory action of vinculin in mAR-stimulated Caco2 cells, we designed siRNAs that could induce downregulation of endogenous vinculin. The siRNA was capable of inducing knockdown of endogenous vinculin to almost 70% of the control levels when introduced into Caco2 cells by transient transfection (Figure 7A). The effect of the siRNA was specific, because an unrelated siRNA had no apparent effect on the protein levels of vinculin (Figure 7A). Analysis of the vinculin and actin cytoskeleton of cells under such conditions of siRNA transient transfection showed a dramatic inhibitory effect on the ability of testosterone-HSA to affect vinculin morphology and actin reorganization (Figure 7B). In contrast, the control siRNA did not interfere with the actin and vinculin responses to testosterone-HSA. In addition, the relative decrease of endogenous vinculin levels led to a significant restoration of the migratory capacity of Caco2 cells upon mAR activation, as indicated by both the Matrigel and Transwell assays (Figure 7C, D). Similar results were obtained in cells pretreated with testosterone-HSA for 2 h prior to assessment of the migration potential by the Matrigel and Transwell assays in the presence or absence of siRNAs against vinculin or the corresponding negative siRNA control (Figure 8). The results of this experiment also suggest that upon testosterone-HSA treatment the observed inhibitory signals for cell migration and adhesion are triggered very early upon mAR activation, when cells are still viable, and cannot be attributed to an artificial response of dying cells measured after 24 h of TAC treatment (as presented in Figure 3). We conclude that vinculin is a critical component downstream of mAR that regulates the responses to actin-cytoskeleton reorganization and migration potential in colon tumor cells.
Figure 7.

Effect of testosterone-HSA on Caco2 cells silenced with vinculin siRNA (A) Caco2 cells transfected with vinculin siRNA or a negative control siRNA were lysed, and equal amounts of total lysates were immunoblotted (IB) with a specific antibody against vinculin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase). The immunoblots were analyzed by densitometry. (B) Confocal laser-scanning microscopic analysis of vinculin and actin in mAR-activated Caco2 cells silenced either with vinculin siRNA or a negative control siRNA. Transfected cells treated with 10−7 mol/L testosterone-HSA for different time periods were cultured in coverslips, fixed and stained with mouse antivinculin, anti–mouse-FITC as secondary antibody, DRAQ5™ for nuclei staining and rhodamine-phalloidin for filamentous actin. Magnification 100×. (C) Representative motility experiment of Caco2 cells transfected or not with vinculin siRNA. Cells were cultured in the presence of 10−7 mol/L testosterone-HSA on the Matrigel-coated upper compartment or Transwell culture chambers, provided with an 8-μm pore-size polycarbonate filter, according to the manufacturer’s instructions. After 24 h, invaded cells, attached to the lower surface of the filter, were stained with DAPI. (D) Quantification of the motility experiments shown in (C) for Caco2 cells silenced with vinculin siRNA and cultured in the presence of 10−7 mol/L testosterone-HSA as described in Figure 3A and C.
Figure 8.
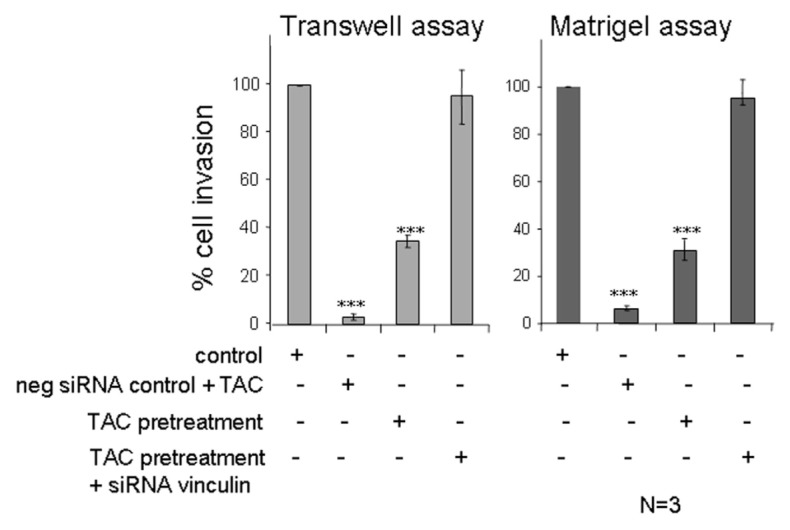
Effect of testosterone-HSA on Caco2 cells silenced with vinculin siRNA. Quantification of control Matrigel and Tran-swell motility experiments with Caco2 cells pretreated with 10−7 mol/L testosterone-HSA for 2 h and silenced with either negative control siRNA or vinculin siRNA. Bars represent the % of cell invasion in control and treated cells; (n = 3), ***P < 0.001.
DISCUSSION
The present paper discloses the novel finding that prosurvival signals are effectively downregulated in mAR-expressing colon tumors following stimulation by TAC in vitro and in vivo. First, phosphorylated kinase Akt, which is constitutively upregulated in colon tumors but not in nontransformed cells (Figure 1A, B; Supplemental Figure 1; and data not shown), was significantly downregulated upon long-term mAR activation by TAC. In line with this result, PI-3K, an upstream regulator of Akt, was dephosphorylated upon long-term TAC treatment (Figure 1D), indicating that the prosurvival PI-3K/Akt signaling is downregulated in mAR-activated Caco2 cells. In addition, the proapoptotic Bad protein was efficiently dephosphorylated and thus activated by TAC following similar kinetics (Figure 1C). These results suggest that, in contrast to nontransformed intestinal IEC06 cells, mAR-expressing colon tumor cells express activated pro-survival signals that may protect them from apoptotic cell death. mAR activation downregulated the activity of these signals via dephosphorylation, a finding consistent with the strong apoptotic regression upon mAR stimulation reported recently (5). These results were confirmed by in vivo experiments in TAC-treated APCMin/+ mice. This mouse model carries defective APC, which results in the spontaneous development of gastrointestinal tumors (25). Indeed, the colon tumor incidence observed in the treated 2- to 6-month-old age-matched animals was significantly reduced by 80% (Figure 2B), corroborating previously reported data obtained in a chemically induced colon tumor model in Balb/c mice (5). Interestingly, histological immunostaining analysis revealed that both p-Akt and p-Bad were effectively downregulated in colon tumor specimens isolated from TAC-treated animals (Figure 2D). These findings collectively provide novel mechanistic evidence, pointing to p-Akt/p-Bad signaling, which may control the mAR-induced antitumorigenic effects in vitro and in vivo. It should be noted that APCMin/+ mutant mice of both sexes were used in this study to avoid conflicting data regarding the sex-related prevalence of colon tumors of APCMin/+ mutant mice. It has been pointed out that the enhanced susceptibility of male mice to intestinal tumor growth (26,27) results instead from the classical androgen receptor, because mAR antitumorigenic effects seem to be independent of classical androgen-receptor signaling (1,5,12,13).
In vitro downregulation of the PI-3K/Akt signaling upon mAR activation has also been reported in DU145 prostate cancer cells (16). In these cells, PI-3K was constitutively activated (13), whereas long-term mAR stimulation by specific agonists induced dephosphorylation of both PI-3K and its downstream effector Akt (16). Taken together, the results observed in prostate and colon tumors indicate that mAR expression is associated with active prosurvival pathways and thus protects cells from apoptotic regression. Activation of these receptors triggers specific signaling to restrict this prosurvival machinery. This assumption is fully supported by the in vitro and in vivo results presented here, which support the argument that the in vitro findings reported previously are not simply a side effect of mAR activation.
Akt has been also shown to play a major role in the invasive potential of colon cancer cells in response to a variety of stimuli, for example, heregulin (28), PAK1 (29) and Sprouty-2 (30). Moreover, inhibition of Akt-dependent pathways has been linked to reduced cell motility in colon cancer (21), whereas mAR stimulation downregulates p-Akt (16 and this work), and mAR-dependent activation has been shown to block cell motility and invasion in prostate cancer cells (1). Thus, we sought to determine an effect of mAR activation on cell motility in colon tumor cells. The present observations further reveal that mAR stimulation modulates specific molecular targets controlling cell motility. Activation of mAR by two distinct nonpermeable TAC (testosterone-HSA and testosterone-BSA) markedly inhibited cell motility as documented by different assays (Figure 3). The possibility that testosterone conjugates may be converted to estrogen and influence the overall interpretation of our results was also considered. However, previous binding studies in colon cancer (5) as well as in prostate cancer cells (2) have clearly indicated that estrogen (and progesterone) displaced radiolabeled testosterone with significantly lower affinity (104- to 102-fold). These findings indicate that even if such a conversion takes place, it can not influence the mAR-induced effects described so far, because of the high androgen selectivity of these membrane receptors. In addition, control experiments showed that neither migration nor apoptotic responses were influenced by the aromatase inhibitor anastrozole (Figure 4), further supporting the androgen specificity of the mAR-induced effects. Finally, because flutamide did not influence the motility effects (Figure 4A) nor the apoptotic responses in colon cancer (5), it is believed that the mAR pool mediating the observed effects is most likely unrelated to a membrane-associated form of the classical, intracellular AR that may be present in the plasma membrane of colon tumor cells. Although the existence of such a form of membrane tethered intra-cellular AR has not been reported in colon cells, experimental data in prostate cells places iARs on the cell membrane (31). In that case, however, and in sharp contrast to what we have observed in our assays in colon tumors, membrane-associated iARs induced cell proliferation (instead of apoptosis), which was efficiently blocked by antiandrogens and antiestrogens (31). In conclusion, our data support the existence of an active, non-AR/ER–related membrane receptor bearing anticancer action in colon cancer cells. This conclusion is in-line with recent findings demonstrating that iARs could not be detected in membrane preparations of Caco2 cells (5).
Inhibition of migration is usually correlated with impaired expression/activation of adhesion molecules and reorganization of focal contact structures, including the actin cytoskeleton (32). Because actin reorganization is a major effect of mAR activation in tumor cells (2,3,5,13), we focused on the molecular mechanism underlying the mAR- induced inhibition of cellular motility in Caco2 cells (Figure 3). Our results demonstrate that vinculin is a main target of mAR activation that may regulate cell motility. Phosphorylation of vinculin (Figure 5A) was an early and persistent event leading to significant morphological changes of Caco2 cells. It was clearly correlated with actin restructuring, as indicated by the visualization of newly organized actin filaments emanating from the vinculin spots on the cell- adhesion contacts (Figure 5B). Vinculin silencing or inhibition of vinculin phosphorylation by specific inhibitors largely reversed actin reorganization and the inhibition of migration. These findings imply that vinculin phosphorylation/activaton upon mAR stimulation regulates cell adhesion and inhibits the migration potential of Caco2 tumor cells. This conclusion is in line with several reports in the literature. Thus, vinculin was shown to be important in regulating adhesion dynamics and cell migration (33), and it was postulated that vinculin may connect early adhesion sites to the actin-driven protrusive machinery (23). According to this model vinculin stabilizes focal adhesions and thereby suppresses cell migration, an effect that is relieved by modifications of inositol phospholipids (34). Although the precise role of vinculin in focal adhesions remains to be elucidated, recent experimental evidence suggests that vinculin overexpression reduces cell migration, whereas vinculin downregulation enhances cell motility (34). This hypothesis is in-line with the results presented in our study, showing inhibition of cell motility ahead of vinculin activation in mAR-stimulated Caco2 cells. Notably, this effect was efficiently reversed by silencing of endogenous vinculin by use of siRNAs. Finally, we noted that the effects of TAC on cell invasion were manifested even after short treatment of cells with this compound (Figure 8). Moreover, these effects were still silenced by siRNAs against vinculin (Figure 8). These results clearly indicate that the inhibitory signals on invasion are activated early upon mAR stimulation, are dependent on vinculin and are present in cells well before they commit to the mAR- induced apoptotic program.
In conclusion, our results provide novel mechanistic insights into mAR- induced antitumorigenic actions. The long-term activity of the prosurvival regulators PI-3K, Akt and Bad is effectively suppressed and the specific molecular target for cell adhesion, vinculin, is modulated upon mAR activation. Because this molecule may adapt signaling pathways involved in apoptosis, cell survival and motility (35,36), we hypothesize that it represents a key signaling effector regulating the mAR-dependent antitumorigenic effects observed in our reported studies. Further experiments are now needed to address the molecular identity of mAR and to evaluate the potential role of these signaling targets for the development of novel antitumorigenic strategies based on specific mAR activation.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by grants from Deutsche Forschungsgemeinschaft (GRK 1302; SFB773; Mercator program) and the Greek Ministry of Health (KESY program).
Footnotes
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Online address: http://www.molmed.org
REFERENCES
- 1.Hatzoglou A, et al. Membrane androgen receptor activation induces apoptotic regression of human prostate cancer cells in vitro and in vivo. J Clin Endocrinol Metab. 2005;90:893–903. doi: 10.1210/jc.2004-0801. [DOI] [PubMed] [Google Scholar]
- 2.Kampa M, et al. The human prostate cancer cell line LNCaP bears functional membrane testosterone receptors that increase PSA secretion and modify actin cytoskeleton. FASEB J. 2002;16:1429–31. doi: 10.1096/fj.02-0131fje. [DOI] [PubMed] [Google Scholar]
- 3.Kallergi G, Agelaki S, Markomanolaki H, Georgoulias V, Stournaras C. Activation of FAK/PI3K/Rac1 signaling controls actin reorganization and inhibits cell motility in human cancer cells. Cell Physiol Biochem. 2007;20:977–86. doi: 10.1159/000110458. [DOI] [PubMed] [Google Scholar]
- 4.Kampa M, et al. Opposing effects of estradiol- and testosterone-membrane binding sites on T47D breast cancer cell apoptosis. Exp Cell Res. 2005;307:41–51. doi: 10.1016/j.yexcr.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 5.Gu S, et al. Functional membrane androgen receptors in colon tumors trigger pro-apoptotic responses in vitro and reduce drastically tumor incidence in vivo. Mol. Cancer. 2009;8:114. doi: 10.1186/1476-4598-8-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benten WP, et al. Testosterone signaling through internalizable surface receptors in androgen receptor-free macrophages. Mol Biol Cell. 1999;10:3113–23. doi: 10.1091/mbc.10.10.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benten WP, et al. Functional testosterone receptors in plasma membranes of T cells. FASEB J. 1999;13:123–33. doi: 10.1096/fasebj.13.1.123. [DOI] [PubMed] [Google Scholar]
- 8.Gatson JW, Kaur P, Singh M. Dihydrotestosterone differentially modulates the mitogen- activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology. 2006;147:2028–34. doi: 10.1210/en.2005-1395. [DOI] [PubMed] [Google Scholar]
- 9.Somjen D, et al. Role of putative membrane receptors in the effect of androgens on human vascular cell growth. J Endocrinol. 2004;180:97–106. doi: 10.1677/joe.0.1800097. [DOI] [PubMed] [Google Scholar]
- 10.Lieberherr M, Grosse B. Androgens increase intracellular calcium concentration and inositol 1,4,5-trisphosphate and diacylglycerol formation via a pertussis toxin-sensitive G-protein. J Biol Chem. 1994;269:7217–23. [PubMed] [Google Scholar]
- 11.Sun YH, Gao X, Tang YJ, Xu CL, Wang LH. Androgens induce increases in intracellular calcium via a G protein-coupled receptor in LNCaP prostate cancer cells. J Androl. 2006;27:671–8. doi: 10.2164/jandrol.106.000554. [DOI] [PubMed] [Google Scholar]
- 12.Papakonstanti EA, Kampa M, Castanas E, Stournaras C. A rapid, nongenomic, signaling pathway regulates the actin reorganization induced by activation of membrane testosterone receptors. Mol Endocrinol. 2003;17:870–81. doi: 10.1210/me.2002-0253. [DOI] [PubMed] [Google Scholar]
- 13.Papadopoulou N, Charalampopoulos I, Alevizopoulos K, Gravanis A, Stournaras C. Rho/ROCK/actin signaling regulates membrane androgen receptor induced apoptosis in prostate cancer cells. Exp Cell Res. 2008;314:3162–74. doi: 10.1016/j.yexcr.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 14.Kampa M, et al. Activation of membrane androgen receptors potentiates the antiproliferative effects of paclitaxel on human prostate cancer cells. Mol Cancer Ther. 2006;5:1342–51. doi: 10.1158/1535-7163.MCT-05-0527. [DOI] [PubMed] [Google Scholar]
- 15.Papadopoulou N, Papakonstanti EA, Kallergi G, Alevizopoulos K, Stournaras C. Membrane androgen receptor activation in prostate and breast tumor cells: molecular signaling and clinical impact. IUBMB Life. 2009;61:56–61. doi: 10.1002/iub.150. [DOI] [PubMed] [Google Scholar]
- 16.Papadopoulou N, et al. Membrane androgen receptor activation triggers down-regulation of PI-3K/Akt/NF-kappaB activity and induces apoptotic responses via Bad, FasL and caspase-3 in DU145 prostate cancer cells. Mol. Cancer. 2008;7:88. doi: 10.1186/1476-4598-7-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardone MH, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 18.Datta SR, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 19.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–7. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 20.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–76. [PMC free article] [PubMed] [Google Scholar]
- 21.Lai TY, et al. 17beta-estradiol inhibits prostaglandin E2-induced COX-2 expressions and cell migration by suppressing Akt and ERK1/2 signaling pathways in human LoVo colon cancer cells. Mol Cell Biochem. 2010;342:63–70. doi: 10.1007/s11010-010-0469-7. [DOI] [PubMed] [Google Scholar]
- 22.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 24.Bailly M. Connecting cell adhesion to the actin polymerization machinery: vinculin as the missing link. Trends Cell Biol. 2003;13:163–5. doi: 10.1016/s0962-8924(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 25.Smith KJ, et al. The APC gene product in normal and tumor cells. Proc Natl Acad Sci U S A. 1993;90:2846–50. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinoi T, et al. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 2007;67:9721–30. doi: 10.1158/0008-5472.CAN-07-2735. [DOI] [PubMed] [Google Scholar]
- 27.Lind GE, et al. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cell lines. Mol. Cancer. 2004;3:28. doi: 10.1186/1476-4598-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshioka T, et al. Significance of integrin alphavbeta5 and erbB3 in enhanced cell migration and liver metastasis of colon carcinomas stimulated by hepatocyte-derived heregulin. Cancer Sci. 2010;11:2011–18. doi: 10.1111/j.1349-7006.2010.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huynh N, Liu KH, Baldwin GS, He H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK-and AKT-dependent pathways. Biochim Biophys Acta. 20101803:1106–13. doi: 10.1016/j.bbamcr.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 30.Holgren C, et al. Sprouty-2 controls c-Met expression and metastatic potential of colon cancer cells: sprouty/c-Met upregulation in human colonic adenocarcinomas. Oncogene. 2010;29:5241–53. doi: 10.1038/onc.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Migliaccio A, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–17. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. 2003;8:d982–96. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- 33.Critchley DR. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem Soc Trans. 2004;32:831–6. doi: 10.1042/BST0320831. [DOI] [PubMed] [Google Scholar]
- 34.Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends Cell Biol. 2006;16:453–60. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez Fernandez JL, et al. Suppression of tumorigenicity in transformed cells after transfection with vinculin cDNA. J Cell Biol. 1992;119:427–38. doi: 10.1083/jcb.119.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subauste MC, et al. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J Cell Biol. 2004;165:371–81. doi: 10.1083/jcb.200308011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.