

Abstract
Patients with NF1 microdeletion develop more neurofibromas at a younger age, and have an increased risk of malignant peripheral nerve sheath tumors (MPNSTs). We postulated that the increased risk of malignancy could be due to inactivation, in addition to NF1, of a second tumor suppressor gene located in the typical 1.4-Mb microdeletion found in most of the microdeleted patients. We investigated the expression of NF1, the other 16 protein-coding genes and the 2 microRNAs located in the 1.4-Mb microdeletion by means of real-time quantitative reverse-transcription polymerase chain reaction (RT-PCR) in a large series of human dermal and plexiform neurofibromas and MPNSTs. Five genes were significantly upregulated: OMG and SUZ12 in plexiform neurofibromas and ATAD5, EVI2A and C17orf79 in MPNSTs. More interestingly, two genes were significantly downregulated (RNF135 and CENTA2) in tumor Schwann cells from MPNST biopsies and in MPNST cell lines. This study points to the involvement of several genes (particularly RNF135 and CENTA2) in the increased risk of malignancy observed in NF1-microdeleted patients.
INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal-dominant neurocutaneous disorder affecting 1 in 3,000 individuals worldwide (1). The NF1 gene, located in chromosome region 17q11.2, was identified by positional cloning; its protein product, neurofibromin, functions as a tumor suppressor (2,3). Main clinical characteristics of NF1 include café au lait spots (CALS), Lish nodules, freckling and neurofibromas. These patients are also at an increased risk of both benign and malignant tumors, and NF1 is thus classified as a tumor predisposition syndrome. The most common tumors are benign peripheral nerve sheath tumors (neurofibromas), which vary greatly in both number and size and may be dermal or plexiform (4).
In contrast to dermal neurofibromas, plexiform neurofibromas can progress to malignant peripheral nerve sheath tumors (MPNSTs) in about 10% of patients with NF1 (5). MPNSTs are resistant to conventional therapies, and their deep-seated position and locally invasive growth hinder complete surgical resection. Both neurofibromas and MPNSTs are heterogeneous tumors mainly composed of Schwann cells (60–80%), together with fibroblasts, mast cells and other cell types. Schwann cells are considered to be the pathogenic cell type of these two tumor types.
Most patients with NF1 have a small mutation in the NF1 gene (point mutation, small deletion, intragenic insertion or duplication). Approximately 5% of NF1 patients have a large rearrangement, classically known as NF1 microdeletion (6–8). Most of these microdeleted patients have a germline 1.4-Mb microdeletion encompassing the entire 350-kb NF1 gene, caused by unequal recombination between two highly homologous segments termed NF1 low copy repeats (NF1-REPs) (9). Genome database analysis indicates that the common NF1 microdeletion region contains at least 16 additional protein-coding genes, 4 pseudogenes and 2 microRNAs (Table 1 and Figure 1) (10). Most of these genes have unknown functions. It is noteworthy that 1 of the 2 microRNAs (has-mir-193a) was recently identified as a tumor suppressor miRNA that is epigenetically silenced in oral squamous cell carcinoma (11).
Table 1.
Genes located in the 1.4-Mb microdeletion.
| Gene Symbola | Alternative symbols | GenBank accession number | Location at 17q11.2 (kb) | Orientation: Sense (+);Antisense (−) | Gene name |
|---|---|---|---|---|---|
| LRRC37B2 | DKFZp667M2411 | NM_207323 | 25,959–25,966 | + | Leucine rich repeat containing 37, member B2 |
| SUZ12Pb | LOC440423, JJAZ1-P | BC041912 | 26,082–26,121 | + | Suppressor of zeste 12 homolog pseudogene |
| CRLF3 | CYTOR4 | NM_015986 | 26,175–26,133 | − | Cytokine receptor-like factor 3 |
| LOC646013b | — | XR_017620 | 26,182–26,183 | + | LOC646013 similar to 40S ribosomal protein S17 |
| ATAD5 | FLJ12735 | NM_024857 | 26,183–26,246 | + | ATPase family, AAA domain containing 5 |
| C17orf42 | FLJ22729 | NM_024683 | 26,257–26,250 | − | Chromosome 17 open reading frame 42 |
| CENTA2 | HSA272195 | NM_018404 | 26,273–26,310 | + | Centaurin, α2 |
| RNF135 | MGC13061 | NM_032322 | 26,322–26,351 | + | Ring finger protein 135 |
| DPRXP4b | — | AC138207 | 26,326–26,327 | + | Divergent-paired related homeobox pseudogene 4 |
| LOC646021 | — | XM_933339 | 26,356–26,396 | + | LOC646021 hypothetical LOC646021 |
| LOC646037 | — | XM_928998 | 26,404–26,399 | − | LOC646037 similar to CG11835-PA |
| NF1 | — | NM_000267 | 26,446–26,725 | + | Neurofibromin 1 |
| OMG | OMGP | NM_002544 | 26,648–26,645 | − | Oligodendrocyte myelin glycoprotein |
| EVI2B | — | NM_006495 | 26,665–26,654 | − | Ecotropic viral integration site 2B |
| EVI2A | — | NM_014210 | 26,672–26,668 | − | Ecotropic viral integration site 2A |
| AK3P1b | — | X60674 | 26,696–26,697 | + | Adenylate kinase 3 pseudogene 1 |
| RAB11FIP4 | Rab11-FIP4 | NM_032932 | 26,742–26,889 | + | RAB11 family interacting protein 4 (class II) |
| MIRN193A | hsa-mir-193a | MI0000487 | 26,911–26,911 | + | MicroRNA 193a |
| MIRN365-2 | hsa-mir-365-2 | MI0000769 | 26,926–26,926 | + | MicroRNA 365-2 |
| C17orf79 | HSA272196 | NM_018405 | 27,210–27,203 | − | Chromosome 17 open reading frame 79 |
| UTP6 | HCA66, C17orf40 | NM_018428 | 27,252–27,214 | − | UTP6, small subunit (SSU) processome component |
| SUZ12 | JJAZ1, KIAA0160 | NM_015355 | 27,288–27,352 | + | Suppressor of zeste 12 homolog (Drosophila) |
| LRRC37B | LOC114659 | NM_052888 | 27,372–27,404 | + | Leucine-rich repeat containing 37B |
Entrez gene symbol.
Pseudogene.
Figure 1.

Schematic representation of the 17q11.2 region. The 1.4-Mb deletion encompasses the entire 350-kb NF1 gene, 16 flanking protein-coding genes, 4 pseudogenes, and 2 microRNAs.
NF1-microdeleted patients have a more severe phenotype than the general NF1 population. In particular, NF1-microdeleted patients show learning disability, facial dysmorphism and cardiovascular malformations (12–14). It has been suggested that OMG and RNF135 haploinsufficiency may be involved in the learning disability, the RNF135 gene in the facial dysmorphism and the SUZ12/JJAZ1 and CENTA2 genes in the cardiovascular malformations (13–15). NF1-microdeleted patients have more neurofibromas, at an earlier age, and are also at an increased risk of developing MPNSTs (9,16–19). De Raedt et al. (16) estimated that the lifetime risk of MPNST in NF1- microdeleted patients is almost twice that observed in the general NF1 population. The increased malignancy risk may be explained by variations in the expression of one or several genes (tumor suppressor genes rather than oncogenes) located in the 1.4-Mb microdeletion. Piddubnyak et al. (20) suggested that reduced expression of HCA66 protein, owing to haploinsufficiency of the UTP6/HCA66 gene, could make NF1-microdeleted patients’ cells less susceptible to apoptosis.
The aim of this study was to identify candidates for involvement in MPNST tumorigenesis among the 18 additional genes (16 protein-coding genes and 2 microRNA genes) located in the 1.4-Mb microdeletion. Changes in the expression of NF1 and of these 18 genes were analyzed by real-time quantitative reverse-transcription polymerase chain reaction (RT-PCR) in a large series of human dermal and plexiform neurofibromas and MPNSTs, as well as in MPNST cell lines.
MATERIALS AND METHODS
Patients and Samples
Samples of 23 dermal neurofibromas, 13 plexiform neurofibromas and 13 MPNSTs were obtained by laser excision (dermal neurofibromas) or surgical excision (plexiform neurofibromas and MPNSTs) from NF1 patients at Henri Mondor Hospital (Creteil, France). The experiments of this work comply with the current French laws. Written informed consent for sample collection and data publication was obtained from all patients or their relatives. The dermal neurofibromas were used as “normal” control samples, since they are not at risk of developing into malignant MPNSTs. Indeed, neurofibromas are heterogeneous benign tumors composed of Schwann cells, fibroblasts, mast cells and other cells and have no “normal” tissue equivalent. Gene expression levels in plexiform neurofibromas and MPNSTs, determined by real-time RT-PCR analysis, were thus expressed relative to the expression levels in dermal neurofibromas. Immediately after surgery, the tumor samples were flash-frozen in liquid nitrogen and stored at −80°C until RNA extraction. The main clinical and histological characteristics of the patients with MPNSTs are shown in Supplemental Table 1.
We also analyzed the seven following MPNST cell lines: NMS-2, NMS-2PC, 88-3, ST88-14, 90-8, S462 and T265. NMS-2 and NMS-2PC were gifts from Dr. Akira Ogose (Niigata University School of Medicine, Niigata, Japan); 88-3, ST88-14 and 90-8 were gifts from Dr. Nancy Ratner (Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA); S462 was a gift from Dr. Lan Kluwe (University Hospital Eppendorf, Hamburg, Germany); and T265 was a gift from Dr. Georges De Vries (Loyola University, Chicago, OH, USA). MPNST cell lines were grown in RPMI medium supplemented with 15% heat-inactivated fetal bovine serum, 10 IU/mL penicillin and 10μg/mL streptomycin.
Normal Schwann cells and fibroblasts were obtained by primary cell culture and differential isolation from normal skin and sciatic nerve biopsies, respectively, and using cell culture and isolation conditions as previously described (21–22). Normal mast cells were obtained by means of cell culture and various specific purification steps from cord blood–derived human cells, as previously described (23).
NF1 Alteration Analysis
NF1 alteration analysis (GenBank reference sequence NM_000267.1, www.ncbi.nlm.nih.gov/GenBank) was performed using a variety of NF1 gene screening methodologies including sequencing (for point mutations) and loss of heterozygosity assessment using a series of 17q11.2-linked microsatellite markers (D17S841, D17S635, D17S1307, D17S2163, D17S1166, IVS38-GT53.0, D17S1800 and D17S798). The primer oligonucleotide sequences for microsatellite markers study are given in Supplemental Table 2.
Table 2.
Oligonucleotide primer sequences.
| Genes | Oligonucleotide | Sequence | PCR product size (bp) |
|---|---|---|---|
| LRRC37B2 | Upper primer | 5′-GCA GAA GTC AAG TGA GCT CAT GAA-3′ | 84 |
| Lower primer | 5′-ATG ACA GTC ACA GAT ATT GCG AAG AT-3′ | ||
| CRLF3 | Upper primer | 5′-GAA TCA TCG GGT GTT CTC TAC TCC A-3′ | 83 |
| Lower primer | 5′-TGT CCC ACA GTT TCA ACT CTG AAT-3′ | ||
| ATAD5 | Upper primer | 5′-GTT TGA TGG CTG CTT TGA AGA AA-3′ | 60 |
| Lower primer | 5′-CTG GCA ACA TTT AGC AGG GAA G-3′ | ||
| C17orf42 | Upper primer | 5′-CTG AGA AAG CTC CTC AAA CCA GAC-3′ | 95 |
| Lower primer | 5′-CAG GCA ATT CTT CGA GTA CCA AA-3′ | ||
| CENTA2 | Upper primer | 5′-CCC TCA ACC GGC TTA CTG CAT C-3′ | 86 |
| Lower primer | 5′-AGG TGT TCA GTG GCA GCC AGT TC-3′ | ||
| RNF135 | Upper primer | 5′-TGC CTG ACC AGA GCC ACC C-3′ | 63 |
| Lower primer | 5′-GAT GGA TGG CCC ACT GAG CA-3′ | ||
| LOC646021 | Upper primer | 5′-CAG AGG CCG GGA CAG TTT CT- 3′ | 98 |
| Lower primer | 5′-TGT GAT CTC AGG TTT TGG ATA GGT T-3′ | ||
| LOC646037 | Upper primer | 5′-TGA TGT TGG GCA CCT CAA CTC C-3′ | 65 |
| Lower primer | 5′-GGC TTT GGC AGG GGG TGA-3′ | ||
| NF1 | Upper primer | 5′-ACA GAG CGT GGC CTA CTT AGC A-3′ | 166 |
| Lower primer | 5′-GGA CCA TGG CTG AGT CTC CTT T-3′ | ||
| OMG | Upper primer | 5′-TCC AGA CCA ATC TTT TGA CCA ACT-3′ | 123 |
| Lower primer | 5′-GCT TTT GTT TCC ATC ATC CAC TTC-3′ | ||
| EVI2B | Upper primer | 5′-CAG GAC GAC TAC GCT CGG CA-3′ | 90 |
| Lower primer | 5′-CAC CAC AGC GTC AGG GGC T-3′ | ||
| EVI2A | Upper primer | 5′-TGT CTC CTG GAA CAA AAG CAA ACT-3′ | 93 |
| Lower primer | 5′-GGT TTC TGC CTG TCT TGT TTT GAA-3′ | ||
| RAB11FIP4 | Upper primer | 5′-AGC CTC AGC CTC TAC GAA GCA A-3′ | 111 |
| Lower primer | 5′-CTT CAG GGC TTC CAT TAG CTC ATC-3′ | ||
| C17orf79 | Upper primer | 5′-TCA AGG GAA TCC ATA TGA TGC TGA-3′ | 72 |
| Lower primer | 5′-CAG CAC ACC CAA GGT TTA AGC TCT-3′ | ||
| UTP6 | Upper primer | 5′-GGC TCC CGG AAT TGG AAC AG-3′ | 82 |
| Lower primer | 5′-TCG GAA GCC TTC TTA ATG ATA GCC T-3′ | ||
| SUZ12 | Upper primer | 5′-TCG CAA CGG ACC AGT TAA GAG AA-3′ | 64 |
| Lower primer | 5′-TGT TCG TTT TGG CCT GCA CA-3′ | ||
| LRRC37B | Upper primer | 5′-CCG GCA TTA AAA TAT CTA GAC ATG-3′ | 87 |
| Lower primer | 5′-TTC CAG TTC AAC AGT CAT CGT GAG-3′ | ||
| MKI67 | Upper primer | 5′-ATT GAA CCT GCG GAA GAG CTG A-3′ | 105 |
| Lower primer | 5′-GGA GCG CAG GGA TAT TCC CTT A-3′ | ||
| TBP | Upper primer | 5′-TGC ACA GGA GCC AAG AGT GAA-3′ | 132 |
| Lower primer | 5′-CAC ATC ACA GCT CCC CAC CA-3′ | ||
| RPLP0 | Upper primer | 5′-GGC GAC CTG GAA GTC CAA CT-3′ | 149 |
| Lower primer | 5′-CCA TCA GCA CCA CAG CCT TC-3′ |
Real-Time RT-PCR for Protein-Coding Genes
The theoretical and practical aspects of real-time quantitative RT-PCR using the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) have been described in detail elsewhere (24). Quantitative values are obtained from the threshold cycle number at which the increase in the fluorescence signal generated by SYBR® Green dye amplicon complex formation (associated with exponential growth of PCR products) passes a fixed threshold above baseline. The precise amount of total RNA added to each reaction mix (based on optical density) and its quality (that is, lack of extensive degradation) are difficult to assess. We therefore also quantified transcripts of two endogenous RNA control genes involved in two cellular metabolic pathways, namely TBP (Gen-Bank accession NM_003194), which encodes the TATA box-binding protein (a component of the DNA-binding protein complex transcription factor II D [TFIID]), and RPLP0 (also known as 36B4; NM_001002), which encodes human acidic ribosomal phosphoprotein P0. We selected TBP and RPLP0 as endogenous controls because the prevalence of their transcripts is low (threshold cycle [Ct] values between 24 and 26 in the tumor samples) and high (Ct values between 18 and 20), respectively. Each sample was normalized on the basis of its TBP (or RPLPO) content. Results, expressed as N-fold differences in target gene expression relative to the TBP (or RPLPO) gene and termed “Ntarget,” were determined as Ntarget = 2ΔCt sample, where the ΔCt value of the sample was determined by subtracting the average Ct value of the target gene from the average Ct value of the TBP (or RPLP0) gene (24,25). The Ntarget values of the samples were subsequently normalized such that the mean of the dermal neurofibromas (used as calibrators) Ntarget values was 1. All PCRs were performed with an ABI Prism 7900 Sequence Detection System (Applied Biosystems) and the SYBR Green PCR Core Reagents Kit (Applied Biosystems). Experiments were performed with duplicates for each data point. All patient samples with a coefficient of variation of Ct values >1% were retested. The nucleotide sequences of the oligonucleotide primers used to amplify TBP, RPLP0, MKI67 and the 17 target genes are shown in Table 2. To avoid amplification of contaminating genomic DNA, 1 of the 2 primers was placed at the junction between two exons. For each primer pair, we performed no-template control and no-reverse-transcriptase control assays, which produced negligible signals (usually Ct >40), suggesting that primer-dimer formation and genomic DNA contamination effects were negligible. The conditions of RNA extraction, cDNA synthesis and PCRs are described elsewhere (24).
Real-Time RT-PCR for Mature miRNAs
MicroRNAs were isolated as described above for the protein-coding genes, and the expression of specific mature miR-NAs (hsa-mir-365-2 and has-mir-193a) was quantified with real-time PCR and Human TaqMan® MicroRNA assay kits according to the manufacturer’s protocol (Applied Biosystems). U44 small nucleolar RNA was used as an internal control to normalize RNA input in the real-time RT-PCR assay. Expression of this endogenous control was measured with the TaqMan Endogenous Control RNU44 kit.
Statistical Analysis
Because the mRNA (and microRNA) levels did not fit a Gaussian distribution, the mRNA (microRNA) levels in each subgroup of samples were expressed as medians and ranges rather than as means and coefficients of variation and relationships between the molecular markers and clinical and biological parameters were tested with the nonparametric Mann–Whitney U test (26). Differences between two populations were considered significant at confidence levels >95% (P < 0.05).
All supplementary materials are available online at www.molmed.org.
RESULTS
NF1 Mutation Analysis
Germline NF1 mutations and loss of heterozygosity assessment in plexiform neurofibromas and MPNSTs of the study NF1 patients are summarized in Supplemental Table 3.
Table 3.
Median mRNA levels (and ranges) of 15 genes and 2 microRNAs in the 1.4-Mb deletion in dermal and plexiform neurofibromas and MPNSTs.
| Genes | Alternative symbols | Dermal neurofibromas (n = 23) | Plexiform neurofibromas (n = 13) | MPNSTs (n = 13) | Pa | Pb |
|---|---|---|---|---|---|---|
| LRRC37B2 | DKFZp667M2411 | 1.00 (0.54–1.90) | 1.16 (0.49–2.71) | 1.22 (0.47–1.89) | NS | NSc |
| CRLF3 | CYTOR4 | 1.00 (0.42–1.88) | 1.25 (0.66–1.97) | 0.76 (0.17–2.32) | NS | NS |
| ATAD5 | FLJ12735 | 1.00 (0.38–2.52) | 1.19 (0.65–1.62) | 3.02 (0.44–9.55) | NS | 1.4 × 10−4 |
| C17orf42 | FLJ22729 | 1.00 (0.62–1.79) | 1.24 (0.53–3.63) | 1.14 (0.27–2.71) | NS | NS |
| CENTA2 | HSA272195 | 1.00 (0.62–2.27) | 0.95 (0.53–4.59) | 0.38 (0.15–1.89) | NS | 0.014 |
| RNF135 | MGC13061 | 1.00 (0.41–1.68) | 0.92 (0.57–1.37) | 0.49 (0.13–1.74) | NS | 0.026 |
| NF1 | — | 1.00 (0.81–1.27) | 1.01 (0.49–1.84) | 0.93 (0.38–2.25) | NS | NS |
| OMG | OMGP | 1.00 (0.52–1.57) | 1.65 (0.51–3.09) | 0.92 (0.32–2.16) | 0.013 | NS |
| EVI2B | — | 1.00 (0.54–1.75) | 0.74 (0.59–1.82) | 0.30 (0.14–1.47) | NS | 6 × 10−4 |
| EVI2A | — | 1.00 (0.46–1.94) | 1.10 (0.46–3.16) | 2.03 (0.44–6.93) | NS | 0.047 |
| RAB11FIP4 | Rab11-FIP4 | 1.00 (0.37–4.12) | 1.23 (0.47–3.23) | 0.99 (0.23–5.07) | NS | NS |
| MIRN193A | hsa-mir-193a | 1.00 (0.64–1.48) | 0.65 (0.38–1.35) | 0.77 (0.40–2.83) | NS | NS |
| MIRN365-2 | hsa-mir-365-2 | 1.00 (0.55–1.95) | 1.49 (0.77–2.35) | 0.84 (0.44–1.89) | NS | NS |
| C17orf79 | HSA272196 | 1.00 (0.35–1.89) | 0.95 (0.18–1.69) | 2.58 (0.86–4.53) | NS | 0.006 |
| UTP6 | HCA66, C17orf40 | 1.00 (0.38–1.51) | 1.09 (0.57–1.71) | 0.83 (0.32–2.51) | NS | NS |
| SUZ12 | JJAZ1, KIAA0160 | 1.00 (0.47–1.81) | 1.90 (1.02–3.83) | 0.96 (0.31–3.31) | 2 × 10−4 | NS |
| LRRC37B | LOC114659 | 1.00 (0.10–2.07) | 0.83 (0.16–1.62) | 0.94 (0.54–1.96) | NS | NS |
| MKI67 | 1.00 (0.30–2.82) | 1.54 (0.88–4.77) | 30.7 (13.6–82.9) | 0.012 | 8 × 10−6 |
Data are median (range) mRNA levels, unless otherwise indicated. Bold data indicate significantly different expression.
Mann–Whitney U test: plexiform neurofibromas versus dermal neurofibromas.
Mann–Whitney U test: MPNSTs versus dermal neurofibromas.
NS, not significant.
RNA Expression of NF1, the Other 16 Protein-Coding Genes and the 2 MicroRNAs
Very low levels of LOC646021 and LOC646037 mRNA—detectable but not reliably quantifiable by real-time quantitative RT-PCR (mainly based on fluorescence SYBR Green methodology; Ct >32)—were observed in dermal and plexiform neurofibromas and MPNSTs.
Table 3 shows the median mRNA levels (and ranges) of the remaining 15 protein-coding genes and the two microRNAs in the three groups of tumor samples. For each gene, mRNA levels were normalized such that the median value of the 23 dermal neurofibromas was 1. Table 3 also shows the mRNA levels of the MKI67 gene encoding Ki-67, a large protein of unknown function classically used as a histopathological marker of cell proliferation.
OMG and SUZ12 were significantly upregulated in the plexiform neurofibromas compared with the dermal neurofibromas, whereas ATAD5, EVI2A and C17orf79 were specifically upregulated in the MPNSTs.
The CENTA2, RNF135 and EVI2B genes were significantly downregulated in MPNSTs but not in plexiform neurofibromas. The other genes, including NF1 and the two microRNAs (hsa-mir-365-2 and has-mir-193a), were not significantly altered in plexiform neurofibromas or MPNSTs. Figure 2 shows the mRNA levels of the three downregulated genes (CENTA2, RNF135 and EVI2B) in each neurofibroma and MPNST sample. The only grade I MPNST (MP6; see Supplemental Table 1) did not show a decreased expression of CENTA2, RNF135 and EVI2B (Figure 2). It is noteworthy that 3 of the 13 patients with MPNSTs (MP1, MP3 and MP9; see Supplemental Table 3) had a known germline microdeletion of NF1. Interestingly, the mRNA levels of CENTA2, RNF135 and EVI2B were low in these three MPNSTs: the RCENTA2, RRNF135 and REVI2B values (calculated as described in Materials and Methods) were respectively 0.15, 0.49 and 0.41 in MP1; 0.28, 0.49 and 0.30 in MP3; and 0.23, 0.42 and 0.37 in MP9 (Figure 2).
Figure 2.

mRNA levels of CENTA2, RNF135 and EVI2B in dermal and plexiform neurofibromas and MPNSTs. The mRNA levels of CENTA2, RNF135 and EVI2B in 23 dermal neurofibromas (light gray bars), 13 plexiform neurofibromas (dark gray bars) and 13 MPNSTs (black bars) were normalized such that the median mRNA level in the dermal neurofibromas was 1. Median values (ranges) are indicated for each tumor subgroup and are represented by a dotted line. *Three MPNST samples from patients with germline NF1 microdeletion (MP1, MP3 and MP9). ‡Only grade I MPNST sample (MP6).
Table 3 shows the mRNA abundance of the 15 protein-coding genes (calculated as described in “Materials and Methods”) relative to the endogenous control (TBP) used to normalize the starting amount and quality of total RNA. Similar results were obtained with a second endogenous control, RPLP0 (data not shown).
mRNA Expression of CENTA2, RNF135 and EVI2B in Normal Human Cells
Neurofibromas and MPNSTs are heterogeneous tumors mainly composed of Schwann cells (60–80%), together with fibroblasts, mast cells and other cells. We have previously observed a depletion of mast cells and fibroblasts during malignant transformation of plexiform neurofibromas into MPNSTs (27,28).
If CENTA2, RNF135 and EVI2B were mast cell or fibroblast specific, then a lower abundance of mast cells or fibroblasts in MPNSTs relative to neurofibromas could explain the observed underexpression of these three genes in MPNSTs. To investigate the cellular specificity of CENTA2, RNF135 and EVI2B expression, we analyzed the mRNA levels of these three genes by means of real-time RT-PCR in normal Schwann cells, fibroblasts and mast cells. CENTA2 and RNF135 were similarly expressed in the three cell types (Figure 3). In contrast, EVI2B was far more weakly expressed in Schwann cells than in fibroblasts, possibly explaining the observed underexpression of this gene in MPNSTs (Table 3).
Figure 3.
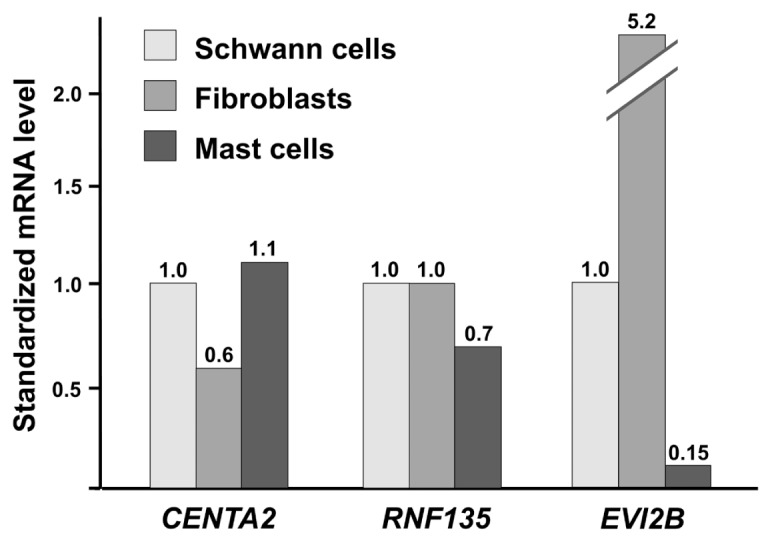
mRNA expression of CENTA2, RNF135 and EVI2B in normal Schwann cells, fibroblasts and mast cells. For each gene, mRNA levels of the samples were normalized such that the value in Schwann cells was 1. The mRNA levels indicated are means of at least three independent experiments.
mRNA Expression of CENTA2 and RNF135 in Seven NF1-Associated MPNST Cell Lines
The expression level of CENTA2 and RNF135 was determined in seven well-characterized NF1-associated MPNST cell lines, namely NMS-2, NMS-2PC, 88-3, ST88-14, 90-8, S462 and T265 (Figure 4).
Figure 4.

mRNA levels of CENTA2 and RNF135 in seven NF1-associated MPNST cell lines. For each gene, mRNA levels were normalized such that the median value in 23 dermal neurofibromas was 1.
CENTA2 was downregulated in all seven MPNST cell lines (more than three-fold less than the median value in the 23 dermal neurofibroma samples), whereas RNF135 was downregulated in only four MPNST cell lines (88-3, 90-8, S462 and T265).
DISCUSSION
Patients with NF1 microdeletions have an increased risk of developing MPNST (9,16–19). We postulated that the increased risk of malignancy might be explained by variations in the expression of one or several genes located in the 1.4-Mb microdeletion. We focused on in-activation of putative tumor suppressor rather than on oncogene activation. We therefore examined the expression of NF1, the other 16 protein-coding genes and the 2 microRNA genes located within the 1.4-Mb deletion by means of real-time quantitative RT-PCR in a large series of dermal and plexiform neurofibromas and MPNSTs.
Several genes were significantly upregulated only in the plexiform neurofibromas (OMG and SUZ12) or only in the MPNSTs (ATAD5, EVI2A and C17orf79). Among these five putative oncogenes, C17orf79 showed the strongest upregulation in the seven NF1-associated MPNST cell lines tested; its expression was two-to nine-fold higher than in the dermal neurofibromas (data not shown). The function of 3 of these 5 genes (ATAD5, EVI2A and C17orf79) is unknown. The other 2 genes (OMG and SUZ12) are good candidates for involvement in NF1 tumorigenesis. It is noteworthy that these 2 genes are upregulated in plexiform neurofibromas but not in MPNSTs, suggesting that they are more specifically involved in the development of plexiform neurofibromas (Table 3). OMG, which is located in an antisense orientation within intron 27b of the NF1 gene, encodes the oligodendrocyte–myelin glycoprotein, 1 of 3 known Nogo receptor ligands (29). However, our observation of OMG mRNA upregulation in plexiform neurofibromas is consistent with a positive effect of OMG on cell proliferation. However, this is difficult to reconcile with evidence suggesting that OMG is a tumor-suppressor gene and that its overexpression blocks mitogenic signaling in NIH3T3 fibroblasts (30).
SUZ12, the second gene found to be upregulated in plexiform neurofibromas, has a known function and is a good candidate oncogene. Indeed, SUZ12 encodes a major polycomb regulator that is preferentially activated during embryonic stem cell differentiation (31,32). SUZ12 is overexpressed in a broad spectrum of human tumors (33). SUZ12 up-regulation is accompanied by the recruitment of DNA methyltransferases and by DNA hypermethylation, resulting in the silencing of important tumor suppressor genes (34). Interestingly, recurrent chromosomal translocation—t(7;17)(p15;q21)—involving SUZ12 (located at 17q21) has been detected in approximately one-half of endometrial stromal sarcomas, malignancies that derive from mesenchymal tissue (35). The authors of this latter study suggested a genetic pathway for progression of a benign precursor to a sarcoma, involving increased cell survival associated with acquisition of a SUZ12 rearrangement.
Our present findings (that is, gene overexpression in plexiform neurofibromas and MPNSTs) are consistent with a model in which the overexpressed genes promote proliferation and thus act as oncogenes. The upregulation of these five putative oncogenes during NF1 tumorigenesis is probably due to a molecular mechanism independent of the NF1 microdeletion. However, we cannot rule out the possibility that the 1.4-Mb region contains an unknown negative regulator of gene transcription (such as an unidentified noncoding RNA gene), which would be inactivated by deletion and upregulate neighboring oncogenes. It is noteworthy that the two microRNAs (hsa-mir-365-2 and has-mir-193a) located in the NF1 microdeleted region and analyzed in this study were normally expressed in the plexiform neurofibromas and MPNSTs.
More interestingly, three genes (RNF135, CENTA2 and EVI2B) were significantly downregulated in MPNSTs, a finding compatible with a role as tumor suppressor genes. EVI2B was expressed more strongly by fibroblasts than by Schwann cells and mast cells, suggesting that the observed underexpression of this gene was due to a lower abundance of a particular cell type (likely fibroblasts in the present case) in MPNSTs, as previously described (28).
Concerning the other two downregulated genes, we observed that CENTA2 was more strongly underexpressed than RNF135 in MPNSTs (Figure 2) and in MPNST cell lines (Figure 4). It is noteworthy that CENTA2 underexpression was recently described in two MPNSTs as compared with four plexiform neurofibromes (36).
The RNF135 gene encodes a putative 432–amino acid protein of unknown function. This protein contains a RING finger domain, a motif present in a variety of functionally distinct proteins and known to be involved in protein-protein and protein-DNA interactions. Interestingly, Douglas et al. (15) recently identified RNF135 mutations in families characterized by overgrowth, learning disability and dysmorphic features and demonstrated that RNF135 haploinsufficiency contributes to several features of the phenotype of NF1-microdeleted patients. The possible role of RNF135 in the increased risk of MPNST is unknown, but it is conceivable that the growth-promoting effects of RNF135 haploinsufficiency are involved.
CENTA2 encodes the recently described protein centaurin-α2 and forms with p42IP4/centaurin-α1, a protein family with strong homology and the same overall protein structure (37). Centaurins are a family of GTPase-activating proteins for the ADP-ribosylation factor (ARF) family of small G proteins (38). P42IP4/centaurin-α1 can regulate extracellular signal-regulated kinase (ERK)-1/2, and this regulation requires phosphatidylinositol 3-kinase–dependent recruitment of p42IP4/centaurin-α1 to the plasma membrane (39). Recently, Venkateswarlu et al. (40) demonstrated that centaurin-α2 is a target for activated phosphatidylinositol 3-kinase, which is recruited, in a sustained manner, to the plasma membrane and thereby negatively regulates ARF6-mediated actin cytoskeleton reorganization. The centaurin α family thus could be involved, like neurofibromin, in the Ras signaling pathway. Finally, CENTA2 haploinsufficiency could be involved in the cardiovascular malformations observed in NF1-microdeleted patients (13).
In conclusion, this expression profiling study of 18 poorly studied genes (16 protein-coding genes and 2 microRNA genes) located in the NF1 microdeletion region in a large series of dermal and plexiform neurofibromas and MPNSTs points to the involvement of several genes, particularly CENTA2 and RNF135, in the increased risk of malignancy observed in NF1-microdeleted patients. The role of these genes in NF1 tumorigenesis now needs to be confirmed in vitro (gain- and loss-of-function experiments in cultured cells) and in animal models.
Supplemental Data
ACKNOWLEDGMENTS
This work was supported by Association pour la Recherche sur le Cancer, Association Neurofibromatoses et Recklinghausen, Ligue Française Contre les Neurofibromatoses, the Clinical Research program (PHRC 2002), INSERM Projet NF1GeneModif and Ministère de L’Enseignement Supérieur et de la Recherche.
Footnotes
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Online address: http://www.molmed.org
REFERENCES
- 1.Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1–6. [PubMed] [Google Scholar]
- 2.Cawthon RM, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 3.Wallace MR, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 4.Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573–7. [PubMed] [Google Scholar]
- 5.Evans DG, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kluwe L, et al. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat. 2004;23:111–6. doi: 10.1002/humu.10299. [DOI] [PubMed] [Google Scholar]
- 7.Mantripragada KK, et al. Identification of novel deletion breakpoints bordered by segmental duplications in the NF1 locus using high resolution array-CGH. J Med Genet. 2006;43:28–38. doi: 10.1136/jmg.2005.033795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wimmer K, et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–76. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- 9.Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9:35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- 10.Jenne DE, et al. Complete physical map and gene content of the human NF1 tumor suppressor region in human and mouse. Genes Chromosomes Cancer. 2003;37:111–20. doi: 10.1002/gcc.10206. [DOI] [PubMed] [Google Scholar]
- 11.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 12.Upadhyaya M, et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998;102:591–7. doi: 10.1007/s004390050746. [DOI] [PubMed] [Google Scholar]
- 13.Venturin M, et al. Mental retardation and cardiovascular malformations in NF1-microdeleted patients point to candidate genes in 17q11.2. J Med Genet. 2004;41:35–41. doi: 10.1136/jmg.2003.014761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu BL, Austin MA, Schneider GH, Boles RG, Korf BR. Deletion of the entire NF1 gene detected by the FISH: four deletion patients associated with severe manifestations. Am J Med Genet. 1995;59:528–35. doi: 10.1002/ajmg.1320590427. [DOI] [PubMed] [Google Scholar]
- 15.Douglas J, et al. Mutations in RNF135, a gene within the NF1 microdeletion region, cause phenotypic abnormalities including overgrowth. Nat Genet. 2007;39:963–5. doi: 10.1038/ng2083. [DOI] [PubMed] [Google Scholar]
- 16.Raedt De, et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–92. doi: 10.1086/374821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kluwe L, Friedrich RE, Peiper M, Friedman J, Mautner VF. Constitutional NF1 mutations in neurofibromatosis 1 patients with malignant peripheral nerve sheath tumors. Hum. Mutat. 2003;22:420. doi: 10.1002/humu.9193. [DOI] [PubMed] [Google Scholar]
- 18.Wu R, et al. Germline mutations in NF1 patients with malignancies. Genes Chromosomes Cancer. 1999;26:376–80. [PubMed] [Google Scholar]
- 19.Pasmant E, et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat. 2010;31:E1506–18. doi: 10.1002/humu.21271. [DOI] [PubMed] [Google Scholar]
- 20.Piddubnyak V, et al. Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients. Cell Death Differ. 2007;14:1222–33. doi: 10.1038/sj.cdd.4402122. [DOI] [PubMed] [Google Scholar]
- 21.Avellana-Adalid V, et al. In vitro and in vivo behaviour of NDF-expanded monkey Schwann cells. Eur J Neurosci. 1998;10:291–300. doi: 10.1046/j.1460-9568.1998.00049.x. [DOI] [PubMed] [Google Scholar]
- 22.Glasow A, et al. Expression of leptin (Ob) and leptin receptor (Ob-R) in human fibroblasts: regulation of leptin secretion by insulin. J Clin Endocrinol Metab. 2001;86:4472–9. doi: 10.1210/jcem.86.9.7792. [DOI] [PubMed] [Google Scholar]
- 23.Royer B, et al. Autocrine regulation of cord blood-derived human mast cell activation by IL-10. J Allergy Clin Immunol. 2001;108:80–6. doi: 10.1067/mai.2001.115753. [DOI] [PubMed] [Google Scholar]
- 24.Bieche I, et al. Identification of CGA as a novel estrogen receptor-responsive gene in breast cancer: an outstanding candidate marker to predict the response to endocrine therapy. Cancer Res. 2001;61:1652–8. [PubMed] [Google Scholar]
- 25.Bieche I, et al. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin Chem. 1999;45:1148–56. [PubMed] [Google Scholar]
- 26.Mann H, Whitney D. On a test of whether one of two random variables is stochastically larger than the other. Annals of Mathematical Statistics. 1947;18:50–60. [Google Scholar]
- 27.Levy P, et al. Molecular profiles of neurofibromatosis type 1-associated plexiform neurofibromas: identification of a gene expression signature of poor prognosis. Clin Cancer Res. 2004;10:3763–71. doi: 10.1158/1078-0432.CCR-03-0712. [DOI] [PubMed] [Google Scholar]
- 28.Levy P, et al. Molecular profiling of malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1, based on large-scale real-time RT-PCR. Mol. Cancer. 2004;3:20. doi: 10.1186/1476-4598-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang KC, et al. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002;417:941–4. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- 30.Habib AA, Gulcher JR, Högnason T, Zheng L, Stefánsson K. The OMG gene, a second growth suppressor within the NF1 gene. Oncogene. 1998;16:1525–31. doi: 10.1038/sj.onc.1201683. [DOI] [PubMed] [Google Scholar]
- 31.Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–56. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 33.Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SUZ12 as a potential molecular target for human cancer therapy. Mol Cancer Ther. 2003;2:113–21. [PubMed] [Google Scholar]
- 34.Reynolds PA, et al. Tumor suppressor P16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- 35.Li H, et al. Effects of rearrangement and allelic exclusion of JJAZ1/SUZ12 on cell proliferation and survival. Proc Natl Acad Sci U S A. 2007;104:20001–6. doi: 10.1073/pnas.0709986104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartelt-Kirbach B, Wuepping M, Dodrimont-Lattke M, Kaufmann D. Expression analysis of genes lying in the NF1 microdeletion interval points to four candidate modifiers for neurofibroma formation. Neurogenetics. 2009;10:79–85. doi: 10.1007/s10048-008-0154-0. [DOI] [PubMed] [Google Scholar]
- 37.Hanck T, Stricker R, Sedehizade F, Reiser G. Identification of gene structure and sub-cellular localization of human centaurin alpha 2, and p42IP4, a family of two highly homologous, Ins 1,3,4,5-P4-/PtdIns 3,4,5-P3-binding, adapter proteins. J Neurochem. 2004;88:326–36. doi: 10.1046/j.1471-4159.2003.02143.x. [DOI] [PubMed] [Google Scholar]
- 38.Randazzo PA, Hirsch DS. Arf GAPs multi-functional proteins that regulate membrane traffic and actin remodelling. Cell Signal. 2004;16:401–13. doi: 10.1016/j.cellsig.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi H, et al. Centaurin-alpha1 is a phosphatidylinositol 3-kinase-dependent activator of ERK1/2 mitogen-activated protein kinases. J Biol Chem. 2006;281:1332–7. doi: 10.1074/jbc.M505905200. [DOI] [PubMed] [Google Scholar]
- 40.Venkateswarlu K, Brandom KG, Yun H. PI-3-kinase-dependent membrane recruitment of centaurin-alpha2 is essential for its effect on ARF6-mediated actin cytoskeleton reorganisation. J Cell Sci. 2007;120:792–801. doi: 10.1242/jcs.03373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.