

Abstract
Restenosis is due to neointimal hyperplasia, which occurs in the coronary artery after percutaneous transluminal coronary angioplasty (PTCA). During restenosis, an impairment of nitric oxide (NO)-dependent pathways may occur. Concomitant hypercholesterolemia may exacerbate restenosis in patients undergoing PTCA. Here, we show that a NO-releasing aspirin derivative (NCX-4016) reduces the degree of restenosis after balloon angioplasty in low-density lipoprotein receptor-deficient mice and this effect is associated with reduced vascular smooth muscle cell (VSMC) proliferation and macrophage deposition at the site of injury. Drugs were administered following both therapeutic or preventive protocols. We demonstrate that NCX-4016 is effective both in prevention and treatment of restenosis in the presence of hypercholesterolemia. These data indicate that impairment of NO-dependent mechanisms may be involved in the development of restenosis in hypercholesterolemic mice. Although experimental models of restenosis may not reflect restenosis in humans in all details, we suggest that a NO-releasing aspirin derivative could be an effective drug in reducing restenosis following PTCA, especially in the presence of hypercholesterolemia and/or gastrointestinal damage.
Atherosclerosis-related diseases represent the major cause of death in the world. Atherosclerosis is the most common cause of acute myocardial infarction. Although novel therapeutic strategies are in development to reduce atherogenesis in the coronary arteries, primary or secondary percutaneous transluminal coronary angioplasty (PTCA) is still the most common method of treatment. However, the development of restenosis after PTCA continues to be a clinical problem despite the use of endovascular stents and/or brachytherapy and despite increasing insights into the pathophysiology of the restenotic process (1–3). Neointimal hyperplasia is regarded as the main factor responsible for lumen renarrowing (1–3). According to early studies in experimental models, antithrombotic therapy is mandatory during PTCA in patients with coronary heart disease (3, 4). In particular, aspirin and other antiplatelet drugs are widely used in patients undergoing PTCA (3, 4). However, major limitations to the clinical use of aspirin are the modest therapeutic effects on restenosis and the associated gastrotoxicity, the single most common adverse effect of the medication (5–8).
Nitric oxide (NO) is a chemical mediator released from the vascular endothelium, which play a pivotal role in maintaining endothelial and vascular smooth muscle cell (VSMC) functions (9, 10). Restenosis has been proposed to result from impaired NO production by the damaged endothelium (11). There is evidence that modulation of NO-dependent pathways may be used to modify the development of restenosis. For example, the administration of l-arginine, the precursor to NO, reduces restenotic lesion formation and monocyte recruitment in cholesterol-fed rabbits (12, 13), whereas l-NAME, a nonselective inhibitor of NO-synthase, stimulates neointimal hyperplasia (14). Thus, increased NO production at the site of balloon injury may exert beneficial effects. An NO-releasing aspirin derivative (NCX-4016) (15) has been developed that represents a tool for studying the effects of simultaneous delivery of NO and aspirin in experimental restenosis. Moreover, NCX-4016 exhibits little or no ulcerogenic activity in several experimental models (6, 7, 15), inhibits caspase activation (7), and has antithrombotic properties (16). Although the extent of contribution of high blood cholesterol to the pathogenesis of restenosis is still a matter of controversy, in the presence of hypercholesterolemia, restenosis can be more severe and is refractory to cholesterol-lowering drugs or apheresis (14, 17–19). Therefore, in this clinical setting, a more effective therapeutic approach is necessary. NCX-4016 possesses two additional actions, namely antiinflammatory and antiplatelet adhesion (20), which are well known to be beneficial in treating restenosis (1–3).
Although the predictive clinical value of the data obtained from the study of animal models for restenosis has been limited (21), encouraging signs are emerging from the development of transgenic murine models of atherosclerosis. Thus, the goal of the study was to investigate the comparative effects of NCX-4016 and aspirin on experimental restenosis in an animal model of familial hypercholesterolemia represented by the low-density lipoprotein (LDL) receptor-deficient mouse (22).
Materials and Methods
Experimental Procedure.
Animal studies were carried out according to the Guidelines of the American Heart Association for Accreditation of Laboratory Animal Care and complied with the Guide for the Care and Use of Laboratory Animals (23), approved by the American Physiology Society. Although LDL receptor-deficient mice have the same genetic defect as humans with familial hypercholesterolemia (24), these transgenic mice, derived from LDL receptor-deficient mice crossed with C57BL/6J mice for ten generations, develop only “moderate” elevated plasma cholesterol levels (250–300 mg/dl) when fed regular mouse chow (22). However, high cholesterol levels are easily achieved by enriched-cholesterol diets (22) that induce extensive atherosclerosis throughout the arterial tree (22, 25). The experiments described here were carried out on male mice (10–12 weeks of age) fed a high-cholesterol and cholate-free diet (21% fat by weight, 0.15% by weight cholesterol and 19.5% by weight casein; #88137, Harlan/Teklad, Madison, WI) for 18 weeks (Table 1).
Table 1.
Average weights and plasma lipids, in LDL receptor-deficient mice fed with midwestern diet for 18 weeks
| Initial weight, g | Final weight, g | Cholesterol, mg/dl | Triglycerides, mg/dl |
|---|---|---|---|
| 27.1 ± 3.5 | 32.5 ± 5.2 | 741 ± 57 | 112 ± 23 |
Data are expressed as mean ± SD.
After anesthesia with ketamine (80 mg/kg body weight, i.p. Ketaset) and xylazine (5 mg/kg body weight, i.p. AnaSed), the carotid arteries were exposed (midline neck incision) under light microscope and ligated distally (6-0 silk ties). According to the procedure proposed by Manka et al. (26), a standard angioplasty microcatheter (1 cm long and 2 mm diameter balloon introduced with a 0.014-inch flexible angioplasty guidewire) was placed into the common carotid artery under fluoroscopic control. Eccentric plaques (mild to moderate intimal thickening) are often present at this site, and reduce the lumen of the vessel of 30–35% (125–150 μm thickness of lesions). This is an important methodological consideration because the rat model of restenosis offers healthy carotid arteries that are free of atherosclerotic lesions (21). This condition is totally different from the clinical condition of PTCA in humans with extensive coronary atherosclerotic lesions (21). To induce endothelial denudation, the balloon was inflated at constant inflation pressure (3 passes at 1.5 atmospheres achieved with an inflation device; Guidant, Santa Clara, CA) for 14–15 sec (artery ratio of 1.2:1). Our preliminary experiments and others (26) revealed that this experimental procedure resulted in a completely denuded surface covered by platelets 30 min after injury and significant neointima formation and medial thickening 14 days after ballon angioplasty. The intervention phase was achieved by using male animals matched for age (2–3 months), body weight, and plasma cholesterol (Table 1) that were treated with low doses of NCX-4016 [2-acetoxybenzoic acid 3-(nitrooxymethyl) phenyl ester (10 and 30 mg/kg); Fig. 1] or higher doses of aspirin (18 and 54 mg/kg) following two different protocols. In the first protocol, drugs were administered 7 days before, and 21 days after balloon injury (Groups 1–4). In the second protocol, drugs were administered only for 21 days after balloon injury (Groups 5–7). A control untreated group was also included.
Figure 1.

Chemical structures of aspirin and NCX-4016.
Morphometric Analysis.
Mice were killed with a lethal dose of sodium pentobarbital 21 days after injury and a 24-gauge needle was placed in the left portion of the aortic arch (ascendent aorta) to achieve in situ perfusion fixation of the carotid arteries at physiologic pressure (100 mm Hg) with phosphate-buffered paraformaldehyde (4%, 0.1 M, pH 7.3) for histology and normal saline for immunohistochemistry assessed by computer-assisted imaging analysis (25, 27, 28). At this time, histologic analysis of the gastrointestinal system indicated both a severe mucosal and ulcerogenic damage induced by aspirin, but not by NCX-4016 (data not shown). These data are in agreement with previously reported findings (6, 7, 15, 16). Carotid segments were immersed in 4% paraformaldehyde. After stepwise dehydration with graded alcohols, specimens were embedded in epoxy-araldit resin (22, 26). Carotid arteries were serially sectioned in 15–20 slices (10 μM) with a rotating diamond-coated knife (Leica, Deerfield, IL), and sections were stained with toluidine blue. The length of the external elastic lamina, the area confined by the internal elastic lamina, and the cross-sectional neointimal area were measured by morphometry (27, 28). The vessel injury score was determined according to the original method devised by Schwartz et al. (29), adapted for mice (22, 26). The percent area stenosis was then calculated. In additional sections, atherosclerotic carotid artery lesions were determined by oil-red O stained sections using computer-assisted imaging (25, 27, 28). Total cell number and number of proliferating cells in the neointima of the arterial cross sections were determined by computer-assisted imaging cell counting after immunohistochemistry of serial sections with α-actin monoclonal mouse antibody (M0858 from Dako), and antibody against proliferating cell nuclear antigen (PCNA, 1:250 dilution; clone PC10 from Dako; the adjacent medial layer of vessels served as positive control) (27, 28). Macrophage-derived foam cells were stained by the F4/80 antibody (1:500 dilution; Accurate Scientific, Westbury, NY) (27, 28). VSMCs were counted only if they stained for VSMC α-actin and the cell nucleus was seen. The mean percentage of VSMCs relative to total cells was determined for each animal. To determine whether the PCNA-positive cells were derived from VSMCs, immunohistochemical double staining (30) with PCNA and VSMC α-actin was performed. Briefly, on the double-labeled slides, each field was scored for the number of PCNA-positive nuclei associated with cytoplasm positive for VSMC. The number of intimal VSMCs and total cell nuclei were counted in eight nonoverlapping fields. The ratio of the total number of double-labeled cells to total number of PCNA-labeled cells was used to indicate percentages of VSMC proliferative activity in the arterial wall. Additional experiments were also performed with F4/80/PCNA double staining. Epitopes recognized by the primary antibody were detected by an avidin-biotin-peroxidase method (27, 28).
Statistical Analysis.
The inhibitory effects on VSMC proliferation and restenosis induced by treatment with NCX-4016 and aspirin were compared by using the Kruskal–Wallis test. Intimal cell density and VSMC content from animals in treatment and control groups were compared by using nonparametric ANOVA analysis with adjustment for multiple measurements from each animal.
Results
Restenosis after balloon dilation of atherosclerotic carotid arteries is reflected as migration and proliferation of VSMCs and accumulation of macrophages. As shown in Fig. 2, it is evident that following balloon injury there is a massive VSMC proliferation concomitant with neointimal formation at the site of lesion in control mice (Fig. 2A). This phenomenon was associated with early intimal remodeling and it was reduced modestly by high doses of aspirin (Fig. 2B) and to a larger extent by lower doses of NCX-4016 (Fig. 2C). The progression of the lesion was accompanied by migration of macrophage into the regions of neointimal hyperplasia (Fig. 3A). Macrophage deposition at the site of injury was not significantly inhibited by aspirin (54 mg/kg, protocol 2; Fig. 3B; −7 ± 3% of reduction in single-label serial arterial sections stained with the F4/80 antibody, P = 0.678 vs. controls), but it was significantly reduced by NCX-4016 (30 mg/kg, protocol 2; Fig. 3C; −45 ± 8% of reduction in serial arterial sections stained with F4/80, P = 0.0023 vs. controls).
Figure 2.
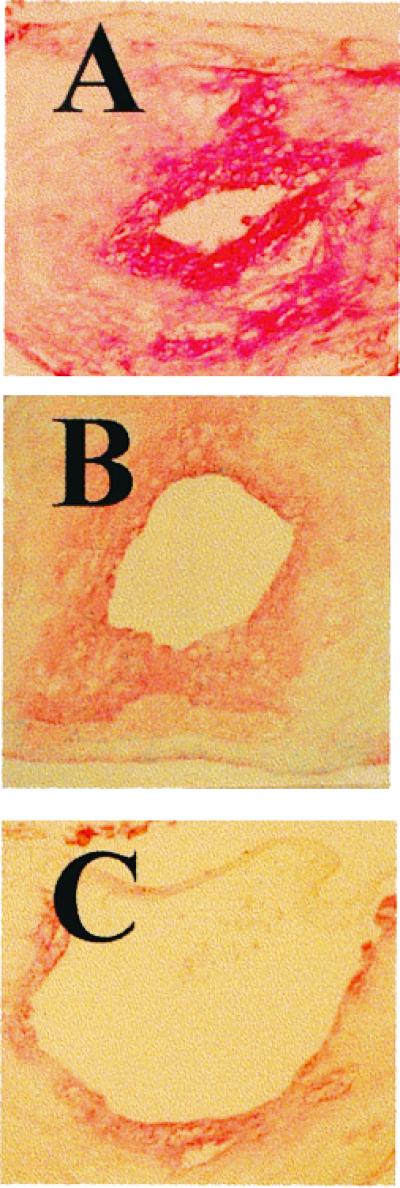
(A) Complicated restenosis following balloon angioplasty, neointimal hyperplasia, macrophage deposition, and VSMC proliferation (VSMCs/PCNA) in carotid artery of control vehicle-treated LDL receptor-deficient mice. (B) Treatment with aspirin (54 mg/kg; protocol 1) reduced intima remodeling and the degree of VSMC proliferation. (C) NCX-4016 (30 mg/kg; protocol 1) showed a more significant reduction of neointimal hyperplasia and VSMC proliferation.
Figure 3.
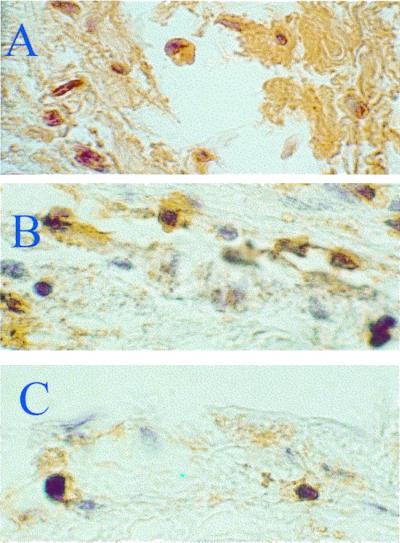
(A) The progression of the lesion was accompanied by migration of macrophages-foam cells in the neointimal hyperplasia. (B) Macrophage deposition was not reduced significantly by aspirin. (C) In contrast, this phenomenon was significantly reduced by NCX-4016.
Apparently, hypercholesterolemia can worsen the extent of restenosis because wild-type mice have a lesser degree of restenosis compared with LDL receptor-deficient mice (intima/media ratio of 56 ± 8% greater than that of wild-type mice, P = 0.0075). Results obtained with protocol 1 and 2 by using 10 and 30 mg/kg of NCX-4016 or 16 and 54 mg/kg of aspirin in each protocol are shown in Table 2. The balloon injury scores related to the treated groups were similar among different protocols (Table 2). Thus, differences among groups were not ascribed to a different degree of balloon injury. This evaluation is an important methodological consideration in the relatively small vessels of murine models. Unfortunately, this evaluation is rarely made or reported in the literature.
Table 2.
Effects on restenosis of NCX-4016 versus aspirin in hypercholesterolemic LDL receptor-deficient mice
| Injury score | Neointimal area, μm2 | % Lumen stenosis | Number of neointimal cells | % Proliferating cells, VSMC/PCNA | Neointimal cell density, n/0.1 mm2 | |
|---|---|---|---|---|---|---|
| Controls (n = 8; balloon injury) | 1.81 ± 0.14 | 34.9 ± 1.8 | 72.9 ± 3.19 | 399.9 ± 19.8 | 48.2 ± 2.0 | 765 ± 82 |
| Group 1 (n = 8; 10 mg/Kg NCX-4016 Protocol 1) | 1.83 ± 0.08 | 18.8 ± 0.8*‡† | 31.4 ± 2.32*‡† | 223.3 ± 13.1*‡† | 14.6 ± 0.5*‡† | 436 ± 41* |
| Group 2 (n = 8; 30 mg/Kg NCX-4016 Protocol 1) | 1.82 ± 0.09 | 16.4 ± 0.5*‡† | 28.9 ± 1.45*‡† | 200.4 ± 11.2*‡† | 11.2 ± 0.3*‡† | 406 ± 32*‡ |
| Group 3 (n = 8; 16 mg/Kg Aspirin Protocol 1) | 1.89 ± 0.09 | 23.8 ± 0.9* | 42.8 ± 3.12* | 298.9 ± 15.6* | 22.8 ± 0.6* | 545 ± 41* |
| Group 4 (n = 8; 54 mg/Kg Aspirin Protocol 1) | 1.84 ± 0.06 | 21.2 ± 0.6*† | 39.6 ± 2.83*† | 272.8 ± 12.5*† | 19.9 ± 0.5*† | 508 ± 29*† |
| Group 5 (n = 8; 10 mg/Kg NCX-4016 Protocol 2) | 1.86 ± 0.08 | 22.8 ± 0.8*‡ | 42.8 ± 2.64*‡ | 284.3 ± 12.7*‡ | 21.8 ± 0.5*‡ | 521 ± 39*‡ |
| Group 6 (n = 8; 30 mg/Kg NCX-4016 Protocol 2) | 1.89 ± 0.09 | 20.9 ± 0.5*‡ | 36.5 ± 3.11*‡ | 252.1 ± 13.3*‡ | 18.4 ± 0.4*‡ | 488 ± 29*‡ |
| Group 7 (n = 8; 16 mg/Kg Aspirin Protocol 2) | 1.86 ± 0.07 | 32.8 ± 1.8 | 64.5 ± 4.95 | 382.5 ± 17.1 | 45.5 ± 1.5 | 679 ± 94 |
| Group 8 (n = 8; 54 mg/Kg Aspirin Protocol 2) | 1.85 ± 0.06 | 29.9 ± 2.6 | 61.5 ± 3.85** | 332.5 ± 14.6** | 31.9 ± 1.6* | 622 ± 78 |
Values are mean ± SE. Protocol 1: 7 days before and 21 days after injury treatment; Protocol 2: 21 days after injury treatment.
, P < 0.01 vs. controls; **, P < 0.05 vs. vehicle-controls;
, P < 0.05 vs. respective aspirin dose in the same protocol;
, P < 0.05 vs. respective treatment between protocols.
The neointimal global area (N/M ratio) was significantly reduced by all treatments as compared with untreated controls (Table 2). However, neointimal hyperplasia and cell density were greater in animals receiving both doses of aspirin alone when compared with those receiving NCX-4016 (Table 2). Double-label immunohistochemistry in serial carotid sections for VSMCs (Table 2) and macrophages (−38 ± 11% of F4/80/PCNA double positive stained sections vs. controls in the 30 mg/kg NCX-4016, Protocol 1 group; P = 0.0087 vs. controls) indicated that both types of cells were reduced by NCX-4016 at the site of lesion. Accordingly, the residual percentage of lumen stenosis was significantly reduced in animals treated with NCX-4016 in both protocols (Table 2). These effects on the degree of restenosis clearly demonstrate the beneficial actions of NCX-4016 on the development of restenosis in the presence of hypercholesterolemia.
When data obtained from the two protocols were analyzed (i.e., drugs administered 7 days before and 21 days after balloon injury or administered only for 21 days after injury), it was evident that further beneficial effects were obtained when drugs were administered beginning 7 days before injury (Table 2). This phenomenon may be due to early effects of NCX-4016 on intracellular signaling pathways, which are involved in neointimal hyperplasia. NCX-4016, at the lower dose in both protocols, reduced all parameters significantly either versus control or aspirin at the highest dose tested (Fig. 4). Moreover, VSMC proliferation was markedly increased after balloon injury in untreated controls (Table 1), and this was associated with collagen I deposition (data not shown).
Figure 4.

Effect of aspirin (30 mg/kg) and NCX-4016 (16 mg/kg) on N/M ratio, percentage of lumen stenosis, and neointimal cell density following the treatments with protocols 1 and 2. White bars are vehicle-controls, black bars are those treated with NCX-4016, and dotted bars are those treated with aspirin. *, P < 0.01 vs. vehicle-controls; #, P < 0.05 vs. aspirin.
Data obtained in this experimental setting with a typical NO-donor agent, S-nitroso-N-acetylpenicillamine (SNAP; 500 μM) (31), showed only a reduction 10 ± 2% in proliferating cells (n = 3, P = 0.456 vs. controls).
Discussion
VSMC proliferation and macrophage activation at the site of injury are involved in the pathogenesis of restenosis and vascular remodeling after angioplasty (1–3). NO inhibits VSMC growth in vitro and experimental neointimal hyperplasia in vivo, indicating a role for NO as a regulator of VSMC proliferation (9, 32, 33). NO is also involved in the control of several other pathophysiological responses, including platelet function and inflammatory cell adhesion, vascular reactivity, and endothelial permeability (9, 32, 33). Thus, a large body of data indicates that NO-release or delivery could be useful in restenosis after PTCA.
The data in this study demonstrate that the reduction in the degree of restenosis elicited by NO-releasing aspirin (NCX-4016) and, to a modest extent, aspirin in hypercholesterolemic LDL receptor-deficient mice subjected to balloon injury is due to reduced VSMC proliferation, cell density, and macrophage deposition. These effects were markedly evident in animals treated with NCX-4016, indicating that this beneficial action was likely due to NO-mediated effects on VSMCs and macrophages at the site of injury. We investigated, by using two different protocols, the impact of NCX-4016 on both prevention (Protocol 1) and treatment (Protocol 2) of experimental restenosis. The therapeutic effects of NCX-4016 were increased following one-week pretreatment prior to balloon injury, indicating that the drug interferes with early signaling mechanisms triggered by the arterial injury. These effects were also evident following the therapeutic protocol where the lower dose of NCX-4016, but not aspirin, significantly inhibited experimental restenosis. Overall, the efficacy of NCX-4016 was significantly greater than aspirin alone and effective at the lower dose tested. High doses of salicylates inhibit human VSMC proliferation in response to platelet-derived growth factor (33), and sulindac, but not aspirin, was effective in preventing restenosis after femoral arterial injury in apolipoprotein E-deficient mice (35). Although aspirin-like drugs have little or no effect on restenosis in humans (3, 4), NO releasing drugs have not been evaluated in restenosis in humans. Although experimental laboratory animal models may not entirely represent the complexity of the human disease (21, 36), from a clinical point of view, these results could be extended to the common clinical conditions of PTCA during hypercholesterolemia and in patients with gastrointestinal restrictions to the use of aspirin (5, 8). Moreover, the presence of atherosclerotic lesions at the site of balloon injury in our model may represent experimental conditions similar to the disease in humans (21, 36). Further benefits in the use this model come from the recent possibility of noninvasive in vivo magnetic resonance imaging of injury-induced neointima formation in the carotid artery of the hypercholesterolemic mouse (37).
Encouraging results come from a very recent large study carried out on 11,500 patients, in which tranilast that restores cytokine-induced NO production was able to reduce clinical, angiographic, and intravascular signs of restenosis after PTCA (38). Thus, the efficacy of NCX-4016 in adversing restenosis may be also due to additional NO-mediated effects on vascular inflammation after PTCA. Tranilast interferes with proliferation and migration of VSMCs induced by platelet-derived growth factor and transforming growth factor-β1. Collagen synthesis in VSMCs is inhibited by tranilast, which also inhibits the release or production of cyclooxygenase-2 and restores cytokine-induced NO production.
We have also observed that macrophage deposition was reduced in neointimal hyperplasia after administration of NCX-4016. This phenomenon was similar to that observed by Niebauer et al. (13) when using local l-arginine delivery in an experimental model of restenosis. The therapeutic response to l-arginine may have been attributed to endogenous NO as l-arginine is converted to NO in vascular endothelial cells. Macrophages also appear to be involved in the restenotic lesions post-PTCA (39, 40) or stenting (40, 41) in humans, suggesting a possible beneficial effect of NO-releasing aspirin by mechanisms involving reduced accumulation of macrophages at sites of injury. In the present study, by using 500 μM S-nitroso-N-acetylpenicillamine (SNAP), a pure NO-donor, we observed a modest reduction of restenosis, indicating that administration of NO alone may be not sufficient to exert a beneficial effect in experimental restenosis. Interestingly, S-nitrosoglutathione, a NO-donor with preferential action on platelets, has been shown to inhibit platelet aggregation following PTCA in patients under aspirin treatment, without altering blood pressure (42). However, the clinical management of pure NO-donors could be difficult due to systemic effects on blood pressure. Our findings are in line with several studies where NO has been shown to serve as vasoprotector, including inhibition of platelet aggregation and leukocyte adherence to the site of injury (43). NO also affects cell cycle, immune system, and angiogenesis in cancer cells (44), but all these networks are also involved in atherogenesis and restenosis (1–4, 9, 10, 45, 46). Further studies at the molecular level are needed to better understand which are the main signaling pathways involved in the protective action of NO-releasing aspirin and its long-term effects on vascular remodeling after balloon injury.
In conclusion, these data show for the first time that a NO-donor drug, NCX-4016, reduces restenosis in hypercholesterolemic mice. This therapeutic effect was significantly greater than that achieved with aspirin and was attained at lower doses. In addition, aspirin is well known to be ulcerogenic in experimental models and in humans (5, 8), whereas NCX-4016 is not (6, 7, 15). NCX-4016, when administered 7 days before balloon injury, showed higher protective effects than treatment after injury alone. These results suggest that NCX-4016 may be clinically useful both in the prevention and treatment of restenosis, particularly in restenosis accompanied by hypercholesterolemia and/or gastrointestinal damage.
Abbreviations
- PTCA
percutaneous transluminal coronary angioplasty
- NCX-4016
NO-releasing aspirin derivative
- LDL
low-density lipoprotein
- VSMC
vascular smooth muscle cell
- PCNA
proliferating cell nuclear antigen
References
- 1.Liu M W, Roubin G S, King S B. Circulation. 1989;79:1374–1387. doi: 10.1161/01.cir.79.6.1374. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz R S, Holmes DR, Jr, Topol E J. J Am Coll Cardiol. 1992;20:1284–1293. doi: 10.1016/0735-1097(92)90389-5. [DOI] [PubMed] [Google Scholar]
- 3.Folts J D, Schafer A I, Loscalzo J, Willerson J T, Muller J E. J Am Coll Cardiol. 1999;33:295–303. doi: 10.1016/s0735-1097(98)00601-9. [DOI] [PubMed] [Google Scholar]
- 4.Lefkovits J, Topol E J. Prog Cardiovasc Dis. 1997;40:141–158. doi: 10.1016/s0033-0620(97)80006-0. [DOI] [PubMed] [Google Scholar]
- 5.Roderick P J, Wilkes H C, Meade P W. Br J Clin Pharmacol. 1993;35:219–226. doi: 10.1111/j.1365-2125.1993.tb05689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace J L, McNight W, Del Soldato P, Baydoun A, Cirino G. J Clin Invest. 1995;96:2711–2718. doi: 10.1172/JCI118338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiorucci S, Antonelli E, Santucci L, Morelli O, Miglietti M, Federici B, Mannucci R, Del Soldato P, Morelli A. Gastroenterology. 1999;116:1089–1106. doi: 10.1016/s0016-5085(99)70012-0. [DOI] [PubMed] [Google Scholar]
- 8.Awtry E H, Loscalzo J. Circulation. 2000;101:1206–1218. doi: 10.1161/01.cir.101.10.1206. [DOI] [PubMed] [Google Scholar]
- 9.Moncada S, Palmer R M, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 10.Ignarro L J, Cirino G, Casini A, Napoli C. J Cardiovasc Pharmacol. 1999;34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 11.Myers P R, Webel R, Thondapu V, Xu X P, Amann J, Tanner M A, Jenkins J S, Pollock J S, Laughlin M H. Int J Cardiol. 1996;55:183–191. doi: 10.1016/0167-5273(96)02684-8. [DOI] [PubMed] [Google Scholar]
- 12.Schwarzacher S P, Lim T T, Wang B, Kernoff R S, Niebauer J, Cooke J P, Yeung A C. Circulation. 1997;95:1863–1869. doi: 10.1161/01.cir.95.7.1863. [DOI] [PubMed] [Google Scholar]
- 13.Niebauer J, Schwarzacher S P, Hayase M, Wang B, Kernoff R S, Cooke J P, Yeung A C. Circulation. 1999;100:1830–1835. doi: 10.1161/01.cir.100.17.1830. [DOI] [PubMed] [Google Scholar]
- 14.Le Tourneau T, Van Belle E, Corseaux D, Vallet B, Lebuffe G, Dupuis B, Lablanche J M, McFadden E, Bauters C, Bertrand M E. J Am Coll Cardiol. 1999;33:876–882. doi: 10.1016/s0735-1097(98)00621-4. [DOI] [PubMed] [Google Scholar]
- 15.Del Soldato P, Sorrentino R, Pinto A. Trends Pharmacol Sci. 1999;20:319–323. doi: 10.1016/s0165-6147(99)01353-x. [DOI] [PubMed] [Google Scholar]
- 16.Wallace J L, Muscara M N, McKnight W, Dicay M, Del Soldato P, Cirino G. Thromb Res. 1999;93:43–50. doi: 10.1016/s0049-3848(98)00134-0. [DOI] [PubMed] [Google Scholar]
- 17.Kanemitsu S, Takekoshi N, Matsui S, Tsugawa H, Ohkubo S, Kitayama M, Matsuda T, Senma J, Masuyama K, Yamagata T, et al. Ther Apheresis. 1998;2:65–70. doi: 10.1111/j.1744-9987.1998.tb00075.x. [DOI] [PubMed] [Google Scholar]
- 18.Lundgren C, Sobel B E, Fujii S. Jpn Heart J. 1995;37:119–126. doi: 10.1536/ihj.37.119. [DOI] [PubMed] [Google Scholar]
- 19.Gurlek A, Dagalp Z, Oral D, Omurlu K, Erol C, Akyol T, Tutar E. J Cardiovasc Risk. 1995;2:51–55. [PubMed] [Google Scholar]
- 20.Lechi C, Andrioli G, Gaino S, Tommasoli R, Zuliani V, Ortolani R, Degan M, Benoni G, Bellavite P, Lechi A, et al. Thromb Haemostasis. 1996;76:791–798. [PubMed] [Google Scholar]
- 21.Johnson G J, Griggs T R, Badimon L. Thromb Haemostasis. 1999;81:835–843. [PubMed] [Google Scholar]
- 22.Palinski W, Napoli C, Reaven P D. In: Contemporary Cardiology, (Harvard Series): Vascular Disease and Injury: Preclinical Research. Simons D I, Rogers C, editors. Totowa, NJ: Humana; 2000. pp. 149–174. [Google Scholar]
- 23.Committee on Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. Bethesda: Natl. Inst. Health; 1985. , NIH Publ. No. 86-23 (DRR/NIH). [Google Scholar]
- 24.Ishibashi S, Goldstein J L, Brown M S, Herz J, Burns D K. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tangirala R K, Rubin E M, Palinski W. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- 26.Manka D R, Wiegman P, Din S, Sanders J M, Green S A, Gimple L W, Ragosta M, Powers E R, Ley K, Sarembock I J. J Vasc Res. 1999;36:372–378. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- 27.Napoli C, Glass C K, Witztum J L, Deutsch R, D'Armiento F P, Palinski W. Lancet. 1999;354:1234–1241. doi: 10.1016/S0140-6736(99)02131-5. [DOI] [PubMed] [Google Scholar]
- 28.Napoli C, Witztum J L, de Nigris F, Palumbo G, D'Armiento F P, Palinski W. Circulation. 1999;99:2003–2010. doi: 10.1161/01.cir.99.15.2003. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz R S, Huber K C, Murphy J G, Edwards W D, Camrud A R, Vlietstra R E, Holmes D R. J Am Coll Cardiol. 1992;19:267–274. doi: 10.1016/0735-1097(92)90476-4. [DOI] [PubMed] [Google Scholar]
- 30.de Nigris F, Youssef T, Ciafre S, Anania V, Condorelli G L, Palinski W, Napoli C. Circulation. 2000;102:2111–2117. doi: 10.1161/01.cir.102.17.2111. [DOI] [PubMed] [Google Scholar]
- 31.Ferrero R, Rodriguez-Pascual F, Miras-Portugal M T, Torres M. Br J Pharmacol. 1999;127:779–787. doi: 10.1038/sj.bjp.0702607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George S E. Coron Artery Dis. 1999;10:295–300. doi: 10.1097/00019501-199907000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar R, Webb R C. J Vasc Res. 1998;35:135–142. doi: 10.1159/000025576. [DOI] [PubMed] [Google Scholar]
- 34.Marra D E, Simoncini T, Liao J K. Circulation. 2000;102:2124–2130. doi: 10.1161/01.cir.102.17.2124. [DOI] [PubMed] [Google Scholar]
- 35.Reis E D, Roque E D, Dansky H, Fallon J T, Badimon J J, Cordon-Cardo C, Shiff S J, Fisher E A. Proc Natl Acad Sci USA. 2000;97:12764–12769. doi: 10.1073/pnas.210394497. . (First Published October 10, 2000; 10.1073/pnas.210394497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuster V, Poon M, Willerson J T. Circulation. 1998;97:16–18. doi: 10.1161/01.cir.97.1.16. [DOI] [PubMed] [Google Scholar]
- 37.Manka D R, Gilson W, Sarembock I J, Ley K, Berr S S. J Magn Reson. 2000;12:790–794. doi: 10.1002/1522-2586(200011)12:5<790::aid-jmri19>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Holmes D, Fitzgerald P, Goldberg S, LaBlanche J M, Lincoff A M, Savage M, Serruys P W, Willerson J T, Granett J R, Chan R, et al. Am Heart J. 2000;139:23–31. doi: 10.1016/s0002-8703(00)90304-1. [DOI] [PubMed] [Google Scholar]
- 39.Moreno P R, Bernardi V H, Lopez-Cuellar J, Newell J B, McMellon C, Gold H K, Palacios I F, Fuster V, Fallon J T. Circulation. 1996;94:3098–3102. doi: 10.1161/01.cir.94.12.3098. [DOI] [PubMed] [Google Scholar]
- 40.Moreno P R, Palacios I F, Leon M N, Rhodes J, Fuster V, Fallon J T. Am J Cardiol. 1999;84:462–466. doi: 10.1016/s0002-9149(99)00334-3. [DOI] [PubMed] [Google Scholar]
- 41.Komatsu R, Ueda M, Naruko T, Kojima A, Becker A E. Circulation. 1998;98:224–233. doi: 10.1161/01.cir.98.3.224. [DOI] [PubMed] [Google Scholar]
- 42.Langford E J, Brown A S, Wainwright R J, de Belder A J, Thomas M R, Smith R E, Radomski M W, Martin J F, Moncada S. Lancet. 1994;344:1458–1460. doi: 10.1016/s0140-6736(94)90287-9. [DOI] [PubMed] [Google Scholar]
- 43.Kibbe M, Billiar T, Tzeng E. Cardiovasc Res. 1999;43:650–657. doi: 10.1016/s0008-6363(99)00130-3. [DOI] [PubMed] [Google Scholar]
- 44.Wink D A, Vodovotz Y, Laval J, Laval F, Dewhirst M W, Mitchell J B. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- 45.Kojda G, Harrison D. Cardiovasc Res. 1999;43:562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 46.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]