

Abstract
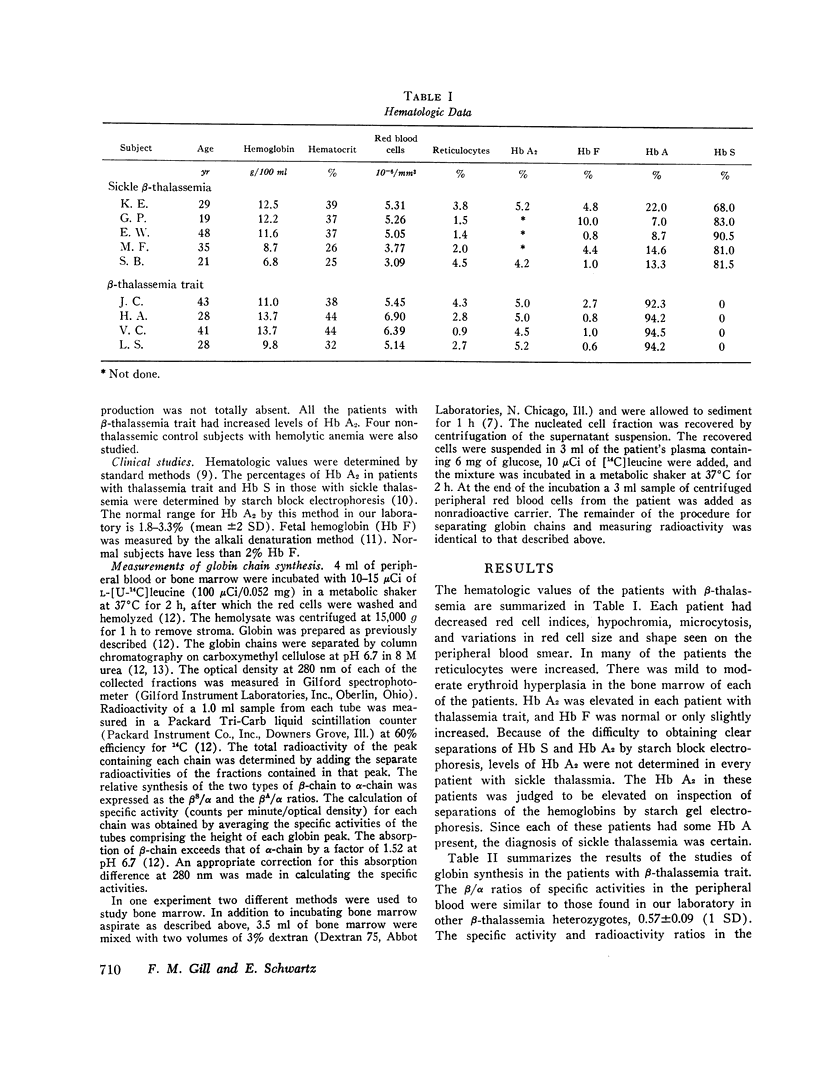
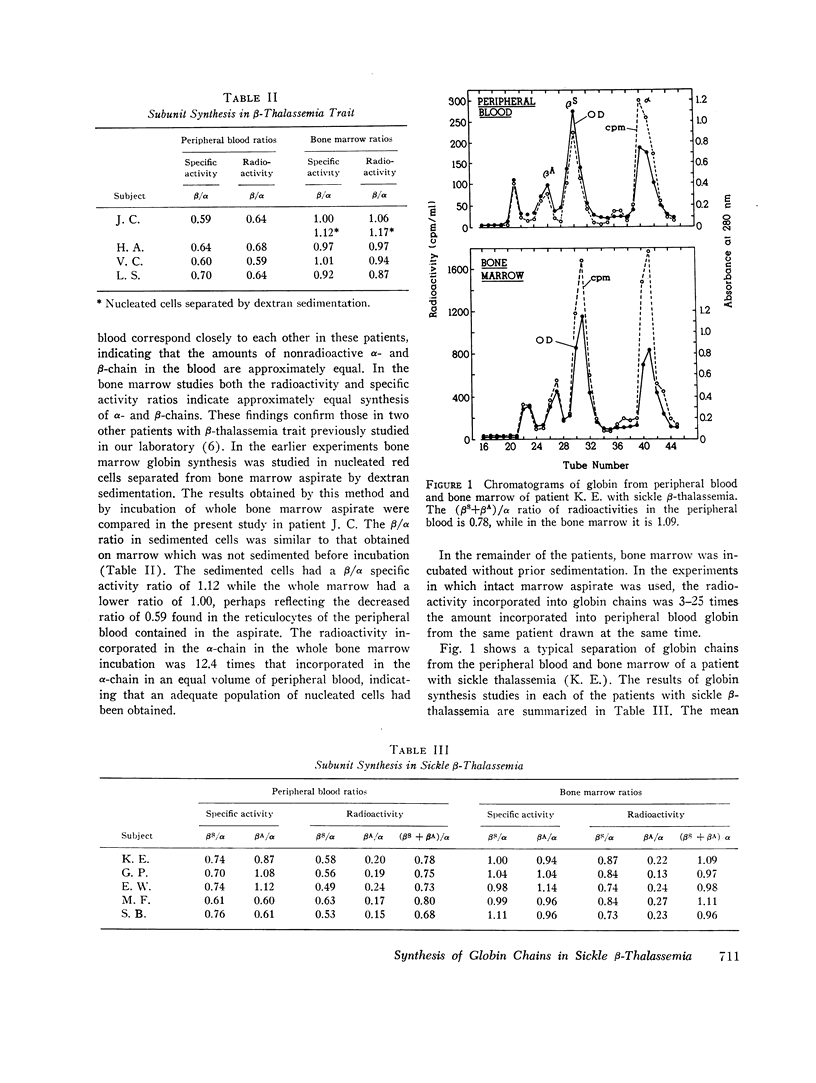
In five patients with sickle β-thalassemia there was balanced α- and β-globin synthesis in the bone marrow and decreased total β-chain synthesis relative to that of α-chain in the peripheral blood. These findings are similar to those in patients with simple β-thalassemia trait. Despite a range of hemoglobin concentrations from 6.8 to 12.5 g/100 ml in the patients with sickle thalassemia, there was no evidence of a significant excess of α-chains in the red cells of the bone marrow which could contribute to the hemolysis and anemia.
In patients heterozygous for β-thalassemia the capacity to synthesize β-chain decreases more rapidly than that for α-chain. In nonthalassemic subjects the rates of β- and α-chain synthesis decrease equally as the red cell matures. The βS- and βA-chains serve as convenient markers for globin synthesis due to the nonthalassemic and thalassemic alleles in patients with sickle β-thalassemia. The unbalanced globin synthesis in the peripheral blood of these patients is explained by the decrease in relative synthesis of βS-chain, in comparison with that of α-chain. This instability is not present in sickle cell trait. The βA-chain synthesis was only unstable in the two patients who had the most marked anemia. The major mechanism for achieving balanced globin production in the bone marrow in the presence of one thalassemic gene appears to be increased synthesis of β-chain due to the nonthalassemic allele. In addition, there may be a decrease of total α-chain synthesis in some patients.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bank A., Braverman A. S., O'Donnell J. V., Marks P. A. Absolute rates of globin chain synthesis in thalassemia. Blood. 1968 Feb;31(2):226–233. [PubMed] [Google Scholar]
- Bank A., O'Donnell J. V., Braverman A. S. Globin chain synthesis in heterozygotes for beta chain mutations. J Lab Clin Med. 1970 Oct;76(4):616–621. [PubMed] [Google Scholar]
- Braverman A. S., Bank A. Changing rates of globin chain synthesis during erythroid cell maturation in thalassemia. J Mol Biol. 1969 May 28;42(1):57–64. doi: 10.1016/0022-2836(69)90486-0. [DOI] [PubMed] [Google Scholar]
- Clegg J. B., Naughton M. A., Weatherall D. J. An improved method for the characterization of human haemoglobin mutants: identification of alpha-2-beta-2-95GLU, haemoglobin N (Baltimore). Nature. 1965 Aug 28;207(5000):945–947. doi: 10.1038/207945a0. [DOI] [PubMed] [Google Scholar]
- Esan G. J., Adesina T. A., Luzzatto L. Synthesis of haemoglobins specified by allelic genes in human heterozygotes. Nat New Biol. 1971 Feb 3;229(5):143–145. doi: 10.1038/newbio229143a0. [DOI] [PubMed] [Google Scholar]
- Friedman S., Oski F. A., Schwartz E. Bone marrow and peripheral blood globin synthesis in an American black family with beta thalassemia. Blood. 1972 Jun;39(6):785–793. [PubMed] [Google Scholar]
- GERALD P. S., DIAMOND L. K. The diagnosis of thalassemia trait by starch block electrophoresis of the hemoglobin. Blood. 1958 Jan;13(1):61–69. [PubMed] [Google Scholar]
- Kan Y. W., Nathan D. G. Mild thalassemia: the result of interactions of alpha and beta thalassemia genes. J Clin Invest. 1970 Apr;49(4):635–642. doi: 10.1172/JCI106274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y. W., Schwartz E., Nathan D. G. Globin chain synthesis in the alpha thalassemia syndromes. J Clin Invest. 1969 Nov;47(11):2512–2522. doi: 10.1172/JCI105933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D. G. Thalassemia. N Engl J Med. 1972 Mar 16;286(11):586–594. doi: 10.1056/NEJM197203162861107. [DOI] [PubMed] [Google Scholar]
- SINGER K., CHERNOFF A. I., SINGER L. Studies on abnormal hemoglobins. II. Their identification by means of the method of fractional denaturation. Blood. 1951 May;6(5):429–435. [PubMed] [Google Scholar]
- Schwartz E. Heterozygous Beta thalassemia: balanced globin synthesis in bone marrow cells. Science. 1970 Mar 13;167(3924):1513–1514. doi: 10.1126/science.167.3924.1513. [DOI] [PubMed] [Google Scholar]
- Weissman S. M., Jeffries I., Karon M. The synthesis of alpha, beta, and delta peptide chains by reticulocytes from subjects with thalassemia or hemoglobin Lepore. J Lab Clin Med. 1967 Feb;69(2):183–193. [PubMed] [Google Scholar]
- Wrightstone R. N., Huisman T. H., van der Sar A. Qualitative and quantitative studies of sickle cell hemoglobin in homozygotes and heterozygotes. Clin Chim Acta. 1968 Dec;22(4):593–601. doi: 10.1016/0009-8981(68)90108-3. [DOI] [PubMed] [Google Scholar]