

Plants of the Hypericum genus (Clusiaceae) have been known around the world for centuries for their medicinal properties. While H. perforatum (St. John’s wort), used to treat moderate depression,[1] is perhaps the most famous of these species, H. Chinense is the most recent member of this family of plants to receive notoriety as the source of biyouyanagin A. A potent anti-HIV agent and a lipopolysaccharide-induced cytokine production inhibitor, this novel natural product was first reported by Tanaka and co-workers in 2005 as possessing structure 1′ (Figure 1a).[2] Following our total synthesis of biyouyanagin A (1) from ent-zingiberene (ent-3) and hyperolactone C (4), through a [2+2] photocycloaddition, and revision of its structure from 1′ to 1 (Figure 1a),[3,4] a second biyouyanagin (B) was isolated from the same species and reported by the same group as possessing structure 2′ (Figure 1b) on the basis of spectroscopic and mass spectrometric analysis.[5] We now report the total synthesis and structural revision of biyouyanagin B (from 2′ to 2, Figure 1b) as well as the synthesis of several new biyouyanagins not yet discovered in nature, despite their possible existence based on biosynthetic considerations.
Figure 1.

Structures of a) biyouyanagin A (1′, originally assigned and 1, revised), ent-zingiberene (ent-3) and hyperolactone C (4) and b) biyouyanagin B (2′, originally assigned and 2, revised).
The reported structure for biyouyanagin B (2′) was characterized by the same general pentacyclic framework as that of its isomer, biyouyanagin A (1), but had opposite configurations at C-4, C-22 and C-24 (see Figure 1). Assuming its biogenesis proceeds through a non-enzymatic [2+2] photocycloaddition, and in accordance to our previous studies,[3] biyouyanagin B requires the union of 4-epi-hyperolactone C (5, Scheme 1) and zingiberene (3, Scheme 1) as opposed to biyouyanagin A, whose precursors were established to be the naturally occurring hyperolactone C (4) and ent-zingiberene(ent-3) (Figure 1a).[3] In an attempt to synthesize the reported structure of biyouyanagin B (structure 2′), we swiftly prepared zingiberene (3, see SI) and 4-epi-hyperolactone C (5)[3b] following our previously developed routes.[3] We then subjected a mixture of them (3:5 ca 4:1) in CH2Cl2 to photoirradiation (UV lamp, quartz cell, 320 nm filter) at 5 °C in the presence of 2′-acetonaphthone, as shown in Scheme 1. As we suspected from inspection of manual molecular models of transition states 3,5-TS-1 and 3,5-TS-2 (Scheme 2), the major product isolated from this [2+2] photocycloaddition was biyouyanagin 6 (52 % yield), and not 2′. The structure of 6 was based on NMR spectroscopic analysis (see NOEs, Figure 2). Furthermore, the spectroscopic data of 6 did not match those reported for natural biyouyanagin B,[5] precipitating a puzzle as to the true structure of this natural product.
Scheme 1.

Attempted synthesis of the originally proposed structure (2′) of biyouyanagin B from zingiberene (3) and 4-epi-hyperolactone C (5). Synthesis of biyouyanagin 6. Reagents and conditions: 5 (1.0 equiv), 3 (4.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2, 320 nm, 5 °C, 12 h, 52 % yield.
Scheme 2.

[2+2] Photocycloaddition series with 4-epi-hyperolactone C (5). Reagents and conditions: 5 (1.0 equiv), 3, 7, ent-7 and ent-3 (4.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2, 320 nm, 5 °C, 12 h; 52 % yield (6); 37 % yield (8); 39 % yield (9); 44 % yield (10).
Figure 2.

Exhibited NOEs of biyouyanagin 6.
Our next move to demystify the structure of biyouyanagin B was to prepare the remaining three stereoisomers of zingiberene [i.e. ent-zingiberene (ent-3),[3] 7-epi-zingiberene (7)[3] and ent-7-epi-zingiberene (ent-7, see SI), see Scheme 2] in order to react each individually with 4-epi-hyperolactone C (5)[3b] in the hope that one of the new photoadducts would match the natural product, biyouyanagin B. The first objective was easily achieved through the previously developed methodology from the appropriate citronellals and organocatalysts.[3] The results of these new [2+2] photocycloadditions and the one described in Scheme 1 are summarized in Scheme 2 for comparison. In each case, and under the standard photoirradiation conditions, a major biyouyanagin was isolated in good yield (6, 52 %; 8, 37 %; 9, 39 %; 10, 44 %). Much to our chagrin, none of them matched the reported natural product. Interestingly though, these studies led to an intriguing observation: two of the new biyouyanagins (i.e. 9 and 10) obtained from 4-epi-hyperolactone C were found to be formed via an “inverted” [2+2] photocycloaddition approach (i.e. Ph and C-3 quaternary center are placed on opposite sides of the central 5-membered ring) as compared to biyouyanagin A (1, Figure 1) and its 24-epi-diastereoisomer (24-epi-1, Scheme 3)[3] (where the Ph and C-3 are found on the same side of the central 5-membered ring).
Scheme 3.

a) [2+2] Photocycloaddition of zingiberene (3) and 7-epi-zingiberene (7) with hyperolactone C (4). Reagents and conditions: 4 (1.0 equiv), 3, 7 (4.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2, 320 nm, 5 °C, 12 h; 54 % yield (11); 4 % yield (12); 37 % yield (13) and 19 % yield (14) b) [2+2] Photocycloaddition of ent-7-epi-zingiberene (ent-7) with hyperolactone C (4).[3]
After eliminating the four structures derived from 4-epi-hyperolactone (5) and zingiberenes 3, ent-3, 7 and ent-7, we reasoned that biyouyanagin B is most likely derived from naturally occurring hyperolactone C (4) in partnership with one of the remaining zingiberenes (i.e. 3 and 7). We, therefore, set out to combine separately 3 and 7[3] with 4.[3] As shown in Scheme 3a, each cycloaddition gave two new biyouyanagins (11, 54 % and 12, 4 %; 13, 37 % and 14, 19 % yield, respectively) as major products, but much to our surprise, again, neither proved to represent the elusive biyouyanagin B structure. Interestingly, however, two of the products (i.e. 12 and 14) derived from hyperolactone C (4) possessed the “inverted” cyclobutane configuration as compared to the other two (i.e. 11 and 13), which resembled biyouyanagin A (1) and its 24-epi-diastereoisomer (24-epi-1). These results were accompanied by another equally revealing observation. Thus, as a consequence of their inverted cyclobutane configuration, the new biyouyanagin structures 12 and 14 exhibited drastic chemical shift changes as compared to biyouyanagin A (1), as shown in Figure 3. Specifically, the lone olefinic proton on the vinyl group (H-2) was found at considerably lower δ value (i.e. 12: δ = 6.10 ppm; 14: δ = 6.10 ppm) as compared to that of 1 (δ = 5.29 ppm) and its 24-epi-diasteroisomer (24-epi-1) (δ = 5.32 ppm), apparently due to the prevailing anisotropic effect in these “inverted” structures. Indeed, manual molecular models clearly show the H-2 proton residing in the vicinity opposite the phenyl group within structures 12 and 14 (see structures in Figure 3). At this stage we took note that, interestingly, natural biyouyanagin B exhibited a similar downfield shift (δ = 6.07 ppm, see 1H NMR spectrum for 2, Figure 3). The other two biyouyanagins (i.e. 11 and 13) exhibited the same downfield shift (11: δ = 5.06 ppm and 13: δ = 5.06 ppm, see Figure 3).
Figure 3.

Expanded 1H NMR olefinic region of biyouyanagins. Proton at C-2 (CH-2) is indicated with an arrow.
Having synthesized all four possible major biyouyanagins from hyperolactone C (4) [i.e. 1 (see Figure 1b),[3] 24-epi-1 (see Scheme 3b),[3] 11–14 (see Scheme 3a)] and not having found the correct structure of biyouyanagin B, and in the face of our newly gathered intelligence regarding the diagnostic 1H NMR chemical shift for H-2, we decided to re-examine the original photocycloaddition of hyperolactone C (4) with ent-zingiberene (ent-3) from which we previously isolated biyouyanagin A (1) (see Scheme 4).[3] Our intention was to look more carefully for minor biyouyanagin-like products. Indeed, upon careful examination of the 1H NMR spectrum of the crude cycloaddition product of the reaction of ent-3 and 4, we detected, in addition to 1, two new minor biyouyanagin-like components whose downfield 1H NMR signals for H-2 were encouraging. Scaling up the reaction and employing a larger excess of the zingiberene partner (ent-3:4 ca 20:1), we were able to isolate milligram quantities of the two new biyouyanagins and elucidate their structures. Delightfully, one of them proved to be identical in all respects with the long sought-after biyouyanagin B (2, 3 % yield). The other one possessed structure 15 (2 % yield) and was named biyouyanagin C. These assignments were based on spectroscopic analysis (see Figure 4 for NOEs of 2 and 15), and were consistent with their relatively low field 1H NMR signals for H-2 (2: δ = 6.07 ppm and 15: δ = 6.07 ppm, see Figure 3). Upon prolonged standing in a mixture of CH2Cl2/hexanes, biyouyanagin B (2) crystallized into colorless needles (m.p. 125–126 °C). Its X-ray crystallographic analysis (see ORTEP representation, Figure 5)[6] confirmed its structure unambiguously, ending the quest for the true structure of biyouyanagin B.
Scheme 4.

Reinvestigation of the [2+2] photocycloaddition of ent-zingiberene (ent-3) and hyperolactone C (4). Synthesis of biyouyanagin A (1), B (2, revised structure) and C (16). Reagents and conditions: 4 (1.0 equiv), ent-3 (20.0 equiv), 2′-acetonaphthone (1.0 equiv), CH2Cl2, 320 nm, 5 °C, 12 h; 51 % yield (1); 3 % yield (2); 2 % yield (15).
Figure 4.

Selected NOEs for biyouyanagin B (2) and C (15).
Figure 5.
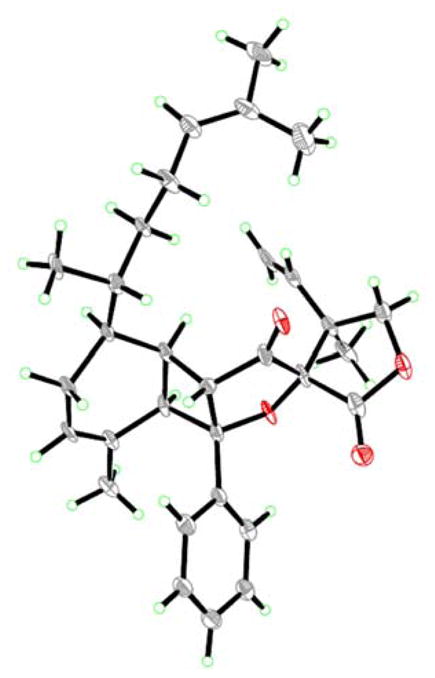
X-ray-derived ORTEP representation of biyouyanagin B (2) with thermal ellipsoids set at 30 % level.
Although there is no conclusive evidence to exclude enzymes participating in the [2+2] cycloaddition step of the biosynthesis of the biyouyanagins, they, as numerous other secondary metabolites, may be formed through spontaneous hetero- or self-assembling mechanisms.[7] It was, therefore, of interest to explore the reactivity of the various components of the biyouyanagin forming [2+2] photocycloaddition. In a combinatorial experiment we employed all four stereoisomers of zingiberene (3, ent-3, 7 and ent-7) against hyperolactone C (4) as a photocycloaddition partner (3:ent-3:7:ent-7:4 ca 5:5:5:5:1) under the standard conditions in order to probe their reactivity. 1H NMR analysis of the resulting product mixture revealed the presence of biyouyanagins A (1, 14 % yield), 24-epi-1 (10 % yield), 11 (15 % yield), 12 (< 1 %), 13 (9 % yield), 14 (3 % yield), B (2, 3 % yield) and C (15, 2 % yield). Assuming each zingiberene stereoisomer reacts with hyperolactone C (4) with both modes of facial approach, and with each of its conjugated olefinic bonds, 16 diastereomeric biyouyanagins are expected (32 if we consider 4-epi-hyperolactone). It is possible that all 16 biyouyanagins were formed in this competitive experiment, albeit some of them in trace amount. Further experimentation and analysis may lead to their isolation in the laboratory and/or from nature. Indeed, it will not be a surprise if some of them, if not all, including the ones synthesized in this study but not found as yet (i.e. 6, 24-epi-1, 8–15), are discovered in the future. In considering the entire set of [2+2] photocycloadditions of zingiberenes with hyperolactone C (4) and 4-epi-hyperolactone C (5), it was interesting to note the high diastereoselectivity with 4-epi-hyperolactone C (5) with all four zingiberenes (see Scheme 2), but not with hyperolactone C (4), which leads to varying diastereo-scrambling (see Schemes 3 and 4).
The described chemistry led to the total synthesis and structural revision of biyouyanagin B and the synthesis of a number of other, as yet undiscovered, but likely to exist, biyouyanagins, including biyouyanagin C.
Supplementary Material
Acknowledgments
We thank Dr. D. H. Huang and Dr. L. Pasterneck for NMR spectroscopic assistance, and Dr. Siuzdak and Dr. R. Chadha for mass spectrometric and X-ray crystallographic assistance respectively. Financial support for this work was provided by the National Institute of Health (USA), Universitá degli di Urbino “Carlo Bo” (graduate fellowship to S.S.), and Natural Sciences and Engineering Research Council of Canada (postdoctoral fellowship to T.R.W.).
Dedicated to Rolf Huisgen on the occasion of his 90th birthday
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Schulz V. Phytomed. 2002;9:468–474. doi: 10.1078/09447110260571742. [DOI] [PubMed] [Google Scholar]
- 2.Tanaka N, Okasaka M, Ishimaru Y, Takaishi Y, Sato M, Okamoto M, Oshikawa T, Ahmed SU, Consentino LM, Lee KH. Org Lett. 2005;7:2997–2999. doi: 10.1021/ol050960w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Nicolaou KC, Sarlah D, Shaw DM. Angew Chem. 2007;119:4792–4795. doi: 10.1002/anie.200701552. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:4708–4711. doi: 10.1002/anie.200701552. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Wu TR, Sarlah D, Shaw DM, Rowcliffe E, Burton DR. J Am Chem Soc. 2008;130:11114–11121. doi: 10.1021/ja802805c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For two other formal total syntheses of biyouyanagin A, see: Hodgson DM, Angrish D, Erickson SP, Kloesges J, Lee CH. Org Lett. 2008;10:5553–5556. doi: 10.1021/ol802334y.Du C, Li L, Li Y, Xie Z. Angew Chem. 2009;121:7993–7996.Angew Chem Int Ed. 2009;48:7853–7856. doi: 10.1002/anie.200902908.
- 5.Tanaka N, Kashiwada Y, Kim SY, Hashida W, Sekiya M, Takaishi Y. J Nat Prod. 2009;72:1447–1452. doi: 10.1021/np900109y. [DOI] [PubMed] [Google Scholar]
- 6.CDCC–773682 contains the supplementary crystallographic data for biyouyanagin B (2). This data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.cdc.cam.ac.uk/data_request/cif.
- 7.For a review on this topic, see: Gravel E, Poupon E. Eur J Org Chem. 2008;14:27–42.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.