

Abstract
Molecular in vivo neuroimaging techniques can be used to measure regional changes in endogenous neurotransmitters, evoked by challenges that alter synaptic neurotransmitter concentration. This technique has most successfully been applied to the study of endogenous dopamine release using positron emission tomography, but has not yet been adequately extended to other neurotransmitter systems. This review focuses on how the technique has been applied to the study of the 5-hydroxytryptamine (5-HT) system. The principles behind visualising fluctuations in neurotransmitters are introduced, with reference to the dopaminergic system. Studies that aim to image acute, endogenous 5-HT release or depletion at 5-HT receptor targets are summarised, with particular attention to studies in humans. Radiotracers targeting the 5-HT1A, 5-HT2A, and 5-HT4 receptors and the serotonin reuptake transporter have been explored for their sensitivity to 5-HT fluctuations, but with mixed outcomes; tracers for these targets cannot reliably image endogenous 5-HT in humans. Shortcomings in our basic knowledge of the mechanisms underlying changes in binding potential are addressed, and suggestions are made as to how the selection of targets, radiotracers, challenge paradigms, and experimental design might be optimised to improve our chances of successfully imaging endogenous neurotransmitters in the future.
Keywords: endogenous neurotransmitter release, positron emission tomography (PET), 5-HT
Evolution of Positron Emission Tomography as a Brain Imaging Technique
Neuroimaging techniques such as positron emission tomography (PET) and single photon emission computed tomography (SPECT) have allowed us to image receptors and transporters in the living human brain. Moreover, PET and SPECT can be used to measure dynamic changes in neurotransmission that occur in response to regional fluctuations in the endogenous neurotransmitter, acutely induced by experimental challenges, either in terms of a physiologic stimulus or as a consequence of pharmacological intervention. This approach has been widely applied to the study of the dopaminergic system in which neuroreceptor mapping using the radiotracers [11C]raclopride or [123I]IBZM have been combined with pharmacological challenges that elicit dopamine release (Laruelle, 2000). Even nonpharmacological, physiologic changes in endogenous dopamine may be measurable (Egerton et al, 2009). These challenge studies have contributed to a vast new understanding of brain dopaminergic mechanisms in schizophrenia (Abi-Dargham, 2004), Parkinson's disease (Brooks, 2006), and stimulant abuse (Narendran and Martinez, 2008).
Despite success with imaging endogenous dopamine, this technique has not been convincingly extended to other receptor systems, such as μ-opioid (Scott et al, 2007) or 5-hydroxytryptamine (5-HT) (Aznavour and Zimmer, 2007). Establishing imaging paradigms to measure the dynamics of different neurotransmitters would be an extremely useful tool for improving our understanding of the brain and could potentially provide new treatments for brain disorders. The 5-HT system is of particular interest because of its involvement in the regulation of mood, sleep, and appetite and consequentially in psychiatric disorders, notably depression.
Herein, we will review the attempts made so far to validate serotonergic radiotracers for their susceptibility to endogenous 5-HT release and suggest reasons for possible failure.
The Principles of Imaging Endogenous Neurotransmitters With PET and SPECT
Measuring neurotransmission by PET or SPECT relies on the differential occupation of target receptors by a neurotransmitter, after fluctuations in its synaptic concentration after a challenge. Therefore, the availability of the target receptor to an exogenous radiotracer will be altered, and the corresponding changes in tracer occupancy can be followed by comparing the binding potential (BP) (or equivalent binding measure), under baseline and challenge conditions.
The BP is a convenient outcome parameter defined as the ratio of Bmax (total receptor density) to KD (radioligand equilibrium dissociation constant), and is a measure of specific binding in relation to one of three possible reference concentrations. It is termed accordingly; BPND, when related to a brain region devoid of the receptor in question, and BPP or BPF, when related to total or free plasma concentrations, respectively (for review, see Innis et al, 2007). Typically, reference tissue models are used; thus, BP can be written as follows:
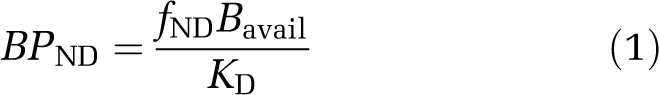 |
where Bavail is the density of receptor sites available for radioligand binding and fND the free fraction of radioligand in the tissue. With slight modification, this equation can also be applied to BPP and BPF (Innis et al, 2007). One explanation for a change in BPND in response to a challenge is described by the competition (or occupancy) model (Figure 1A). Here, a direct competition between the neurotransmitter and the radioligand for occupancy of the receptor results in reduced BPND after neurotransmitter release and increased BPND after neurotransmitter depletion. Any change in BPND can be described by comparing BPND under baseline and challenge conditions (Equations (2), (3) to (4)) according to the competition equation, assuming that KD, Bavail, and FNT themselves are not altered by the challenge. This can be equally applied to measures of BPP and BPF as follows:
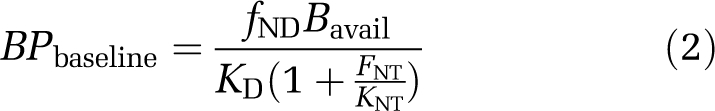 |
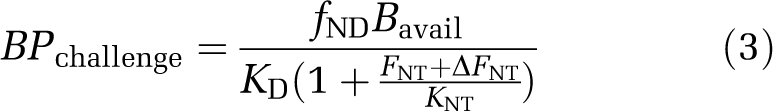 |
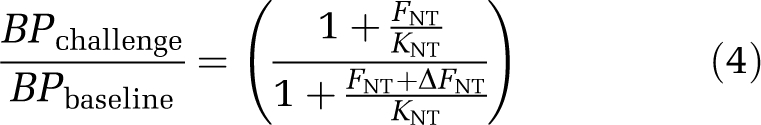 |
Figure 1.

Schematic representations of the competition and internalisation models. (A) The competition (occupancy) model. If two ligands with affinity for the same receptor are simultaneously present in the synapse, such as endogenous neurotransmitter and radioligand, it can be assumed that they will directly compete with one another for occupation of their target, and under basal conditions, this will reach equilibrium. If a challenge is introduced to alter synaptic levels of the neurotransmitter, displacement will occur resulting in corresponding changes in BP. This predicts that increases in endogenous neurotransmitter will result in higher occupancy levels, reduced receptor availability for the radioligand, and a corresponding decrease in BP. The reverse would be predicted in response to neurotransmitter depletion. The competition model is explained by Equations 1, 2, 3 to 4. The model assumes that the defining parameters for BP are not altered by the change in endogenous competition itself. Given that equilibrium is not achieved during challenge paradigms, that affinities can be altered by endogenous neurotransmitter, e.g., by cooperative binding (Frankle et al, 2009; Taly et al, 2009), and that Bavail can change by receptor-trafficking mechanisms, this cannot be guaranteed. Modified from Laruelle (2000). (B) The internalisation model. Agonist-induced receptor internalisation regulates the cell-surface expression of GPCRs and thus the occurrence of cell signalling processes. Receptors are internalised by vesicles, into the endosomal compartment for recycling back to the membrane or for degradation, as required. This model predicts that under the stimulated condition, neurotransmitter release will increase the incidence of agonist-induced internalisation, therefore reducing BP by virtue of a reduction in the affinity of the tracer for the internalised receptor or a reduction in Bavail or both. This implies either that the tracer shows reduced affinity for internalised receptors compared with those on the cell surface, perhaps because of unfavourable vesicular or endosomal binding conditions, or that the internalised receptor compartment is somehow inaccessible to the tracer because of low lipophilicity for example. The latter explanation is perhaps less likely, given the tracer's ability to cross the blood–brain barrier and the lack of correlation between radiotracer lipophilicity and its sensitivity to neurotransmitter. Some evidence supports the former hypothesis; in the case of D2, in vitro studies have shown that raclopride affinity is reduced in intracellular versus cell-surface receptors (Guo et al, 2010). Under the depleted condition, decreases in neurotransmitter release could permit recruitment of internalised receptors back to the cell surface, resulting in increased Bavail or increased affinity and thus an increased BP. The model could be even more complex; BP could increase or decrease by virtue of altered internal affinity with or without a change in Bavail. If internalisation is involved, consideration for the regulatory properties of the neurotransmitter system under investigation and susceptibility of the target to translocation, as well as the binding properties of cell surface versus internalised receptors will be crucial in determining a radiotracer's ability to image neurotransmission. As receptor activation also depends on the extent to which the receptor is coupled to the G protein, the availability of G protein and associated accessory proteins may also be important. Modified from Laruelle (2000). BP, binding potential; GPCR, G protein-coupled receptor.
Therefore, the following factors determine the ability of a tracer to detect fluctuations in synaptic neurotransmitter concentration: affinity of the neurotransmitter for its receptor (KNT), basal neurotransmitter concentration (FNT), and the magnitude of change in neurotransmitter concentration (ΔFNT) after a challenge. Accordingly, receptor occupancy elicited by the changed neurotransmitter concentration is determined by Equations (5) and (6), where the susceptibility of a neurotransmitter to be displaced is highest if ΔFNT is much larger than the sum of the baseline neurotransmitter concentration and the affinity of the neurotransmitter for the receptor:
If the competition model applies, the probability of measuring a robust signal change in response to a pharmacological challenge is independent of radiotracer characteristics, and solely depends on the affinity of the endogenous neurotransmitter for the target receptor, because the baseline and evoked change in neurotransmitter concentrations will apply for all targets.
vA critical review of neuroimaging of the dopamine system has questioned the validity of the competition model to provide an adequate explanation of imaging data obtained in pharmacological challenge experiments (Laruelle, 2000). The direction of change in the BP response was consistent with the competition model for some (such as raclopride, IBZM (iodobenzamide), NPA (N-propyl-norapomorphine)) but not for all (such as spiperone, NMSP (N-methylspiperone)) D2 tracers. In addition, a temporal uncoupling of dopamine elevation and tracer displacement was observed, as measured by microdialysis; the duration of the BP response outlasted that of the transient increase in dopamine levels and a delay existed between maximal change in BP and peak dopamine release (Carson et al, 2001; Endres et al, 1997; Laruelle et al, 1997). The latter could be explained by the slow dissociation of radiotracers from the receptor in vivo, but the temporal characteristics of the former observation may more closely match those of agonist-induced receptor internalisation. This mechanism regulates the cell-surface expression of G protein-coupled receptors, such as the D2 receptor (Bernard et al, 2006; Koenig and Edwardson, 1997). Trafficking is bidirectional, potentially allowing a reciprocal mechanism to upregulate the cell-surface expression in response to dopamine depletion. Therefore, the internalisation model was proposed as an alternative (or additive) to the competition model (Figure 1B). This model suggests that a reduction in receptor affinity upon internalisation decreases BP rather than a change in Bavail and there is some preliminary evidence to support this theory (Guo et al, 2010; Skinbjerg et al, 2010).
Regardless of the mechanism, an important factor governing the likelihood of visualising a BP response after a challenge is the extent to which the receptors are available for neurotransmitter binding versus radiotracer. Accessibility may differ because of differential receptor-affinity states. If the receptor of interest is a G protein-coupled receptor, relative availability is partly determined by the proportion of receptors in the high-affinity state (RHIGH), according to the ternary complex model (Figure 2). This predicts that because the majority of radiotracers are antagonists, they will have access to a greater proportion of receptors than will endogenous neurotransmitters (agonists), thus reducing the signal-to-noise ratio, as compared with equal access. The ratio may be increased using an agonist tracer. Accessibility may also differ because of cellular compartmentalisation, e.g., through the existence of intracellular receptor pools or as a result of internalisation (Figure 1B). By way of example, the differential in vivo availability of D2 receptors to dopamine has been estimated by comparing images produced by the antagonist tracer [11C]raclopride with the agonist [11C]NPA under basal and challenge conditions (Figure 3, Abi-Dargham et al, 2000; Laruelle, 2000; Narendran et al, 2004). The model confirms the improved signal-to-noise ratio of an agonist versus antagonist tracer, in line with the ternary complex model. A maximal proportion of only 38% is available for dopamine binding relative to the tracer in vivo (whether an antagonist or an agonist), which may explain why a ceiling effect is observed with a maximum ∼40% reduction in BP in the human brain, regardless of the magnitude of change in dopamine. The model also points to the existence of a small proportion of receptors that are ‘protected' from occupation by dopamine in vivo by compartmentalisation (Figures 1B and 3).
Figure 2.

The ternary complex model. The actions of drugs are determined by two fundamental properties; namely affinity (the propensity of a ligand to bind) and efficacy (its propensity to induce signalling events). All ligands able to bind to a receptor possess affinity, but only agonists possess the ability to elicit receptor function and are therefore said to have efficacy. (A) The ternary complex model for agonist interaction at a GPCR states that the receptor must be bound to two other components for agonism to occur: agonist (A) and the associated G protein (G). The receptor exists in two different states: uncoupled (R) and G protein coupled (RG). The agonist-bound state (ARG) allows receptor activation to occur. (B) The extended ternary complex model explains the existence of different affinity states of the same receptor in vivo. This theory is an extension of the simple model and postulates that partially activated receptor conformation(s) (R*) exist in equilibrium with the ground state (R) and the activated G protein-coupled (AR*) conformations, and that each state has differing affinity for its endogenous (or otherwise) agonist. Agonists bind with greatest affinity to the partially activated (G protein coupled) R* state, known as the high-affinity state (RHIGH) and with lower affinity to the uncoupled ground state (R), known as the low-affinity state (RLOW). Antagonists bind with equal affinity to all states, which explains why agonists label a smaller fraction of the same receptor compared with antagonists. This model also accounts for the characteristic known as constitutive or intrinsic activity, whereby the presence of the agonist is not necessarily required for receptor activity to occur. GPCR, G protein-coupled receptor.
Figure 3.

Pharmacological models of the D2 receptor. (A) Original and (B) extended pharmacological model of D2 receptors in vivo. (Panel A) D2 receptors exist in different affinity states for agonists according to the ternary complex model for GPCRs (Figure 2): RHIGH and RLOW, with each contributing 50% of the total receptor population (derived from in vitro data; Sibley et al, 1982). Under baseline conditions, 10% of RHIGH are occupied by dopamine (Abi-Dargham et al, 2000), leaving 40% available to further DA occupancy. It is assumed that DA does not bind effectively to D2 receptors that are in the low-affinity configuration. Thus, 90% are available to antagonist tracer and of these, only 44% are available to DA, representing the ‘ceiling' extent to which increases in extracellular DA can displace the radiolabelled tracer after a challenge. (Panel B) A fraction of RHIGH sites appear protected from DA occupation, suggesting a subdivision of receptor populations into synaptic and nonsynaptic compartments. The differential effect of an agonist tracer ([11C]NPA) versus an antagonist tracer ([11C]raclopride), suggested that a higher proportion (73%) of D2 are RHIGH, with 38% of these susceptible to endogenous competition and only 27% in the RLOW state. Modified from Narendran et al, 2004. GPCR, G protein-coupled receptor.
Development of Radiotracers to Image Endogenous 5-HT
The successful application of the technique relies on the use of suitable radiotracers. From the dopaminergic literature, the relative merits of radioligands that render them sensitive to displacement or internalisation by endogenous dopamine are unclear (Laruelle, 2000). An ideal tracer would be sensitive to both increases in neurotransmitter concentration (corresponding to release) and decreases (caused by depletion). Baseline BP values should be large and noiseless enough in brain regions of interest (ROIs) to allow for calculation of decreases, and test–retest reproducibility should be high.
A summary of all 5-HT receptor targets, their current ligand availability, and receptor/transporter densities in the human brain are provided in Table 1. Many represent suitable targets for whole-brain imaging techniques; PET ligands have been developed for 6 of the 15 targets. Of these, four have been formally evaluated for their susceptibility to image endogenous 5-HT in humans, namely the 5-HT1A, 5-HT2A, and 5-HT4 receptors and the 5-HT transporter, serotonin reuptake transporter (SERT). Another target, the 5-HT1B receptor, is under investigation.
Table 1. 5-HT receptor properties and available selective radioligands.
| Target | Nomenclature (transduction) | 5-HT affinity Ki, nM (pKi) | Highest Bmax fmol/mg protein (region) | Reference | Selective ligands | Selective radioligand | PET tracer | |
|---|---|---|---|---|---|---|---|---|
| GPCRs | 5-HT1A | (Gi/o) | 0.2–400 (9.7-6.4) | 860–3,140* (hipp) | ||||
| 300–1,910* (ctx) | (Burnet et al, 1997) | ✓ | ✓ | ✓ | ||||
| 5-HT1B | (Gi/o) | 1–40 (9.0-7.4) | 400–500* (g pall) | |||||
| 396* (occ ctx) | (Varnas et al, 2001) | ✓ | ✓ | ✓ | ||||
| 5-HT1D | (Gi/o) | 1–10 (9.0-8.0) | 226 (g pall) | (Miller and Teitler, 1992) | ✓ | (✓) | (✓) | |
| 5-ht1e | (Gi/o) | 6.3–10 (8.2-8.0) | 224 (put) | (Miller and Teitler, 1992) | — | — | — | |
| 5-HT1F | (Gi/o) | 10–20 (8.0-7.7) | 79 (rat wb) | (Wainscott et al, 2005) | ✓ | ✓ | — | |
| 5-HT2A | (Gq/11) | 4–1,000 (8.4-6.0) | 570 (ctx) | (Rosel et al, 2000) | ✓ | ✓ | ✓ | |
| 5-HT2B | (Gq/11) | 4–12 (8.4-7.9) | ND | — | ✓ | — | — | |
| 5-HT2C | (Gq/11) | 2.5–158 (8.6-6.8) | 688 (pig ch plx) | (Leonhardt et al, 1992) | ✓ | ✓ | — | |
| 5-HT4 | (Gs) | 100–1,259 (7.0-5.9) | 223 (stri) | (Bonaventure et al, 2000) | ✓ | ✓ | ✓ | |
| 5-ht5a | (?) | 126–199 (6.9-6.7) | ND | — | ✓ | — | — | |
| 5-ht5b | (?) | — | ND | — | — | — | — | |
| 5-HT6 | (Gs) | 32–158 (7.5-6.8) | 215 (stri) | (Hirst et al, 2000) | ✓ | ✓ | ✓ | |
| 5-HT7 | (Gs) | 0.3–8 (9.6-8.1) | 68 (thal) | (Thomas et al, 2002) | ✓ | ✓ | — | |
| Ion channel | 5-HT3 | — | 316–631 (6.5-6.2)# | 465 (pig ctx) | (Fletcher and Barnes, 1996) | ✓ | ✓ | — |
| Transporter | SERT | — | 1,000–1,200 (6.0-5.9)* | 587 (ctx) | (Stanley et al, 1982) | |||
| 682 (thal) | (Rosel et al, 1997) | ✓ | ✓ | ✓ | ||||
ch plx, choroid plexus; ctx, cortex; g pall, globus pallidus; GPCR, G protein-coupled receptor; hipp, hippocampus; ND, not determined; occ, occipital; PET, positron emission tomography; put, putamen; SERT, serotonin reuptake transporter; stri, striatum; thal, thalamus; wb, whole brain; 5-HT, 5-hydroxytryptamine.
For comprehensive reviews of 5-HT receptors, see Barnes and Sharp (1999); Torres et al (2003); and Tricklebank and Middlemiss (2004). According to the nomenclature agreed by IUPHAR (International Union of Basic and Clinical Pharmacology), the term ‘receptor' is only applied to entities for which operational, structural, and signal transduction information is available; thus, 5-ht1E, 5-ht5A, and 5-ht5B have lowercase letters indicating that no function has yet been attributed to them. Ki values are taken from the IUPHAR GPCR database (http://www.iuphar-db.org), except #values are from the Ki database; http://kidb.cwru.edu/pdsp.php. Bmax values are taken from the literature and are from the human brain, unless otherwise stated. Units are equivalent fmol/mg protein, * denotes a simple conversion from fmol/mg tissue assuming that 10% of tissue weight is protein. Bmax values vary between tracers, species, and laboratories, but only one such example is provided here. For comparison, the D2 receptor Bmax in the caudate is ∼100 fmol/mg protein (Ruiz et al, 1992; von Euler et al, 1990).
✓Indicates that selective compounds are available.
(✓) Ligands also have 5-HT1B affinity. ‘Selective ligands' refer to availability of compounds with published selectivity for the given target relative to other receptors. ‘Selective radioligands' refer to availability of selective radiolabelled ligands for use in vitro and/or in vivo, and ‘PET tracers' refer to availability of in vivo tracers that have shown promise in human subjects using PET.
A summary of published studies that determine tracer sensitivity to endogenous 5-HT is provided in Table 2. The majority have used PET paradigms in humans, nonhuman primates, or small animals. Ex vivo radioligand-binding experiments have also been included. The following sections discuss the findings and relative merits of each study in turn, with particular reference to human studies.
Table 2. Studies investigating the susceptibility of receptor radioligands to manipulation of endogenous 5-HT.
| Receptor | Radioligand | Challenge | Species (n) | Protocol | BP effect | Effect on 5-HT or surrogate | Reference |
|---|---|---|---|---|---|---|---|
| 5-HT1A | [3H]-WAY100635 | Fenfluramine (3 mg/kg i.p., 5 min prior) | Mouse (n=9–11) | Ex vivo (D) | None | — | Rice et al (2001) |
| MDMA (10 mg/kg i.p., 5 min prior) | TOS; 45 min | — | |||||
| Cocaine (20 mg/kg i.p., 5 min prior) | — | ||||||
| p-Chloroamphetamine (10 mg/kg i.p., 5 min prior) | — | ||||||
| p-Chlorophenylalanine (150 mg/kg, twice daily for 4 days prior) | ↓57–62% (TL) | ||||||
| [11C]-WAY100635 | 5,7-Dihydroxytryptamine (150 μg i.v. infusion) | Rat (n=6–7) | Ex vivo (D) | None | ↓>65% (TL) | Maeda et al (2001) | |
| Reserpine (5 mg/kg, s.c., 24 h prior) | TOS; 20 min | — | |||||
| Fenfluramine (10 mg/kg i.p., 10 min prior) | ↑9-fold | ||||||
| [11C]-WAY100635 | Pindolol (0.005-0.1 mg/kg i.v., 10 min prior) | Rat (n=4–5) | PET, bolus | ↑hipp, Fctx | — | Hirani et al (2000) | |
| [11C]-WAY100635 | Fenfluramine (10 mg/kg i.p., 30 min prior) | Rat (n=6–7) | PET, bolus | ↓10–20% hipp | ↑15-fold | Hume et al (2001) | |
| None Fctx | ↑4.5-fold | ||||||
| [11C]-WAY100635 | Fenfluramine (10 mg/kg i.v., 20 min post) | Rat (n=3–4) | PET, bolus | None | ↑9-fold | Maeda et al (2001) | |
| [11C]-WAY100635 | Tryptophan depletion/infusion | Human (n=4) | PET bolus | None | — | Rabiner et al (2002) | |
| [18F]-FCWAY | Paroxetine (10 mg/kg i.p., 20 min or 1 h prior) | Mouse (n=?, awake) | Ex vivo (D) | None | — | Jagoda et al (2006) | |
| [18F]-FPWAY | Paroxetine (10 mg/kg i.p., 10 min post) | Mouse (n=5–6, awake) | Ex vivo (A) | None | — | Jagoda et al (2006) | |
| Fenfluramine (20 mg/kg i.p., 10 min post) | TOS; 30 min | ||||||
| Paroxetine (10 mg/kg i.p., 20 min prior or post) | Mouse (n=5–6, awake) | Ex vivo (D) | None | — | |||
| Fenfluramine (20 mg/kg i.p., 20 min post) | TOS; 30 min | ||||||
| Fenfluramine (10 mg/kg i.p., 20 min post) | Rat (n=5, awake) | Ex vivo (D) | None | — | |||
| Paroxetine (10 mg/kg i.p., 20 min post) | TOS; 60 min | ||||||
| Fenfluramine (10 mg/kg i.v., 20 min post) | |||||||
| [18F]-FPWAY | Paroxetine (5 mg/kg i.v., 90 min post) | Monkey (n=4) | PET bolus infusion | ↑7–13% ctx | — | Giovacchini et al (2005) | |
| ↓8–27% raphe | |||||||
| [18F]-MPPF | Fenfluramine (10 mg/kg i.v., 20 min post) | Rat (n=9–10, awake) | Ex vivo (D) | None | — | Jagoda et al (2006) | |
| TOS; 60 min | |||||||
| [18F]-MPPF | Fenfluramine (10 mg/kg i.p., 30 min prior) | Rat (n=5–7) | Ex vivo (A) | ↓20% hipp | ↑30-fold | Udo de Haes et al (2005) | |
| TOS; 30 min | |||||||
| [18F]-MPPF | Citalopram (10 μmol/kg) + ketanserin (100 nmol/kg s.c., 30 min prior) | Rat (n=5–7) | Ex vivo (A) | None | ↑10-fold | Udo de Haes et al (2005) | |
| TOS; 30 min | |||||||
| [18F]-MPPF | Fenfluramine (1, 2 and 10 mg/kg i.v., 20 min post) | Rat (n=4) | β-Probe | ↓25% hipp | ↑20% | Zimmer et al (2002) | |
| ↓60% hipp | ↑50% | ||||||
| ↓100% hipp | ↑15-fold | ||||||
| [18F]-MPPF | p-EPA (5 mg/kg i.p., 4 h prior) | Rat (n=9) | β-Probe | ↑hipp | ↓60% | Zimmer et al (2003) | |
| [18F]-MPPF | Raphe stimulation (plus clomipramine) 10, 20 and 30 min | Rat (n=4) | β-Probe | ↓27–53% hipp | ↑1.5-, 2- and 2.8-fold | Rbah et al (2003) | |
| [18F]-MPPF | 8-OH-DPAT (0.5 mg/kg i.v., 15 min prior) | Rat (n=4–5) | β-Probe | ↓30% raphe | ↓34% 5-HT1A surface density | Zimmer et al (2004) | |
| [18F]-MPPF | Fluoxetine (10 mg/kg i.p., 1 h prior) | Rat (n=5) | β-Probe | ↓35% raphe | ↓36% 5-HT1A surface density | Riad et al (2004) | |
| [18F]-MPPF | Fluoxetine (5 mg/kg i.v., 30 min prior) | Cat (n=3) | PET bolus | ↓34% raphe | — | Aznavour et al (2006) | |
| [18F]-MPPF | Fluoxetine (20 mg p.o., 5 h prior) | Human (n=8) | PET bolus | ↓44% raphe | — | Sibon et al (2008) | |
| [18F]-MPPF | Fenfluramine (10 mg/kg i.v., 90–130 min post) | Monkey (n=5, awake) | PET bolus | None | ↑35-fold | Udo de Haes et al (2005) | |
| [18F]-MPPF | Tryptophan depletion versus tryptophan infusion | Human (n=6) | PET bolus | None | ↓67% and ↑10-fold plasma tryptophan | Udo de Haes et al (2002) | |
| [18F]-MPPF | Tryptophan depletion | Human (n=8, patients) | PET bolus | None | ↓86% plasma tryptophan | Praschak-Rieder et al (2004) | |
| [18F]-MPPF | Sleep (versus wake) | Human (n=14, patients) | PET bolus | ↑12% whole brain | — | Derry et al (2006) | |
| ↑ctx and others | |||||||
| [11C]-CUMI-101 | Citalopram (4 mg/kg, i.v.) | Baboon (n=2) | PET bolus | ↓raphe and others | — | Milak et al (2008b) | |
| 5-HT2A | [3H]-NMSP | Fenfluramine (3 mg/kg i.p., 5 min prior) p-chloroamphetamine (10 mg/kg i.p., 5 min prior) | Mouse (n=6–8) | Ex vivo (D) | None | — | Rice et al (2001) |
| Paroxetine (2 mg/kg i.p., 5 min prior) | TOS; 60 min | ||||||
| [11C]-MDL100907 | Fenfluramine (10 mg/kg i.p., 30 min prior) | Rat (n=5) | PET, bolus | None | ↑5-HT2A c-fos and Arc expression | Hirani et al (2003) | |
| [18F]-setoperone | Paroxetine (20 mg p.o., 4.5 h prior) | Human (n=5) | PET bolus | None | — | Meyer et al (1999) | |
| [18F]-setoperone | Tryptophan depletion | Human (n=10) | PET bolus | ↓8% ctx and others | ↓72% plasma tryptophan | Yatham et al (2001) | |
| [18F]-deutero-altanserin | Fenfluramine (1.5 mg/kg i.v., 6 h post) | Baboon (n=2) | PET bolus infusion | None | — | Staley et al (2001) | |
| [18F]-altanserin | Citalopram (0.25 mg/kg i.v., 1 h post) plus pindolol (<25 mg p.o. daily for 4 days prior) | Human (n=7) | PET bolus infusion | None | ↑Plasma prolactin | Pinborg et al (2004) | |
| [18F]-altanserin | Ketamine (0.05 mg/kg i.v., 2 h post plus 0.2 mg/kg/h infusion) | Human (n=3) | PET bolus infusion | None | — | Matusch et al (2007) | |
| [18F]-altanserin | Clomipramine (25 mg i.v., bolus 10 min prior, plus infusion for 20 min post) | Human (n=11, patients) | PET bolus | ↓14–23% in Vt (ctx) | — | Larisch et al (2003) | |
| 5-HT4 | [11C]-SB207145 | Citalopram (0.25 mg/kg i.v., 30 min prior) plus pindolol (<25 mg p.o. daily for 3 days prior) | Human (n=6/7) | PET bolus | None | ↑Plasma prolactin | Marner et al (2010) |
| SERT | [11C]-DASB | TCP (15 mg/kg i.p., 1 h prior) | Rat (n=5–6) | Ex vivo (D) | ↓Various | — | Lundquist et al (2005) |
| TOS; 60 min | |||||||
| [11C]-DASB | TCP (10 mg/kg i.p., 4 h prior) | Cat (n=2) | PET bolus | ↓21–40% various | — | Ginovart et al (2003) | |
| [11C]-DASB | TCP (15 mg/kg i.v., 30 min post) | Rat (n=2–4) | PET, bolus infusion | ↓40% thal | — | Lundquist et al (2007) | |
| TCP (2 mg/kg i.v., 1 h prior) | Monkey (n=3) | PET, bolus | ↓30% (Vt) thal | Lundquist et al (2007) | |||
| [11C]-DASB | 5-HTP (20 mg/kg i.v., 30 min prior) | Monkey (n=5, awake) | PET bolus | ↓35–40% stri | ↑<250-fold | Yamamoto et al (2007) | |
| ↓10–15% various | ↑8-fold | ||||||
| [11C]-DASB | Tryptophan depletion | Baboon (n=2, 14/9 scans) | PET bolus | ↓30% various | ↓65% plasma tryptophan | Milak et al (2005) | |
| [11C]-DASB | Tryptophan depletion | Human (n=8) | PET bolus | None | ↓>70% plasma tryptophan | Talbot et al (2005) | |
| [11C]-DASB | Tryptophan depletion | Human (n=14) | PET bolus | None | ↓90% plasma tryptophan | Praschak-Rieder et al (2005) | |
| 5-HT1B | [11C]-AZ10419369 | Fenfluramine (1 and 5 mg/kg i.v., 20 min post) | Monkey (n=3) | PET bolus | ↓33 and 52% Octx | — | Finnema et al (2010) |
| ↓20 and 54% g pall | |||||||
| ↓21 and 46% midbrain | |||||||
| [11C]-P943 (GSK-174722) | (+)Fenfluramine (0.8 and 2.5 mg/kg i.v. infusion, 15 min prior) | Baboon (n=3) | PET bolus | ↓22 and 47% (VND) | — | Dr E Rabiner, personal communication |
Abbreviations: Octx, occipital cortex; Fctx, frontal cortex; g pall, globus pallidus; hipp, hippocampus; i.p., intraperitoneal; i.v., intravenous; p-EPA, p-ethynylphenylalanine; p.o., oral; s.c., sub-cutaneous; stri, striatum; TCP, tranylcypromine; thal, thalamus; TL, tissue level.
All data presented are provided in the cited references. Challenge includes drug, dose, route of administration and time administered relative to the initial bolus or bolus-infusion dose of radiotracer. The numbers of subjects of each species used in each experiment are provided and number of scans, where appropriate. All animal experiments were performed under anaesthesia unless otherwise stated. The protocol indicates the imaging procedure used; ex vivo studies (A; autoradiography, D; regional dissection) and the time of sacrifice (TOS); all used a bolus injection of radiotracer followed by whole brain perfusion under anaesthesia at various intervals after injection (TOS; 20 min to 1 h). BP effect represents the approximate effect on BP, total distribution volume (Vt) or equivalent parameter as determined by PET, or specific binding as determined by ex vivo radioligand binding. Effect on 5-HT or surrogate column indicates the effect of the challenge on synaptic 5-HT concentrations as measured by in vivo microdialysis in the same brain region, species and publication as the stated change in BP. Where microdialysis was not used, alternative surrogate markers of central 5-HT levels are noted.
Are 5-HT1A Ligands Sensitive to Endogenous 5-HT?
Studies With [11C]WAY100635
[11C]WAY100635 is currently the most used 5-HT1A PET tracer in vivo (for review, see Kumar and Mann, 2007). Using several radioligand-binding techniques, its sensitivity to 5-HT elevation and depletion was investigated after pharmacological challenge in mice and rats (Hirani et al, 2000; Hume et al, 2001; Maeda et al, 2001; Rice et al, 2001). A range of potent releasing and depleting agents were administered to manipulate synaptic 5-HT concentrations, but none altered binding in any brain region, despite significant reductions in tissue 5-HT levels, with two exceptions. In response to high doses of pindolol, [11C]WAY100635 BP was reduced as expected (reflecting receptor occupancy), but lower doses increased BP in the postsynaptic regions of the frontal cortex and hippocampus (Hirani et al, 2000). The authors posited that decreases in endogenous 5-HT release could be responsible. Their follow-up study combined microdialysis with rat PET using fenfluramine to increase 5-HT (Hume et al, 2001). Small decreases in hippocampal BP were recorded, but no significant change was observed in the frontal cortex where increases in 5-HT were of a smaller magnitude.
The single human study included pilot data obtained from four subjects who had undergone acute dietary tryptophan depletion (to deplete central 5-HT levels; (Carpenter et al, 1998; Williams et al, 1999) versus tryptophan infusion (to increase central 5-HT levels) before tracer injection (Rabiner et al, 2002). No difference in BP was evident in any region of the tested ROIs.
Studies With [18F]-WAY Analogues
In a concerted effort to design more sensitive probes, some analogues of WAY100635 were successfully 18F-labelled (McCarron et al, 2004; Sandell et al, 2001). Three of these have been tested for their vulnerability to detect changes in 5-HT, namely [18F]FPWAY, [18F]FCWAY, and [18F]FBWAY, which became better known as [18F]MPPF (Passchier et al, 2000).
Both [18F]FPWAY and [18F]FCWAY were studied using ex vivo techniques in rodents with various manipulations, but no change in BP in response to challenge was observed under any condition that could be attributed to changes in specific 5-HT1A-binding signal (Jagoda et al, 2006). In contrast, a monkey PET study observed a slow and steady decrease in raphe BP and a transient increase in cortical BP when [18F]FPWAY was administered under a bolus-infusion paradigm with paroxetine (Giovacchini et al, 2005), indicating possible detection of a change in endogenous 5-HT. To our knowledge, no challenge studies have been performed in humans.
Studies With [18F]MPPF
The development of [18F]MPPF led a multidisciplinary group to perform a sequence of elegant experiments to determine its sensitivity to endogenous 5-HT (Aznavour and Zimmer, 2007). The kinetics of [18F]MPPF was monitored with a β-sensitive intracerebral probe, detecting emitted β-particles within a 1.1 mm range in the rat brain. Probes were inserted into the ROI and reference region void of 5-HT1A receptors. The methodology is experimentally powerful because it can be combined with microdialysis in the same animal under challenge conditions.
They first determined whether [18F]MPPF could detect increases in 5-HT in the rat. After fenfluramine challenge, dose-dependent increases in 5-HT and simultaneous decreases in [18F]MPPF BP were recorded in the hippocampus, and a linear relationship between the two was established, consistent with [18F]MPPF displacement from 5-HT1A receptors by 5-HT (Zimmer et al, 2002). This finding was supported by another study using ex vivo autoradiography (Udo de Haes et al, 2005). Electrical stimulation of the raphe nucleus, at high firing rates and in combination with a 5-HT reuptake blocker, could also produce modest, dose-dependent reductions in hippocampal BP corresponding to regional increases in 5-HT (Rbah et al, 2003). Conversely, reductions in 5-HT evoked by the tryptophan hydroxylase inhibitor p-ethynylphenylalanine significantly increased hippocampal binding (Zimmer et al, 2003). These data were in accordance with the competition model in the hippocampal regions. However, by combining β-probe measurements with immunoelectron microscopy, receptor internalisation was shown to have a role in the BP response in raphe regions (Zimmer et al, 2004). First, acute 5-HT1A agonists were shown to internalise autoreceptors in the raphe but not the somatodendritic receptors of the hippocampus (Riad et al, 2001). Subsequently, a single dose of the selective serotonin reuptake inhibitor (SSRI) fluoxetine was shown to evoke the same response; reductions in raphe 5-HT1A plasma membrane receptors corresponded to remarkably similar and relatively substantial (30%) decreases in [18F]MPPF binding, which were suggested to be mediated by 5-HT elevation afforded by SERT inhibition (Riad et al, 2004). Autoradiographical data confirmed that the total number of 5-HT1A receptors had not changed and combining fluoxetine with 8-OH-DPAT (8-hydroxy-N,N-dipropyl-2-aminotetralin) treatment did not enhance receptor internalisation, suggesting that a fixed proportion of G protein-coupled receptors can be internalised, and that this pool is the same whether mobilised by an exogenous pharmacological agent or by endogenous 5-HT. A follow-up PET study in cats showed similar results (Aznavour et al, 2006).
One could reasonably expect to visualise changes of this magnitude in human PET studies; therefore, this paradigm was applied to human subjects. Acute fluoxetine was administered in a randomised, double-blind paradigm 5 hours before a [18F]MPPF PET scan in eight healthy volunteers (Sibon et al, 2008). An average 44% reduction in [18F]MPPF BP was observed in the raphe with no other significant changes. Although test–retest variability was relatively high (23% to 36%) and questions remain over the legitimacy of raphe delineation methods, the findings are convincing because of the strengths of the study design, its double-blind, randomised, and placebo-controlled crossover procedures, as well as its agreement with the aforementioned animal studies. No attempts were made to decrease 5-HT levels, e.g., by tryptophan depletion regimes.
In the only experiment to indicate that PET imaging might be sensitive to physiologic changes in 5-HT, a study indicated that sleep could increase [18F]MPPF BP in narcoleptic patients, relative to wakefulness (Derry et al, 2006). On the basis of microdialysis studies showing state-dependent alterations in 5-HT levels (Portas et al, 1998), it was hypothesised that sleep could reduce 5-HT levels and thereby increase [18F]MPPF BP. Global increases in BP were detected in all ROIs in the ‘sleep' condition, particularly cortical, with more pronounced changes in good compared with poor sleepers. However, several experimental problems and confounding factors overshadow this study, including the use of a patient group, the necessity for withdrawal of (possibly confounding) medication, the possibility of an order effect, and the lack of specific information relating to how much sleep was obtained, of what type, and when. Replication and rectification are required if these results are to be considered convincing.
Data opposing the hypothesis that [18F]MPPF is sensitive to alterations in endogenous 5-HT came from several groups. Fenfluramine and a challenge combining uptake inhibition with modulation of feedback at 5-HT2C receptors (citalopram plus ketanserin) were found not to alter [18F]MPPF BP (Jagoda et al, 2006; Udo de Haes et al, 2005). Of most significance were the findings from a fenfluramine challenge study using monkey PET (Udo de Haes et al, 2006). By combining microdialysis with [18F]MPPF PET in a bolus-infusion protocol, this exemplary study showed that a substantial dose of intravenous fenfluramine did not produce significant or consistent effects on [18F]MPPF BP in the cortex, hippocampus, or any other region, despite increasing 5-HT concentrations by 35-fold in the prefrontal cortex. Two human studies also showed no change in [18F]MPPF BP in response to acute tryptophan depletion. In the first study, scans were compared under tryptophan depletion and infusion conditions in healthy volunteers. The BP did not change in cortical areas and there was only a nonsignificant reduction in raphe BP (Udo de Haes et al, 2002). Unfortunately, this study suffers from the absence of an ideal control group, but its findings were supported by those from the other study in recovered depressed patients on citalopram (Praschak-Rieder et al, 2004). Depressed patients exhibit different 5-HT receptor physiology at baseline compared with normal controls and were believed likely to display larger-magnitude responses to changes in 5-HT after depletion, thus more likely to show a BP response. However, no change in [18F]MPPF BP was seen in any ROI, despite an 85% reduction in plasma tryptophan and 6 out of 8 patients experiencing transient depressive relapse.
Are 5-HT2A Tracers Sensitive to 5-HT Manipulation?
Several selective antagonist radioligands are available with which to study the 5-HT2A receptor by PET and other methods. So far, the nonselective tracers [18F]setoperone and [3H]NMSP and the selective tracers [18F]altanserin and [11C]MDL100907 have been tested for their susceptibility to image endogenous 5-HT.
Using ex vivo dissection in mice, [3H]NMSP was found to be insensitive to 5-HT-releasing agents (Rice et al, 2001). Although fenfluramine significantly decreased [3H]NMSP binding (>50% in the cortex), this was believed not to reflect a specific increase in 5-HT levels owing to the lack of attenuation by paroxetine and a failed replication study with p-chloroamphetamine. Similarly, a lack of [11C]MDL100907 displacement was shown in an elegant rat PET study after fenfluramine challenge (Hirani et al, 2003). In vivo activation of 5-HT2A receptors by 5-HT was confirmed by appropriate cortical increases in c-fos, arc and 5-HT2A mRNA, but these were not associated with any change in [11C]MDL100907 binding.
In humans, [18F]setoperone was shown to be insensitive to 5-HT elevation induced by paroxetine (Meyer et al, 1999), but contrary to expectations, tryptophan depletion reduced cortical BP (Yatham et al, 2001). The lack of a biological model for this finding renders interpretation difficult.
Three publications have suggested that [18F]altanserin (or its deuterated analogue) is insensitive to changes in 5-HT. First, an exemplary PET study in humans administered [18F]altanserin under a bolus-infusion protocol with citalopram challenge (Pinborg et al, 2004). Pindolol was administered 3 days earlier to prevent 5-HT1A autoinhibition and to maximise 5-HT elevation. Subjective experience of hot flushes and increased plasma prolactin levels confirmed increased 5-HT function, but no changes in [18F]altanserin BP occurred under any challenge condition. Another study showed that [18F]altanserin was not sensitive to changes in 5-HT induced by ketamine in healthy volunteers (Matusch et al, 2007), although ketamine is primarily an N-methyl-aspartic acid antagonist and D2 agonist with secondary effects on the 5-HT system (Aghajanian and Marek, 2000). In a bolus-infusion PET study in baboons, the deuterated analogue of [18F]altanserin was administered with intravenous fenfluramine challenge (Staley et al, 2001) and again, no change in [18F]deuteroaltanserin BP was observed.
In the only study to suggest a possible sensitivity of [18F]altanserin, clomipramine challenge was used in remitted depressed patients in which six subjects had received previous SSRI medication and five had not (Larisch et al, 2003). A global analysis showed cortical decreases in BP, but cerebellar test–retest variability was large and clomipramine has some binding affinity for the 5-HT2A and 5-HT2C sites (Hyttel, 1994), which may explain the reductions in binding.
Are 5-HT4 Tracers Sensitive to 5-HT Manipulation?
The radioligand [11C]SB207145 is currently the only well-validated PET tracer for imaging cerebral 5-HT4 receptors in humans (Marner et al, 2009). In a challenge protocol similar to that described in the study by Pinborg et al (2004) using pindolol plus citalopram, [11C]SB207145 BP was not altered in healthy volunteers (n=6/7), despite increases in plasma prolactin levels (Marner et al, 2010). As a power analysis of [11C]SB207145 showed that detection of a difference of 15% necessitates inclusion of four or five individuals, examined twice, a decrease <5% could have been overlooked, but such a small difference was deemed irrelevant.
Are SERT Tracers, Such as, [11C]DASB Sensitive to 5-HT Manipulation?
Among several good PET ligands for imaging SERT in the human brain, the hitherto most used emerged from the diarylsulphides, which is a class of SSRI. [11C]DASB is the only one to have been investigated for its sensitivity to endogenous 5-HT. There are few pharmacological agents that increase 5-HT levels, which also lack SERT affinity; hence, the options for manipulation of 5-HT are somewhat limited. The studies described below have used three different techniques to alter synaptic 5-HT: tranylcypromine, a nonselective monoamine oxidase inhibitor, 5-hydroxytryptophan (5-HTP), the precursor to 5-HT, and tryptophan depletion.
In the only ex vivo study, a dose-dependent decrease in [11C]DASB binding was detected in rats in response to tranylcypromine challenge (Lundquist et al, 2005), in which decreases in uptake were observed across the brain. This finding was first replicated in a PET study in cats using a similar challenge (Ginovart et al, 2003). Cats were scanned before and after tranylcypromine treatment and a decrease in [11C]DASB binding was found in the midbrain, thalamus, striatum, and cortex. Two further animal PET studies supported these findings: a bolus-infusion protocol in rats and a bolus protocol in monkeys (Lundquist et al, 2007). Tranylcypromine decreased [11C]DASB thalamic binding in monkeys and reduced BP by 40% in the rat thalamus. Although possible regional cerebral blood flow (rCBF) changes were detected in monkey experiments, the differential time course suggested that they were not related to the BP reduction that was believed to coincide with 5-HT elevation.
In the only study to combine imaging SERT with microdialysis, global decreases in [11C]DASB BP were shown after a 5-HTP challenge in conscious monkeys (Yamamoto et al, 2007), alongside regional decreases that correlated with areas of maximal 5-HT elevation and with regional activity of amino acid decarboxylase (the 5-HTP to 5-HT converting enzyme).
Another three studies imaged SERT binding after 5-HT depletion. The first study was conducted in baboons, suggesting that tryptophan depletion could alter [11C]DASB BP (Milak et al, 2005). There was no control group, only a direct comparison with baseline, but paradoxical decreases in BP of between 27 and 33% were detected in several regions. The authors posited that the results were in agreement with the proposed regulation of SERT trafficking by 5-HT in vivo, suggesting that an extracellular reduction in 5-HT could promote SERT internalisation, corresponding to a reduction in cell-surface expression (Ramamoorthy and Blakely, 1999).
Two further studies were published simultaneously (Praschak-Rieder et al, 2005; Talbot et al, 2005). Both were crossover studies in healthy volunteers showing no significant effect of tryptophan depletion on [11C]DASB BP, despite recording an 85% reduction in plasma levels of tryptophan (Talbot et al, 2005).
Summary of Published Findings
Overall, in spite of numerous attempts, a radiotracer of equivalent value to the study of 5-HT as [11C]raclopride has been to dopamine has not yet been identified.
Only four publications claim to have shown radiotracer sensitivity to changes in endogenous 5-HT concentrations in human subjects. Two pointed to the sensitivity of [18F]MPPF binding to 5-HT1A receptors (Derry et al, 2006; Sibon et al, 2008); one in response to fluoxetine, in which changes were restricted to the raphe, for which difficulties associated with visualisation and analytical reproducibility are yet to be resolved (Kalbitzer et al, 2009) and the other following sleep, in which methodological issues preclude any definitive conclusions. The other two studies were also hampered by methodological problems and/or the lack of a biological explanation. On the positive side, radiotracers insensitive to acute fluctuations in 5-HT levels are advantageous in that they are ideal ligands to accurately determine regional receptor densities in which BP values are not associated with confounding effects, such as diurnal 5-HT fluctuations.
Data obtained in rodent and nonhuman primates were more mixed; the presence or absence of a response tended to correlate with the magnitude of change in 5-HT and positive outcomes confirmed the anticipated increases or decreases in binding, but effects were not consistent between tracers. [18F]MPPF, the 5-HT1A antagonist tracer, could successfully be used in animal paradigms to detect internalisation of 5-HT1A receptors in the raphe, but the problems of imaging small nuclei may limit its utility here. Unfortunately, [18F]MPPF was not reliably sensitive to 5-HT in larger brain structures; the most comprehensive PET study in monkeys showed no change despite the biggest recorded 5-HT elevation (Udo de Haes et al, 2006). Consistent reductions in [11C]DASB BP were observed across species in response to large-magnitude 5-HT elevation, but this may be of limited use if such potent serotonergic challenges obtained in (anaesthetised) animals cannot be translated safely into humans. The tested 5-HT2A receptor antagonist tracers do not seem useful for imaging changes in endogenous 5-HT.
Of the few positive findings in animal studies, most could sensibly be explained by the simple competition model, whereby changes in synaptic 5-HT are reflected in its occupation of the target to produce corresponding changes in BP (Figure 1). There are some notable exceptions. First, the strong evidence using immunoelectron microscopy support the hypothesis that 5-HT1A autoreceptors in the raphe will undergo rapid agonist-induced internalisation, whereas postsynaptic receptors localised in the hippocampus and cortex do not (Aznavour et al, 2006; Riad et al, 2004). This is further supported by the differential expression of G proteins (Mannoury la Cour et al, 2006). Therefore, any reductions in 5-HT1A tracer BP observed in the hippocampal and cortical regions may be best explained by competition, whereas changes recorded in the raphe may be more likely to represent 5-HT-induced internalisation, although either hypothesis could be applied. Mixed findings could thus be explained by a combination of the two mechanisms (e.g. Giovacchini et al, 2005). Other exceptions are the paradoxical effects of tryptophan depletion on SERT and 5-HT2A binding in which the direction of change in BP is contrary to the competition model (Milak et al, 2005; Yatham et al, 2001). A lack of replication and a poor understanding of 5-HT receptor behaviour under tryptophan depletion conditions in vivo make interpretation difficult.
Discussion Points
Several important questions are raised by these studies. First, if the observed changes in BP in vivo are a genuine reflection of altered endogenous 5-HT, are the findings supported by known 5-HT receptor pharmacology? Second, what are the reasons for inconsistency or failure to visualise a change in BP when 5-HT levels are clearly altered? Do the predominantly negative findings point to a fundamental difference between the serotonergic and dopaminergic systems? In which case, do we have sufficient information about the biological underpinning of the BP response and of the serotonergic system to understand how this might be overcome? Are we simply targeting the wrong receptors or are our serotonergic challenges insufficient? Alternatively, are we suffering from a lack of consistency, statistical power, or rigour in experimental design?
5-HT Receptor Characteristics: A Comparison With Dopamine
Using dopamine as an example, Figure 3 shows how findings from experimental challenge studies, drug-receptor theory, and known receptor pharmacology can be combined to model the interaction between tracer and endogenous agonist under baseline and challenge conditions (Laruelle, 2000). The original model was updated after a direct comparison between an agonist and an antagonist PET tracer, estimating that the proportion of D2 receptors available to dopamine in vivo was 38%, which fits with the maximal change in BP response recorded after a dopamine challenge (Narendran et al, 2004). Similar definitive studies that would provide comparable estimations of in vivo availability of 5-HT are not yet possible, but in vitro pharmacological data provide initial clues. Are there differences between the two receptor systems that might explain the lack of sensitivity of 5-HT tracers compared with, e.g., raclopride?
Factors governing the likelihood of observing a BP response for any neurotransmitter system primarily affect the availability of a receptor population to the radiotracer compared with the neurotransmitter. In addition to those factors previously identified from the competition equation, other variables may include receptor-affinity states, localisation, regulatory properties, and susceptibility to undergo internalisation. If one or more of these factors are particularly unfavourable for any given 5-HT receptor, visualising fluctuations in endogenous 5-HT may not be possible, regardless of the potency of the challenge or the quality of the tracer.
Factors Affecting BP Response: the Competition Model
The competition equation can be used to identify where the main differences between the two systems may arise. Under baseline conditions, occupancy by dopamine of the synaptic D2 receptor has been estimated to be 10% of the total population (Abi-Dargham et al, 2000) and in the striatum, baseline extracellular dopamine concentration is ∼50 nmol/L (Zetterstrom et al, 1983).
Basal receptor occupancy has not yet been formally established for 5-HT, but can be estimated (Equation 6). The assumption that extracellular neurotransmitter concentrations accurately reflect those of the synapse cannot be relied upon (Endres et al, 1997), but microdialysis can give an indication of the concentration available for competition with the tracer. Microdialysis studies herein estimate the basal extracellular 5-HT concentration to a range between 0.04 and 3.9 nmol/L (average 0.8 nmol/L). Our best guess of in vivo affinity of 5-HT can be extrapolated from the in vitro affinity values (Table 1) and depends on the functional state of the receptor (Figure 2). Assuming optimal conditions, i.e., high basal 5-HT concentration (FNT=3.9 nmol/L; Maeda et al, 2001), high 5-HT affinity (KNT=50 nmol/L; Khawaja et al, 1995), and 100% of receptors in RHIGH, crude estimations of the expected change in BP can be made for any given change in 5-HT. By inserting in Equation 4, it can be seen that a 15-fold increase in 5-HT (from 3.9 to 58.5 nmol/L; ΔFNT) could produce a 50% reduction in BP for 5-HT1A receptors, which is well within the limit of detection for PET. However, using the average value of 0.8 nmol/L for basal 5-HT concentration, this figure decreases to a maximal 20% reduction in BP. In the likely case of a lower proportion of RHIGH receptors and a smaller change in 5-HT, the BP ratio rapidly decreases beyond the limit of detection for any PET system, given individual test–retest variability. Similar estimations can be performed for the 5-HT2A and 5-HT4 receptors and SERT with less favourable outcomes owing to the lower affinity of 5-HT for these targets. According to the competition equation, 5-HT receptors with the highest affinity for 5-HT should provide the best opportunities as targets for new radioligand developments. Although the reported Ki values are quite variable, Table 1 identifies the 5-HT7 and 5-HT1B receptors as promising targets.
The competition model assumes that all receptors are available for binding. They do not necessarily have to be in the RHIGH state, but must be accessible; one assumes a certain affinity for 5-HT, which is reduced between RHIGH and RLOW (Table 1). In vitro data would suggest that ∼20% to 40% of 5-HT1A receptors (Gozlan et al, 1995; Khawaja et al, 1995; Nenonene et al, 1994) and 40% to 60% of 5-HT2A receptors (Branchek et al, 1990; Lopez-Gimenez et al, 2001) are RHIGH, compared with 70% estimated for D2 receptors (Figure 3B).
If nonaccessible 5-HT receptor pools are also large, then in vivo availability of receptors to 5-HT may be substantially lower than that predicted in vitro. Nonaccessible pools can arise through intracellular receptor localisation in which receptors remain available to the radiotracer but unavailable to 5-HT. If radiotracer affinity is maintained in the intracellular environment, it will not be subject to competition with 5-HT here, thus reducing the pool susceptible to change. This may be an important factor for 5-HT2A receptors for which a significant internalised receptor pool exists (80% to 90% Cornea-Hebert et al, 2002). It may also be relevant for [11C]DASB studies; a proportion of SERT is associated with the membranes of intracellular organelles with vesicular structure (Torres et al, 2003). However, if radiotracer affinity is altered within the intracellular compartments as has been suggested for D2 receptors (Guo et al, 2010), the influence on BP may be more or less pronounced.
These estimations were made on the basis of the competition model fit with the data herein and with findings from the literature. The fact that the majority of 5-HT1A studies were negative is consistent with in vitro data, suggesting that only a small proportion of 5-HT1A receptors is RHIGH and the finding that the presence or absence of a BP response tended to correlate with those experiments obtaining the highest magnitude of change in 5-HT. Studies using radiolabelled 5-HT have failed to efficiently label 5-HT2A receptors because of very low affinity (Kristiansen et al, 2005; Lopez-Gimenez et al, 2001; Sleight et al, 1996), rendering the probability of imaging a BP response by competition less likely. In addition, if 80% are intracellular under basal conditions, even if all of the remaining cell-surface receptors are RHIGH, this leaves only a fraction of the total available to 5-HT in vivo, consistent with the lack of effect on BP in challenge experiments.
For a similar reason, the very low affinity of 5-HT for SERT also renders a response less likely. Consistent with these observations, only a very large-magnitude 5-HT elevation could be visualised by [11C]DASB. However, positive results were consistent, suggesting that either the low affinity was overcome by the size of the change or it could point to a difference between SERT and G protein-coupled receptor interaction with 5-HT. It would be prudent to test the model in human subjects after a 5-HT elevating challenge to complete the picture.
Factors Affecting BP Response: the Internalisation Model
We have observed that if the competition model is entirely responsible for changes in BP, the presence or absence of a BP response will depend heavily on basal occupancy and the magnitude of change in extracellular 5-HT. However, if receptor internalisation is involved, other factors may prove more important. For example, the susceptibility of the target to translocation by receptor-trafficking mechanisms may dictate the size and/or the likelihood of a change, and therefore will be dependent on the efficacy of 5-HT for its receptors rather than its affinity.
The fact that 5-HT1A receptors show differential sensitivity to internalisation depending on regional expression is now established, but little is known about the reciprocal trafficking of 5-HT1A receptors back to the membrane after reinsertion or de novo synthesis. For example, it is possible that increases in [18F]MPPF BP after p-ethynylphenylalanine and sleep could be attributed to receptor translocation rather than competition, given the longer time frames involved in these challenges (Derry et al, 2006; Zimmer et al, 2003). Changes in SERT could be explained by receptor-trafficking mechanisms through reciprocal regulation by endogenous 5-HT in vivo (Blakely et al, 1998; Ramamoorthy and Blakely, 1999; Ramamoorthy et al, 1998; Zahniser and Doolen, 2001). With a time frame of ∼30 minutes, this would be feasible within a 5-hour tryptophan depletion paradigm but it remains to be seen whether temporal characteristics of trafficking are consistent with the observed increases in BP. This is further complicated by the fact that the competition model adequately describes reductions in [11C]DASB BP after tranylcypromine challenge, which would be contrary to the direction of SERT translocation. The 5-HT2A receptors have some complex and unusual regulatory properties (for review, see Gray and Roth, 2001). Data published on trafficking of the 5-HT2A receptor are sparse, but suggest that it may be sensitive to 5-HT concentration. 5-Hydroxytryptamine can stimulate receptor internalisation (Bhattacharyya et al, 2002; Schmid et al, 2008), but mianserin (an antagonist), dopamine, and certain dopaminergic antagonists can also reduce 5-HT2A density (Bhattacharyya et al, 2006; Blackshear and Sanders-Bush, 1982; Hensler and Truett, 1998). In addition, many 5-HT2A ligands previously assumed to be antagonists are in fact inverse agonists (Aloyo et al, 2009; Berg et al, 2005). The significance of these findings is unclear, but may have implications for interpretation of imaging data if receptor internalisation or trafficking is involved in the BP response.
A more comprehensive understanding of the in vivo regulatory properties of 5-HT receptors and SERT is required if we are to evaluate the differential roles had by competition and receptor trafficking in any change in BP. In the absence of this information, observations can be validated by other means. For example, combining microdialysis with dynamic imaging has several benefits; primarily, it not only provides an indication of the magnitude of change in synaptic neurotransmitter concentrations after the challenge but it also permits temporal correlations to be drawn between changes in neurotransmitter and changes in BP. Where dynamic reductions in BP correlate with temporally related increases in release, competition can be safely assumed and vice versa. If not, they may be better correlated with receptor-trafficking events. This has been shown within the dopaminergic literature as described previously (Laruelle, 2000). Unfortunately, although microdialysis and other measures were used to good effect in some studies (Table 2), none specifically reported the temporal relationship between 5-HT release and changes in BP.
Improving Ligand Characteristics
The properties required for an ideal brain imaging tracer for use in vivo are many (Pike, 1993), and successful tracers are usually designed to produce the best quality images of specific brain proteins (Elfving et al, 2007). However, to indirectly image the concentration of an endogenous ligand at the target receptor, rather than the receptor itself, may require an entirely different approach to radiotracer design. The relative merits of radioligands such as raclopride, IBZM, and NPA that render them sensitive to displacement by endogenous dopamine in which other D2 ligands are not remains unclear. It does not seem to involve affinity, pharmacological action (agonist versus antagonist), or lipophilicity, but may involve chemical structure (Laruelle, 2000; Ginovart et al, 2006).
Specific binding of a radiotracer is a function of its affinity for the site relative to the density (Bmax) of that binding site. As receptor affinity is a ratio between the rate of association (Kon) and the rate of dissociation (Koff), if Bmax continues to decrease, eventually the PET signal will deteriorate because of slow Koff kinetics. It has been suggested that faster dissociation kinetics might influence sensitivity to endogenous 5-HT (Endres and Carson, 1998; Laruelle, 2000). No systematic study of whether fast Koff correlates with neurotransmitter ‘displaceability' has been conducted, and often pharmacokinetic experiments that would facilitate such a comparison have not been conducted.
Many published articles discuss the relative merits of ‘lower-affinity' ligands for imaging endogenous neurotransmitter release, citing that they may be ‘more susceptible' to displacement than higher-affinity ligands (Endres and Carson, 1998; Seeman et al, 1989; Skaddan et al, 2002). A similar argument was made for the development of ‘moderate-affinity' fluorinated WAY analogues ligands (Jagoda et al, 2006; Zimmer et al, 2002). However, as can be seen from Equation (4), the probability of a radiotracer being sensitive to competition relies on target properties and not on the absolute affinity of the radioligand. A shortcoming of Equation (4) is that it builds on equilibrium conditions, which may not be present in a dynamic PET scan. For a within-scan challenge (under a bolus-infusion experiment), this may be of some importance.
Development of Agonist Tracers
Whichever target is visualised and regardless of the mechanism, the proportion of receptors susceptible to 5-HT in vivo will virtually always be smaller than the total number, especially if the tracer is an antagonist, resulting in a low signal-to-noise ratio and a small chance of imaging a change in endogenous neurotransmitter. This is particularly likely to be the case for receptors with low basal 5-HT occupation, a small proportion of RHIGH, or in which a proportion of receptors somehow are unavailable for competition under basal conditions.
One could specifically target those receptors in RHIGH by using an agonist tracer rather than an antagonist. Theoretically, agonist tracers should be more sensitive to extracellular changes in an endogenous agonist, because they only compete with receptors in the high-affinity state that are also susceptible to neurotransmitter binding. This was confirmed for D2 agonist tracers that indeed had better sensitivity to endogenous dopamine (Narendran et al, 2004). In addition, using an agonist tracer to image RHIGH receptors that are coupled to their particular G protein would provide a better representation of ‘functional' receptors. This strategy could prove successful, regardless of whether competition or internalisation is involved in the BP response.
Development of 5-HT receptor agonist tracers for this very purpose is already underway. [11C]CUMI-101 is the first putative agonist radiotracer to be developed for the 5-HT1A receptor and has adequate PET characteristics in both baboons and humans (Kumar et al, 2008; Milak et al, 2008a, 2008c). Indications of its possible sensitivity to changes in endogenous 5-HT in baboons were recently presented, reporting significant reductions in BP after intravenous citalopram (Table 2, Milak et al, 2008b). However, displacement was also observed in brain areas with low 5HT1A receptor density, i.e., in areas of supposed nonspecific binding only, which questions whether this is a true 5-HT displacement.
Another group has developed 5-HT1A agonist ligand [18F]F15599 (Lemoine et al, 2009). It is yet to prove its utility as a PET tracer in human subjects but produces good images in the rat and cat brains, and reliable in vivo data confirm that it behaves as a full agonist. The group plans to produce higher-affinity analogues to achieve higher contrast images. Similar programmes are ongoing for the development of agonist tracers for the 5-HT2A receptor, in which a promising candidate, [11C]Cimbi-5 has been tested in the pig brain (Ettrup et al, 2009a, 2009b). It remains to be seen whether these agonist analogues are susceptible to 5-HT displacement in vivo.
Development of Tracers for Novel Targets
Tracer development programmes have been ongoing to produce PET or SPECT tracers for 5-HT1B and 5-HT6 receptors (Table 1). Both targets possess adequate Bmax in the brain and several in vivo tracers have emerged.
5-HT1B Tracers
Although the majority of 5-HT1B ligands share pharmacology with the 5-HT1D receptor, it is now generally accepted that in the human brain, because the majority of receptors are 5-HT1B, any overlapping binding to 5-HT1D is considered negligible. The 5-HT1B receptors may represent a good target for PET tracer development generally; it displays a high Bmax and has been successfully mapped by autoradiography in the human brain, providing a comparator for binding densities and localisation (Varnas et al, 2001). The 5-HT1B receptors have a relatively high affinity for 5-HT (∼1 nmol/L, Millan et al, 2002), and studies have suggested that up to 50% of receptors are in the high-affinity state (Granas et al, 2001; Millan et al, 2002) pointing to the potential for ∼50% reductions in binding. In addition, studies in the rat whole brain tissue suggest that it is likely that the majority of receptors are G protein coupled because of the similarity between agonist- and antagonist-labelled receptor densities (Audinot et al, 1997; Bruinvels et al, 1993; Millan et al, 2002; Offord et al, 1988). The 5-HT1B receptors also share similarities with D2 receptors that might confer sensitivity to the endogenous neurotransmitter; they are presynaptically located, coupled to inhibitory G proteins regulating 5-HT and other transmitter release (Sari, 2004). Importantly, many serotonergic challenge compounds do not appear to interfere with 5-HT1B receptors, pharmacologically (NIMH Psychoactive Drug Screening Programme; Millan et al, 2001).
Two selective 5-HT1B PET ligands have shown particular promise in early studies (Table 2). [11C]AZ10419369 was reported to be a selective 5-HT1B antagonist suitable for PET studies in both humans and nonhuman primates (Pierson et al, 2008; Varnas et al, 2010). In parallel, [11C]-P943 (GSK-174722), another 5-HT1B antagonist, was also shown to be a suitable PET tracer in nonhuman primates, healthy volunteers, and patients (Gallezot et al, 2010; Hu et al, 2010; Murrough et al, 2010; Nabulsi et al, 2010). Subsequently, binding of [11C]AZ10419369 was shown to be reduced after intravenous fenfluramine administration in cynomolgus monkeys (Finnema et al, 2010), while [11C]-P943 binding was reduced after intravenous fenfluramine, citalopram, and amphetamine in baboons (Dr E Rabiner, personal communication), suggesting sensitivity to increases in endogenous 5-HT (Table 2). More recently, [11C]AZ10419369 binding was similarly reduced in a bolus-infusion paradigm (Sjoerd Finnema, personal communication). The authors of these studies propose that a competition between tracer and 5-HT represents the most likely explanation for reductions in binding, although 5-HT1B receptors are susceptible to 5-HT-induced internalisation (Janoshazi et al, 2007). Both tracers could be susceptible to confounding effects of anaesthetics, challenge-induced rCBF changes, and/or pharmacological interaction. In addition, the possibility of displaceable binding in the cerebellum needs to be addressed. Further studies are required to resolve these issues and to determine whether these tracers are sensitive to 5-HT manipulation in human subjects.
Other Targets
Some preliminary data suggest that a 5-HT6 PET tracer may be viable; evaluation of [11C]GSK215083 in human subjects suggests specific binding in the caudate and putamen (Martarello et al, 2008; Parker et al, 2008), while [11C]GSK224558 shows similar characteristics in the porcine brain but with relatively slow kinetics (Huiban et al, 2006). No investigations into their susceptibility to 5-HT displacement have yet been conducted.
Development of tracers for the remaining 5-HT targets has so far been hampered by low Bmax in the brain (5-HT1F, 5-HT3, 5-HT7, 5-HT2B, 5-HT2C), a lack of selective ligands (5-HT1D), and/or insufficient pharmacological or functional data (5-ht1e, 5-ht5a, 5-ht5b).
It could be argued that targeting only one receptor is not necessary if the aim is to measure 5-HT displacement rather than receptor density. Nonselective ligands such as the D2/D3 agonist tracer [11C](+)PHNO (Willeit et al, 2008) and M1/M2 muscarinic acetylcholinergic ligand [11C]3-PPB (Nishiyama et al, 2001; Tsukada et al, 2004) seem to be able to show dopamine and acetylcholine release, respectively. Although multisite pharmacology may increase the chances of released 5-HT displacing the radiotracer, it may render the task of identifying a challenge paradigm without introducing confounding pharmacology very difficult.
Nature of the Serotonergic Challenge
Choosing an appropriate serotonergic challenge that is properly validated is essential for determining the susceptibility of a radiotracer to endogenous 5-HT. The choice is primarily dictated by the radiotracer, e.g., a reuptake inhibitor cannot be used against a SERT tracer. Various serotonergic challenge studies have been undertaken herein. Most are not realistic for human use. Fenfluramine, despite being an efficient way to increase 5-HT in animals, cannot readily be administered to human subjects in high doses (Connolly et al, 1997), although one study has now published preliminary findings using an oral dose to increase brain 5-HT (Hasler et al, 2009). The SSRIs are a safe option and intravenous preparations of citalopram can be obtained for use in infusion paradigms (Marner et al, 2010; Pinborg et al, 2004). Unfortunately, the well-validated method of acute tryptophan depletion does not seem to produce sufficient changes in 5-HT to be visualised by currently available radiotracers (Table 2). Alternative options in humans are somewhat limited: 5-HTP and tryptophan could be used intravenously (Charig et al, 1986; den Boer and Westenberg, 1990) or further 5-HT elevation might be achieved safely by introducing augmenting agents to SSRIs, such as pindolol (Pinborg et al, 2004), ketanserin (Cremers et al, 2004; Udo de Haes et al, 2005), or risperidone (Huang et al, 2006), with antiemetic administration at higher doses to avoid nausea and caution over possible drug interactions and serotonin syndrome.
Validation of the Serotonergic Challenge
Surrogate serotonergic markers and other physiologic parameters indicate whether changes in central 5-HT have actually occurred and are a necessary inclusion to validate findings and to aid in data interpretation, especially where an understanding of the basic biological underpinning of the response is lacking.
Simultaneous microdialysis is the most powerful tool but if it is not possible or practical, surrogate markers of neurotransmitter release, depletion, or receptor activation can serve to validate or disprove the model. Few of the studies mentioned in this review included parallel measures alongside imaging techniques to measure 5-HT (Table 2). Good examples include those combining microdialysis and immunoelectron microscopy to help distinguish between internalisation and competition mechanisms (Riad et al, 2004), and the demonstration that regional reductions in [11C]DASB BP correlated with regional activity of the 5-HTP-converting enzyme, local increases in 5-HT production, and the magnitude of subsequent 5-HT elevation after 5-HTP challenge (Yamamoto et al, 2007). Markers such as immediate early gene expression in rats, or plasma prolactin, cortisol levels, and the subjective experience of hot flushes in humans were suggestive of serotonergic function despite the absence of a BP response (Hirani et al, 2003; Pinborg et al, 2004).
Experimental Considerations
Study design is of paramount importance when performing dynamic imaging studies, particularly when a challenge paradigm is involved. A brief glance at Table 2 shows a great deal of variance in species, challenge doses, routes of administration, dose timing relative to tracer, the interval between tracer injections, and tissue collection. One could argue that an ideal tracer would be sensitive enough to detect change regardless of these variables, but equivocal findings within the 5-HT literature could in part be explained by these inconsistencies. The timing of challenge administration in relation to radiotracer may be crucial herein; e.g., when timings of fenfluramine were designed to ensure maximal concentration of 5-HT at the point of tracer injection, significant reductions in BP were observed (Hume et al, 2001; Udo de Haes et al, 2005). Table 3 summarises the main limitations of current approaches to measuring endogenous neurotransmitters by PET, including those imposed by experimental design, pharmacological challenge, and receptor system, along with potential solutions for addressing these shortcomings. Some are encountered in PET and/or animal studies generally; therefore, further discussion is restricted to those most relevant to design or interpretation of human PET challenge studies.
Table 3. Limitations of current approaches, consequences, and possible solutions.
| Limitation | Example | Consequence | Solution? |
|---|---|---|---|
| Experimental design | |||
| Lack of consistency | Challenge: timing, species, dose, route, Tracer: timing relative to challenge and/or tissue collection | Lack of replication or validation of current findings. Difficulties with data interpretation | Develop shared protocols, standardise methodologies |
| Lack of statistical power | Number of subjects is low, test–retest variability is high, and effect size is small | Power is insufficient to detect a significant change even if a change is occurring | Take steps to minimize test–retest and experimental variability, undertake properly powered studies, share facilities to increase throughput |
| Effect of anaesthetics | Certain anaesthetics interfere with 5-HT system (Mukaida et al, 2007; Seeman and Kapur, 2003; Tokugawa et al, 2007) | Possible confound. Complicates extrapolation to humans and between experimental designs and species | Systematic studies required to optimise anaesthetic and discount confounds (Elfving et al, 2003) |
| Nature of challenge | |||
| Multisite pharmacology | The challenge compound or its metabolite has possible affinity for the target receptor | A direct effect on binding rather than through 5-HT release/depletion cannot be ruled out | Measure brain and/or plasma concentration of compounds. Perform relevant displacement studies |
| rCBF changes | The challenge compound induces blood flow changes | Apparent change in BP after challenge is actually confounding effect of blood flow | Control for, measure or simulate rCBF changes and peripheral clearance |
| Toxicity in humans | Challenge doses that increase 5-HT in animals may be too toxic for use in humans | Sufficient 5-HT elevation unachievable to be measured with current tracers | Probes must become an order of magnitude more sensitive; investigate alternative targets or agonist tracers |
| Lack of challenges | Many 5-HT releasers unavailable for human use, regulatory constraints | Less scope for the use of optimal challenge | New challenges required with fewer safety concerns |
| Species differences | |||
| In response to tracer or challenge | Variation in regional brain anatomy, receptor pharmacology, tracer, and challenge pharmacokinetics, dose window | Changes in BP in response to challenge may be present in one species but not in others; could lead to inappropriate go: no-go decisions | Translational approach required for tracer and challenge validation; between in vitro and in vivo, preclinical and clinical findings |
| Knowledge gaps | |||
| Lack of surrogate markers | Current human markers of central 5-HT activity are variable and poorly validated | Alterations in central 5-HT function after a challenge cannot be verified | Alternative surrogate measures should be sought and validated |
| Inappropriate binding parameters | Binding parameters often obtained under optimal rather than physiologic binding conditions | Temperature, intracellular, extracellular, and vesicular environment greatly affect binding with implications for interpreting BP response | Perform binding experiments under conditions closely reflecting in vivo environment |
| Use of recombinant systems | Receptor-trafficking mechanisms, receptor regulation by 5-HT, tracer affinity, selectivity, and activity assays | Data from cell lines/in vitro models difficult to reliably extrapolate to the native brain tissue, or in vivo situation | Perform validation studies in native tissue or in vivo systems wherever possible |
| Lack of tracer pharmacology | Few tracers tested for affinity against other receptors | Binding at other receptors may complicate the interpretation of BP signal | Routine screening of compounds to determine binding profile e.g., Cerep, NIMH |
| Lack of receptor pharmacology | Few receptors tested for distribution and binding density in the human brain | PET distribution and signal hard to interpret | Postmortem brain studies required |
| Lack of receptor physiology | In vivo data relating to receptor-trafficking mechanisms or regulation by 5-HT are sparse | Relative involvement of competition and internalisation mechanisms for change in BP cannot be assessed | In vivo models of receptor trafficking required, e.g., Skinbjerg et al, 2010 |
| Understanding of BP response | Basic biological underpinning of BP response to challenge is not known | Flaws in both competition and internalisation models; inappropriate interpretation of results | Combination of above studies to dissect differential role had by competition and internalization |
BP, binding potential; PET, positron emission tomography; rCBF, regional cerebral blood flow; 5-HT, 5-hydroxytryptamine.
PET Protocol Design
The differences between PET protocols may affect whether potential changes in BP can be visualised. A tracer can be delivered either as two bolus injections, i.e., control and challenge (Logan et al, 1991) or as a single bolus plus infusion (Endres et al, 1997). In general, the bolus plus infusion approach has the advantage that the challenge can be introduced at equilibrium, its effect on neurotransmitter release is determined in a single study, rCBF changes represent less of a problem, and so does test–retest variability.
Species Differences
Species differences in response to radiotracer and challenge can occur for many reasons (see Table 3) and are a problem for translation of all PET data from animals to humans and vice versa. A good example is provided by recent evidence that dopamine could displace up to 80% of the D2 agonist [11C](+)PHNO binding in the cat brain (Ginovart et al, 2006), which threatened to undermine the widely reputed theoretical ∼40% maximal displacement (Narendran et al, 2004). However, subsequent human studies have shown that this effect may be specific to cats (Willeit et al, 2008).
Challenge Limitations
The addition of a pharmacological challenge to a PET protocol introduces many opportunities for variance and the possibility of confounding effects (Table 3). Mixed pharmacology of the compound or its metabolites may cause particular problems especially if high doses are required. For example, the potential for interaction of clomipramine with 5-HT1A and 5-HT2A receptors (Hyttel, 1994), norfenfluramine with 5-HT2A receptors (Rothman and Baumann, 2002) and amphetamine, a proposed metabolite of tranylcypromine, with SERT (Sherry et al, 2000; Youdim et al, 1979) could have implications for certain challenges. The assumption that a direct effect of the releasing agent on the target receptor is unlikely because of ‘low affinity' cannot be relied upon, unless the actual brain concentration of these compounds and their metabolites is measured. Brain levels could be easily measured in animals and plasma levels in humans to aid in the interpretation of results.
Both pharmacological and physiologic challenges can affect rCBF with implications for tracer BP, especially where changes are not uniformly distributed across the brain (Jagoda et al, 2006). The susceptibility of a tracer to blood flow changes depends on K1, i.e., its ability to cross the blood–brain barrier. If brain penetration is high, then changes in CBF will have a greater effect on BP than for lower brain penetrability. Higher-affinity ligands are more susceptible, especially where receptor density is high, allowing brain accumulation in a nonreversible manner. If no details of rCBF changes are provided, it is difficult to assess what their effects might be (Holthoff et al, 1991; Logan et al, 2007).
Statistical Power
The concept of statistical power is often overlooked in small studies and this increases the risk of both type I and type II errors (Knudsen, 2009). For an 80% chance (0.8 power) of detecting a 10% change in BP (similar to that observed for [11C]raclopride in response to amphetamine challenge) in which s.d. is also 10%, a study of paired design requires inclusion of 10 subjects (Student's paired t-test, two-tailed). With the exception of some ex vivo and β-probe studies, in which smaller variance and larger responses permit smaller subject numbers, few of the PET studies cited achieved sufficient statistical power to reliably detect such a change in BP assuming these parameters. For [18F]MPPF, a realistic variance in BP is ∼20% in the human hippocampus (Praschak-Rieder et al, 2004), requiring n=34 subjects in a paired comparison to detect a 10% change. Where larger changes in BP are expected, subject numbers can be reduced accordingly, e.g., n=8 (Sibon et al, 2008). [11C]DASB binding is generally associated with lower variability, thus with n=14 and a variance of 17% in the human thalamus (Praschak-Rieder et al, 2005), a study would have sufficient power to detect a realistic 14% change in BP, whereas n=8 and a variance of 28% (Talbot et al, 2005) would require a 32% change in BP to reach significance. Test–retest variability must also be considered within any power calculation if separate scans on different days are to be performed.
This has important implications for human data. It is perhaps not surprising that significant changes were not reported in response to challenge studies in which relatively small changes in BP are expected and test–retest variability is high. Larger numbers of subjects would be required to detect changes of this magnitude. Therefore, we must develop better tracers, improve technology, and investigate novel 5-HT imaging targets to minimise this problem. We may also have to accept that larger and unavoidably more expensive studies are necessary to detect changes in BP in humans.
Knowledge Gaps
Aside from experimental issues, the imaging community is suffering from significant gaps in basic biological understanding of the nature of the BP response: how it is manifested, how a change should be interpreted, and what is the functional significance. The pharmacological properties of tracers and the receptor target (such as pharmacokinetics, Bmax, affinity, regulatory mechanisms, localisation) have important implications for in vivo dynamic imaging. It is apparent that a fundamental lack of basic pharmacology, particularly in vivo data, hinders the interpretation of existing data. Choosing a suitable target and developing sensitive ligands would be facilitated if these parameters were well characterised. Existing knowledge gaps are highlighted in Table 3, alongside other limitations to current approaches. Particular problems arise from the majority of binding studies being conducted under ‘optimal' experimental conditions rather than those most likely to reflect the in vivo environment of different cellular compartments, the use of recombinant systems, and the lack of translation between in vitro and in vivo models, and between animals and humans. Often, useful binding parameters are not available; e.g., few PET tracers are tested against a battery of potential challenge compounds in the brain tissue or against other receptors. The absence of both agonist and antagonist in vivo tracers for most targets precludes an assessment of receptor availability to endogenous 5-HT and applicability of the ternary complex model. In vivo data relating to receptor-trafficking mechanisms and their regulation by 5-HT are also sparse; often such experiments are performed in recombinant systems (Allen et al, 2008; Janoshazi et al, 2007). Given these gaps in understanding, it is essential to perform some basic studies to address these issues, in vivo wherever possible. Examples of how such issues might be investigated have emerged for the study of dopamine interaction at the D2 receptor (Guo et al, 2010; Quelch et al, 2010; Skinbjerg et al, 2010), raising important questions for the imaging community.
Can Physiologic Changes in 5-HT Ever be Visualised by PET?
One of the ultimate aims of this methodology is to visualise changes in BP in response to physiologic challenges, so that small changes in neurotransmission can be measured, reflecting realistic perturbations. Microdialysis studies have shown that in excess of 15-fold increases in 5-HT are required to alter BP with currently available tracers. Increases of this magnitude are probably unachievable in human subjects. Physiologic changes in 5-HT are likely to be much smaller; therefore, if we are to visualise them, it is clear that either the tracers must become an order of magnitude more sensitive, or we need to investigate an alternative target, the pharmacology of which is more favourable to the competition and/or internalisation model. New data with the 5-HT1B ligands tend to suggest that smaller increases in 5-HT might be adequate to visualise significant reductions in tracer BP in the nonhuman primate brain. However, it remains to be seen whether these results can be translated into human subjects using equivalent (or less potent) serotonergic challenges. Similarly, agonist radiotracers seem to provide an opportunity for further development.
Conclusions and Recommendations
In the past, we have relied heavily on serendipity: the hope that current radiotracers designed to measure receptor density would also prove satisfactory for measuring fluctuations in endogenous 5-HT. However, this has not proved effective. After a decade of unsuccessful testing, dedicated ligand development programmes are required to focus on producing ligands sensitive to endogenous neurotransmitters. This will have to involve a high level of translational science within a multidisciplinary setting. Collaborative efforts such as the METPETS consortium (Measuring Endogenous Transmitters using PET and SPECT) hope to facilitate this process.
Good experimental design could go some way in improving the chances of imaging changes in endogenous 5-HT. Development of shared protocols, standardisation of methodologies, and shared use of facilities could significantly increase statistical power and throughput and improve outcome. In particular, we would recommend that future studies obtain test–retest data, so that properly powered challenge studies can be conducted to enable sensible interpretation of any (in)significant change in BP alongside the level of within- and between-subject variability. Wherever possible, future animal imaging studies should be combined with simultaneous microdialysis, so that changes in 5-HT can be specifically correlated with those of BP and in particular to elucidate the temporal relationship between the two. We may have already reached the maximum potency of a pharmacological serotonergic challenge that can be safely used in humans; the incidence of 5-HT-related side effects may limit the dose; therefore, smaller changes in 5-HT can be expected, requiring larger numbers of subjects to achieve sufficient power to detect a change in BP. The issue of validation of such challenges in human subjects remains unresolved; new surrogate markers of 5-HT are required.
Even if all these experimental improvements are introduced, only a better understanding of the pharmacology of the BP response will allow sensible data interpretation. Significant gaps in our knowledge remain to be addressed. Therefore, measures outlined above should be pursued in parallel with basic characterisation of the target, tracer, and challenge using a multidisciplinary approach. It may be that we have to revisit our interpretation of findings from PET studies in light of future advances in our understanding of BP.
The evidence from the 5-HT literature strongly suggests that imaging endogenous 5-HT may be more difficult than dopamine. There may be pharmacological reasons why the 5-HT1A, 5-HT2A, and 5-HT4 receptors and SERT are not ideal targets: it seems likely that basal receptor occupancy is low compared with dopamine, pointing to the need for good agonist tracers to reduce the ratio of sites available to 5-HT relative to the tracer and thus improve signal. Such tracer development programmes are now underway. The 5-HT1B receptors may be an alternative target for PET tracer development. We eagerly anticipate future publications in the hope that these tracers will pave the way for a deeper understanding of serotonergic neurotransmission.
There are many lessons to be learned from the studies published so far in the 5-HT field and these can be extended to the study of any receptor system. It seems certain that optimisation of the target, tracer, technology, experimental protocols, and challenge paradigm to answer the specific question of imaging endogenous neurotransmitters will provide the best chance for successful imaging in future.
Acknowledgments
We acknowledge the METPETS (Measuring Endogenous Neurotransmitters by PET and SPECT) consortium members for sharing their recent research, much of which has been presented herein, and their thoughtful insight and helpful discussion. The METPETS consortium is an integrated, collaborative network of European neuroscientists who have come together with the specific aim of developing tracers to image neurotransmitter release in the human brain. See http://www.p1vital.com/consortium_metpets.html. METPETS members include the following: David J Nutt, Robin J Tyacke, and Louise M Paterson (Imperial College London), Gitte M Knudsen (Copenhagen University Hospital), Fokko Bosker (University of Groningen), Adriaan Lammertsma (VU University Amsterdam), Ilan Rabiner, Roger Gunn, and Marc Laruelle (GlaxoSmithKline), Christer Halldin (Karolinska Institutet), Rupert Lanzenberger (Medical University of Vienna), Peter Talbot (University of Manchester), Franciszek Saczewski (Medical University of Gdansk), Jarmo Hietela (Turku University), Gerry Dawson (P1vital). We also acknowledge the Danish Council for Strategic Research, the Medical Research Council, H Lundbeck A/S and the EuroBioFund for providing the financial support that enabled this review to be written.
David J. Nutt has provided consultancy services to Pfizer, GSK, Novartis, Organon, Cypress, Lilly, Janssen, Lundbeck, BMS, Astra Zeneca, Servier, Hythiam, and Sepracor, has received honoraria from Wyeth, Reckitt-Benkiser, and Cephalon; and has received grants or clinical trials payments from MSD, GSK, Novartis, Servier, Janssen, Lundbeck, Pfizer, Wyeth, and Organon. Louise Paterson's salary was partially covered by H. Lundbeck A/S (unrestricted educational grant). Gitte M. Knudsen and Robin J Tyacke have no financial disclosures.
References
- Abi-Dargham A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol. 2004;7 (Suppl 1:S1–S5. doi: 10.1017/S1461145704004110. [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci USA. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian GK, Marek GJ. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain Res Brain Res Rev. 2000;31:302–312. doi: 10.1016/s0165-0173(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Allen JA, Yadav PN, Roth BL. Insights into the regulation of 5-HT2A serotonin receptors by scaffolding proteins and kinases. Neuropharmacology. 2008;55:961–968. doi: 10.1016/j.neuropharm.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloyo VJ, Berg KA, Spampinato U, Clarke WP, Harvey JA. Current status of inverse agonism at serotonin2A (5-HT2A) and 5-HT2C receptors. Pharmacol Ther. 2009;121:160–173. doi: 10.1016/j.pharmthera.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Audinot V, Lochon S, Newman-Tancredi A, Lavielle G, Millan MJ. Binding profile of the novel 5-HT1B/1D receptor antagonist, [3H]GR 125,743, in guinea-pig brain: a comparison with [3H]5-carboxamidotryptamine. Eur J Pharmacol. 1997;327:247–256. doi: 10.1016/s0014-2999(97)89668-9. [DOI] [PubMed] [Google Scholar]
- Aznavour N, Rbah L, Riad M, Reilhac A, Costes N, Descarries L, Zimmer L. A PET imaging study of 5-HT(1A) receptors in cat brain after acute and chronic fluoxetine treatment. Neuroimage. 2006;33:834–842. doi: 10.1016/j.neuroimage.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Aznavour N, Zimmer L. [18F]MPPF as a tool for the in vivo imaging of 5-HT1A receptors in animal and human brain. Neuropharmacology. 2007;52:695–707. doi: 10.1016/j.neuropharm.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- Berg KA, Harvey JA, Spampinato U, Clarke WP. Physiological relevance of constitutive activity of 5-HT2A and 5-HT2C receptors. Trends Pharmacol Sci. 2005;26:625–630. doi: 10.1016/j.tips.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Bernard V, Decossas M, Liste I, Bloch B. Intraneuronal trafficking of G-protein-coupled receptors in vivo. Trends Neurosci. 2006;29:140–147. doi: 10.1016/j.tins.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Puri S, Miledi R, Panicker MM. Internalization and recycling of 5-HT2A receptors activated by serotonin and protein kinase C-mediated mechanisms. Proc Natl Acad Sci USA. 2002;99:14470–14475. doi: 10.1073/pnas.212517999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Raote I, Bhattacharya A, Miledi R, Panicker MM. Activation, internalization, and recycling of the serotonin 2A receptor by dopamine. Proc Natl Acad Sci USA. 2006;103:15248–15253. doi: 10.1073/pnas.0606578103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshear MA, Sanders-Bush E. Serotonin receptor sensitivity after acute and chronic treatment with mianserin. J Pharmacol Exp Ther. 1982;221:303–308. [PubMed] [Google Scholar]
- Blakely RD, Ramamoorthy S, Schroeter S, Qian Y, Apparsundaram S, Galli A, DeFelice LJ. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biol Psychiatry. 1998;44:169–178. doi: 10.1016/s0006-3223(98)00124-3. [DOI] [PubMed] [Google Scholar]
- Bonaventure P, Hall H, Gommeren W, Cras P, Langlois X, Jurzak M, Leysen JE. Mapping of serotonin 5-HT(4) receptor mRNA and ligand binding sites in the post-mortem human brain. Synapse. 2000;36:35–46. doi: 10.1002/(SICI)1098-2396(200004)36:1<35::AID-SYN4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Branchek T, Adham N, Macchi M, Kao HT, Hartig PR. [3H]-DOB(4-bromo-2,5-dimethoxyphenylisopropylamine) and [3H] ketanserin label two affinity states of the cloned human 5-hydroxytryptamine2 receptor. Mol Pharmacol. 1990;38:604–609. [PubMed] [Google Scholar]
- Brooks DJ. Dopaminergic action beyond its effects on motor function: imaging studies. J Neurol. 2006;253 (Suppl 4:IV8–IV15. doi: 10.1007/s00415-006-4003-5. [DOI] [PubMed] [Google Scholar]
- Bruinvels AT, Palacios JM, Hoyer D. Autoradiographic characterisation and localisation of 5-HT1D compared to 5-HT1B binding sites in rat brain. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:569–582. doi: 10.1007/BF00166939. [DOI] [PubMed] [Google Scholar]
- Burnet PW, Eastwood SL, Harrison PJ. [3H]WAY-100635 for 5-HT1A receptor autoradiography in human brain: a comparison with [3H]8-OH-DPAT and demonstration of increased binding in the frontal cortex in schizophrenia. Neurochem Int. 1997;30:565–574. doi: 10.1016/s0197-0186(96)00124-6. [DOI] [PubMed] [Google Scholar]
- Carpenter LL, Anderson GM, Pelton GH, Gudin JA, Kirwin PD, Price LH, Heninger GR, McDougle CJ. Tryptophan depletion during continuous CSF sampling in healthy human subjects. Neuropsychopharmacology. 1998;19:26–35. doi: 10.1016/S0893-133X(97)00198-X. [DOI] [PubMed] [Google Scholar]
- Carson RE, Channing MA, Vuong B, Watabe H, Herscovitch P, Eckelman MC.2001Amphetamine-induced dopamine release: duration of action assessed with [11C]-raclopride in anesthetised monkeys Physiological Imaging of the Brain with PET(Gjedde A HS, Knudsen G, Paulson O, eds)San Diego, CA: Academic Press; 205–209. [Google Scholar]
- Charig EM, Anderson IM, Sen JMR, Nutt DJ, Cowen PJ. L-Tryptophan and prolactin release: evidence for interaction between 5-HT1 and 5-HT2 receptors. Hum Psychopharmacol. 1986;1:93–97. [Google Scholar]
- Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- Cornea-Hebert V, Watkins KC, Roth BL, Kroeze WK, Gaudreau P, Leclerc N, Descarries L. Similar ultrastructural distribution of the 5-HT(2A) serotonin receptor and microtubule-associated protein MAP1A in cortical dendrites of adult rat. Neuroscience. 2002;113:23–35. doi: 10.1016/s0306-4522(02)00146-x. [DOI] [PubMed] [Google Scholar]
- Cremers TI, Giorgetti M, Bosker FJ, Hogg S, Arnt J, Mork A, Honig G, Bogeso KP, Westerink BH, den Boer H, Wikstrom HV, Tecott LH. Inactivation of 5-HT(2C) receptors potentiates consequences of serotonin reuptake blockade. Neuropsychopharmacology. 2004;29:1782–1789. doi: 10.1038/sj.npp.1300474. [DOI] [PubMed] [Google Scholar]
- den Boer JA, Westenberg HG. Behavioral, neuroendocrine, and biochemical effects of 5-hydroxytryptophan administration in panic disorder. Psychiatry Res. 1990;31:267–278. doi: 10.1016/0165-1781(90)90096-n. [DOI] [PubMed] [Google Scholar]
- Derry C, Benjamin C, Bladin P, le Bars D, Tochon-Danguy H, Berkovic SF, Zimmer L, Costes N, Mulligan R, Reutens D. Increased serotonin receptor availability in human sleep: evidence from an [18F]MPPF PET study in narcolepsy. Neuroimage. 2006;30:341–348. doi: 10.1016/j.neuroimage.2005.09.052. [DOI] [PubMed] [Google Scholar]
- Egerton A, Mehta MA, Montgomery AJ, Lappin JM, Howes OD, Reeves SJ, Cunningham VJ, Grasby PM. The dopaminergic basis of human behaviors: a review of molecular imaging studies. Neurosci Biobehav Rev. 2009;33:1109–1132. doi: 10.1016/j.neubiorev.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfving B, Bjornholm B, Knudsen GM. Interference of anaesthetics with radioligand binding in neuroreceptor studies. Eur J Nucl Med Mol Imaging. 2003;30:912–915. doi: 10.1007/s00259-003-1171-8. [DOI] [PubMed] [Google Scholar]
- Elfving B, Madsen J, Knudsen GM. Neuroimaging of the serotonin reuptake site requires high-affinity ligands. Synapse. 2007;61:882–888. doi: 10.1002/syn.20443. [DOI] [PubMed] [Google Scholar]
- Endres CJ, Carson RE. Assessment of dynamic neurotransmitter changes with bolus or infusion delivery of neuroreceptor ligands. J Cereb Blood Flow Metab. 1998;18:1196–1210. doi: 10.1097/00004647-199811000-00006. [DOI] [PubMed] [Google Scholar]
- Endres CJ, Kolachana BS, Saunders RC, Su T, Weinberger D, Breier A, Eckelman WC, Carson RE. Kinetic modeling of [11C]raclopride: combined PET-microdialysis studies. J Cereb Blood Flow Metab. 1997;17:932–942. doi: 10.1097/00004647-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Ettrup A, Palner M, Gillings N, Nagren K, Keller S, Sibomana M, Rasmussen L, Madsen J, Begtrup M, Knudsen GM.2009a[11C]-CIMBI5: a novel 5-HT2A agonist PET tracer J Nucl Med 50(Suppl 249019223420 [Google Scholar]
- Ettrup A, Palner M, Gillings N, Nagren K, Rasmussen K, Keller S, Sibomana M, Begtrup M, Madsen J, Knudsen GM. [11C]-CIMBI5: a novel 5-HT2A agonist PET tracer. J Cereb Blood Flow Metab. 2009b;29:S365. [Google Scholar]
- Finnema SJ, Varrone A, Hwang TJ, Gulyas B, Halldin C, Farde L. Fenfluramine induced serotonin release decreases [11C]AZ10419369 binding to 5-HT1B receptors in the primate brain. Synapse. 2010;64:573–577. doi: 10.1002/syn.20780. [DOI] [PubMed] [Google Scholar]
- Fletcher S, Barnes NM. Distribution and characterisation of the [3H](S)-zacopride labelled 5-HT3 receptor in pig forebrain. Brain Res. 1996;729:281–284. [PubMed] [Google Scholar]
- Frankle WG, Cho RY, Narendran R, Mason NS, Vora S, Litschge M, Price JC, Lewis DA, Mathis CA. Tiagabine increases [11C]flumazenil binding in cortical brain regions in healthy control subjects. Neuropsychopharmacology. 2009;34:624–633. doi: 10.1038/npp.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallezot JD, Nabulsi N, Neumeister A, Planeta-Wilson B, Williams WA, Singhal T, Kim S, Maguire RP, McCarthy T, Frost JJ, Huang Y, Ding YS, Carson RE. Kinetic modeling of the serotonin 5-HT(1B) receptor radioligand [(11)C]P943 in humans. J Cereb Blood Flow Metab. 2010;30:196–210. doi: 10.1038/jcbfm.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, Houle S, Kapur S, Wilson AA. Binding characteristics and sensitivity to endogenous dopamine of [11C]-(+)-PHNO, a new agonist radiotracer for imaging the high-affinity state of D2 receptors in vivo using positron emission tomography. J Neurochem. 2006;97:1089–1103. doi: 10.1111/j.1471-4159.2006.03840.x. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Wilson AA, Meyer JH, Hussey D, Houle S. [11C]-DASB, a tool for in vivo measurement of SSRI-induced occupancy of the serotonin transporter: PET characterization and evaluation in cats. Synapse. 2003;47:123–133. doi: 10.1002/syn.10155. [DOI] [PubMed] [Google Scholar]
- Giovacchini G, Lang L, Ma Y, Herscovitch P, Eckelman WC, Carson RE. Differential effects of paroxetine on raphe and cortical 5-HT1A binding: a PET study in monkeys. Neuroimage. 2005;28:238–248. doi: 10.1016/j.neuroimage.2005.05.042. [DOI] [PubMed] [Google Scholar]
- Gozlan H, Thibault S, Laporte AM, Lima L, Hamon M. The selective 5-HT1A antagonist radioligand [3H]WAY 100635 labels both G-protein-coupled and free 5-HT1A receptors in rat brain membranes. Eur J Pharmacol. 1995;288:173–186. doi: 10.1016/0922-4106(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Granas C, Nordquist J, Mohell N, Larhammar D. Site-directed mutagenesis of the 5-HT1B receptor increases the affinity of 5-HT for the agonist low-affinity conformation and reduces the intrinsic activity of 5-HT. Eur J Pharmacol. 2001;421:69–76. doi: 10.1016/s0014-2999(01)01027-5. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Paradoxical trafficking and regulation of 5-HT(2A) receptors by agonists and antagonists. Brain Res Bull. 2001;56:441–451. doi: 10.1016/s0361-9230(01)00623-2. [DOI] [PubMed] [Google Scholar]
- Guo N, Guo W, Kralikova M, Jiang M, Schieren I, Narendran R, Slifstein M, Abi-Dargham A, Laruelle M, Javitch JA, Rayport S. Impact of D2 receptor internalization on binding affinity of neuroimaging radiotracers. Neuropsychopharmacology. 2010;35:806–817. doi: 10.1038/npp.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler F, Kuznetsova OF, Krasikova RN, Cservenyak T, Quednow BB, Vollenweider FX, Ametamey SM, Westera G. GMP-compliant radiosynthesis of [18F]altanserin and human plasma metabolite studies. Appl Radiat Isot. 2009;67:598–601. doi: 10.1016/j.apradiso.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Hensler JG, Truett KA. Effect of chronic serotonin-2 receptor agonist or antagonist administration on serotonin-1A receptor sensitivity. Neuropsychopharmacology. 1998;19:354–364. doi: 10.1016/S0893-133X(98)00037-2. [DOI] [PubMed] [Google Scholar]
- Hirani E, Opacka-Juffry J, Gunn R, Khan I, Sharp T, Hume S. Pindolol occupancy of 5-HT(1A) receptors measured in vivo using small animal positron emission tomography with carbon-11 labeled WAY 100635. Synapse. 2000;36:330–341. doi: 10.1002/(SICI)1098-2396(20000615)36:4<330::AID-SYN10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Hirani E, Sharp T, Sprakes M, Grasby P, Hume S. Fenfluramine evokes 5-HT2A receptor-mediated responses but does not displace [11C]MDL 100907: small animal PET and gene expression studies. Synapse. 2003;50:251–260. doi: 10.1002/syn.10268. [DOI] [PubMed] [Google Scholar]
- Hirst WD, Minton JA, Bromidge SM, Moss SF, Latter AJ, Riley G, Routledge C, Middlemiss DN, Price GW. Characterization of [(125)I]-SB-258585 binding to human recombinant and native 5-HT(6) receptors in rat, pig and human brain tissue. Br J Pharmacol. 2000;130:1597–1605. doi: 10.1038/sj.bjp.0703458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthoff VA, Koeppe RA, Frey KA, Paradise AH, Kuhl DE. Differentiation of radioligand delivery and binding in the brain: validation of a two-compartment model for [11C]flumazenil. J Cereb Blood Flow Metab. 1991;11:745–752. doi: 10.1038/jcbfm.1991.131. [DOI] [PubMed] [Google Scholar]
- Hu J, Henry S, Gallezot JD, Ropchan J, Neumaier JF, Potenza MN, Sinha R, Krystal JH, Huang Y, Ding YS, Carson RE, Neumeister A. Serotonin 1B receptor imaging in alcohol dependence. Biol Psychiatry. 2010;67:800–803. doi: 10.1016/j.biopsych.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Ichiwaka J, Li Z, Dai J, Meltzer HY. Augmentation by citalopram of risperidone-induced monoamine release in rat prefrontal cortex. Psychopharmacology (Berl) 2006;185:274–281. doi: 10.1007/s00213-005-0206-1. [DOI] [PubMed] [Google Scholar]
- Huiban M, Passchier J, Martarello L, Cunningham VJ, Jakobsen S, Gee AD.2006[11C]GSK224558 as a potential PET ligand for the delineation of 5-HT6 receptors Neuroimage 31T12–T43.16858762 [Google Scholar]
- Hume S, Hirani E, Opacka-Juffry J, Myers R, Townsend C, Pike V, Grasby P. Effect of 5-HT on binding of [(11)C] WAY 100635 to 5-HT(IA) receptors in rat brain, assessed using in vivo microdialysis nd PET after fenfluramine. Synapse. 2001;41:150–159. doi: 10.1002/syn.1069. [DOI] [PubMed] [Google Scholar]
- Hyttel J. Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs) Int Clin Psychopharmacol. 1994;9 (Suppl 1:19–26. doi: 10.1097/00004850-199403001-00004. [DOI] [PubMed] [Google Scholar]
- Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- Jagoda EM, Lang L, Tokugawa J, Simmons A, Ma Y, Contoreggi C, Kiesewetter D, Eckelman WC. Development of 5-HT1A receptor radioligands to determine receptor density and changes in endogenous 5-HT. Synapse. 2006;59:330–341#. doi: 10.1002/syn.20246. [DOI] [PubMed] [Google Scholar]
- Janoshazi A, Deraet M, Callebert J, Setola V, Guenther S, Saubamea B, Manivet P, Launay JM, Maroteaux L. Modified receptor internalization upon coexpression of 5-HT1B receptor and 5-HT2B receptors. Mol Pharmacol. 2007;71:1463–1474. doi: 10.1124/mol.106.032656. [DOI] [PubMed] [Google Scholar]
- Kalbitzer J, Svarer C, Frokjaer VG, Erritzoe D, Baare WF, Madsen J, Hasselbalch SG, Knudsen GM. A probabilistic approach to delineating functional brain regions. J Nucl Med Technol. 2009;37:91–95. doi: 10.2967/jnmt.108.054056. [DOI] [PubMed] [Google Scholar]
- Khawaja X, Evans N, Reilly Y, Ennis C, Minchin MC. Characterisation of the binding of [3H]WAY-100635, a novel 5-hydroxytryptamine1A receptor antagonist, to rat brain. J Neurochem. 1995;64:2716–2726. doi: 10.1046/j.1471-4159.1995.64062716.x. [DOI] [PubMed] [Google Scholar]
- Knudsen GM. Testing for radioligand sensitivity to endogenous neurotransmitter release. Eur J Nucl Med Mol Imaging. 2009;36:472–474. doi: 10.1007/s00259-009-1067-3. [DOI] [PubMed] [Google Scholar]
- Koenig JA, Edwardson JM. Endocytosis and recycling of G protein-coupled receptors. Trends Pharmacol Sci. 1997;18:276–287. doi: 10.1016/s0165-6147(97)01091-2. [DOI] [PubMed] [Google Scholar]
- Kristiansen H, Elfving B, Plenge P, Pinborg LH, Gillings N, Knudsen GM. Binding characteristics of the 5-HT2A receptor antagonists altanserin and MDL 100907. Synapse. 2005;58:249–257. doi: 10.1002/syn.20205. [DOI] [PubMed] [Google Scholar]
- Kumar JS, Mann JJ. PET tracers for 5-HT(1A) receptors and uses thereof. Drug Discov Today. 2007;12:748–756. doi: 10.1016/j.drudis.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Kumar JS, Milak MS, Prabhakaran J, Majo VJ, Simpson NR, Jurewicz A, Savenkoval L, Mann JJ, Parsey RV.2008[11C]CUMI-101, an agonist radiotracer for imaging the 5-HT1A receptor in vivo in human with positron emission tomography J Nucl Med 49(Suppl 179P18077526 [Google Scholar]
- Larisch R, Klimke A, Hamacher K, Henning U, Estalji S, Hohlfeld T, Vosberg H, Tosch M, Gaebel W, Coenen HH, Muller-Gartner HW. Influence of synaptic serotonin level on [18F]altanserin binding to 5HT2 receptors in man. Behav Brain Res. 2003;139:21–29. doi: 10.1016/s0166-4328(01)00412-0. [DOI] [PubMed] [Google Scholar]
- Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423–451. doi: 10.1097/00004647-200003000-00001. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Iyer RN, al-Tikriti MS, Zea-Ponce Y, Malison R, Zoghbi SS, Baldwin RM, Kung HF, Charney DS, Hoffer PB, Innis RB, Bradberry CW. Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse. 1997;25:1–14. doi: 10.1002/(SICI)1098-2396(199701)25:1<1::AID-SYN1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Lemoine L, Verdurand M, Vacher B, Blanc E, Le Bars D, Newman-Tancredi A, Zimmer L. [(18)F]F15599, a novel 5-HT(1A) receptor agonist, as a radioligand for PET neuroimaging. Eur J Nucl Med Mol Imaging. 2009;37:594–605. doi: 10.1007/s00259-009-1274-y. [DOI] [PubMed] [Google Scholar]
- Leonhardt S, Gorospe E, Hoffman BJ, Teitler M. Molecular pharmacological differences in the interaction of serotonin with 5-hydroxytryptamine1C and 5-hydroxytryptamine2 receptors. Mol Pharmacol. 1992;42:328–335. [PubMed] [Google Scholar]
- Logan J, Alexoff D, Kriplani A. Simplifications in analyzing positron emission tomography data: effects on outcome measures. Nucl Med Biol. 2007;34:743–756. doi: 10.1016/j.nucmedbio.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Logan J, Dewey SL, Wolf AP, Fowler JS, Brodie JD, Angrist B, Volkow ND, Gatley SJ. Effects of endogenous dopamine on measures of [18F]N-methylspiroperidol binding in the basal ganglia: comparison of simulations and experimental results from PET studies in baboons. Synapse. 1991;9:195–207. doi: 10.1002/syn.890090306. [DOI] [PubMed] [Google Scholar]
- Lopez-Gimenez JF, Villazon M, Brea J, Loza MI, Palacios JM, Mengod G, Vilaro MT. Multiple conformations of native and recombinant human 5-hydroxytryptamine(2a) receptors are labeled by agonists and discriminated by antagonists. Mol Pharmacol. 2001;60:690–699. [PubMed] [Google Scholar]
- Lundquist P, Roman M, Syvanen S, Hartvig P, Blomquist G, Hammarlund-Udenaes M, Langstrom B. Effect on [11C]DASB binding after tranylcypromine-induced increase in serotonin concentration: positron emission tomography studies in monkeys and rats. Synapse. 2007;61:440–449. doi: 10.1002/syn.20382. [DOI] [PubMed] [Google Scholar]
- Lundquist P, Wilking H, Hoglund AU, Sandell J, Bergstrom M, Hartvig P, Langstrom B. Potential of [11C]DASB for measuring endogenous serotonin with PET: binding studies. Nucl Med Biol. 2005;32:129–136. doi: 10.1016/j.nucmedbio.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Maeda J, Suhara T, Ogawa M, Okauchi T, Kawabe K, Zhang MR, Semba J, Suzuki K. In vivo binding properties of [carbonyl-11C]WAY-100635: effect of endogenous serotonin. Synapse. 2001;40:122–129. doi: 10.1002/syn.1033. [DOI] [PubMed] [Google Scholar]
- Mannoury la Cour C, El Mestikawy S, Hanoun N, Hamon M, Lanfumey L. Regional differences in the coupling of 5-hydroxytryptamine-1A receptors to G proteins in the rat brain. Mol Pharmacol. 2006;70:1013–1021. doi: 10.1124/mol.106.022756. [DOI] [PubMed] [Google Scholar]
- Marner L, Gillings N, Comley RA, Baare WF, Rabiner EA, Wilson AA, Houle S, Hasselbalch SG, Svarer C, Gunn RN, Laruelle M, Knudsen GM. Kinetic modeling of 11C-SB207145 binding to 5-HT4 receptors in the human brain in vivo. J Nucl Med. 2009;50:900–908. doi: 10.2967/jnumed.108.058552. [DOI] [PubMed] [Google Scholar]
- Marner L, Gillings N, Madsen K, Erritzoe D, Baare WF, Svarer C, Hasselbalch SG, Knudsen GM. Brain imaging of serotonin 4 receptors in humans with [11C]SB207145-PET. Neuroimage. 2010;50:855–861. doi: 10.1016/j.neuroimage.2010.01.054. [DOI] [PubMed] [Google Scholar]
- Martarello L, Parker C, Cunningham VJ, Searle GE, Rabiner EA, Gee AD, Davy M, Laruelle M.2008First evaluation in humans of [11C]GSK215083 as a probe to image the 5-HT6 receptors J Nucl Med 49(Suppl 179P18077526 [Google Scholar]
- Matusch A, Hurlemann R, Rota Kops E, Winz OH, Elmenhorst D, Herzog H, Zilles K, Bauer A. Acute S-ketamine application does not alter cerebral [18F]altanserin binding: a pilot PET study in humans. J Neural Transm. 2007;114:1433–1442. doi: 10.1007/s00702-007-0751-3. [DOI] [PubMed] [Google Scholar]
- McCarron JA, Pike VW, Halldin C, Sandell J, Sovago J, Gulyas B, Cselenyi Z, Wikstrom HV, Marchais-Oberwinkler S, Nowicki B, Dolle F, Farde L. The pyridinyl-6 position of WAY-100635 as a site for radiofluorination–effect on 5-HT1A receptor radioligand behavior in vivo. Mol Imaging Biol. 2004;6:17–26. doi: 10.1016/j.mibio.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Meyer JH, Cho R, Kennedy S, Kapur S. The effects of single dose nefazodone and paroxetine upon 5-HT2A binding potential in humans using [18F]-setoperone PET. Psychopharmacology (Berl) 1999;144:279–281. doi: 10.1007/s002130051004. [DOI] [PubMed] [Google Scholar]
- Milak MS, Kumar JSD, Prabhakaran J, Majo VJ, Mann JJ, Parsey RV. [11C]CUMI-101, an agonist 5-HT1A PET tracer in human: preliminary test-retest data. Neuroimage. 2008a;41 (Suppl 2:T96. [Google Scholar]
- Milak MS, Kumar JSD, Severance AJ, Ogden RT, Prabhakaran J, Majo VJ, Mann JJ, Parsey RV. Altered binding in baboons of [11C]CUMI-101, a 5-HT1A agonist PET tracer, in response to intravenous citalopram. Neuroimage. 2008b;41 (Suppl 2:T162. [Google Scholar]
- Milak MS, Ogden RT, Vinocur DN, Van Heertum RL, Cooper TB, Mann JJ, Parsey RV. Effects of tryptophan depletion on the binding of [11C]-DASB to the serotonin transporter in baboons: response to acute serotonin deficiency. Biol Psychiatry. 2005;57:102–106. doi: 10.1016/j.biopsych.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Milak MS, Severance AJ, Ogden RT, Prabhakaran J, Kumar JS, Majo VJ, Mann JJ, Parsey RV. Modeling considerations for 11C-CUMI-101, an agonist radiotracer for imaging serotonin 1A receptor in vivo with PET. J Nucl Med. 2008c;49:587–596. doi: 10.2967/jnumed.107.046540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Gobert A, Lejeune F, Newman-Tancredi A, Rivet JM, Auclair A, Peglion JL. S33005, a novel ligand at both serotonin and norepinephrine transporters: I. Receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. J Pharmacol Exp Ther. 2001;298:565–580. [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Lochon S, Touzard M, Aubry S, Audinot V. Specific labelling of serotonin 5-HT(1B) receptors in rat frontal cortex with the novel, phenylpiperazine derivative, [3H]GR125,743. A pharmacological characterization. Pharmacol Biochem Behav. 2002;71:589–598. doi: 10.1016/s0091-3057(01)00716-x. [DOI] [PubMed] [Google Scholar]
- Miller KJ, Teitler M. Quantitative autoradiography of 5-CT-sensitive (5-HT1D) and 5-CT-insensitive (5-HT1E) serotonin receptors in human brain. Neurosci Lett. 1992;136:223–226. doi: 10.1016/0304-3940(92)90054-b. [DOI] [PubMed] [Google Scholar]
- Mukaida K, Shichino T, Koyanagi S, Himukashi S, Fukuda K. Activity of the serotonergic system during isoflurane anesthesia. Anesth Analg. 2007;104:836–839. doi: 10.1213/01.ane.0000255200.42574.22. [DOI] [PubMed] [Google Scholar]
- Murrough JW, Henry S, Hu J, Gallezot JD, Planeta-Wilson B, Neumaier JF, Neumeister A.2010Reduced ventral striatal/ventral pallidal serotonin(1B) receptor binding potential in major depressive disorder Psychopharmacol(in press) [DOI] [PMC free article] [PubMed]
- Nabulsi N, Huang Y, Weinzimmer D, Ropchan J, Frost JJ, McCarthy T, Carson RE, Ding YS. High-resolution imaging of brain 5-HT 1B receptors in the rhesus monkey using [11C]P943. Nucl Med Biol. 2010;37:205–214. doi: 10.1016/j.nucmedbio.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendran R, Hwang DR, Slifstein M, Talbot PS, Erritzoe D, Huang Y, Cooper TB, Martinez D, Kegeles LS, Abi-Dargham A, Laruelle M. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (−)-N-[11C]propyl-norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]-raclopride. Synapse. 2004;52:188–208. doi: 10.1002/syn.20013. [DOI] [PubMed] [Google Scholar]
- Narendran R, Martinez D. Cocaine abuse and sensitization of striatal dopamine transmission: a critical review of the preclinical and clinical imaging literature. Synapse. 2008;62:851–869. doi: 10.1002/syn.20566. [DOI] [PubMed] [Google Scholar]
- Nenonene EK, Radja F, Carli M, Grondin L, Reader TA. Heterogeneity of cortical and hippocampal 5-HT1A receptors: a reappraisal of homogenate binding with 8-[3H]hydroxydipropylaminotetralin. J Neurochem. 1994;62:1822–1834. doi: 10.1046/j.1471-4159.1994.62051822.x. [DOI] [PubMed] [Google Scholar]
- Nishiyama S, Tsukada H, Sato K, Kakiuchi T, Ohba H, Harada N, Takahashi K. Evaluation of PET ligands (+)N-[(11)C]ethyl-3-piperidyl benzilate and (+)N-[(11)C]propyl-3-piperidyl benzilate for muscarinic cholinergic receptors: a PET study with microdialysis in comparison with (+)N-[(11)C]methyl-3-piperidyl benzilate in the conscious monkey brain. Synapse. 2001;40:159–169. doi: 10.1002/syn.1038. [DOI] [PubMed] [Google Scholar]
- Offord SJ, Ordway GA, Frazer A. Application of [125I]iodocyanopindolol to measure 5-hydroxytryptamine1B receptors in the brain of the rat. J Pharmacol Exp Ther. 1988;244:144–153. [PubMed] [Google Scholar]
- Parker CA, Cunningham VJ, Martarello L, Rabiner EA, Searle GE, Gee AD, Davy M, Johnson CN, Ahmed M, Gunn RN, Laruelle M. Evaluation of the novel 5-HT6 receptor radioligand, [11C]GSK215083 in human. Neuroimage. 2008;41 (Suppl 2:T20. [Google Scholar]
- Passchier J, van Waarde A, Pieterman RM, Elsinga PH, Pruim J, Hendrikse HN, Willemsen AT, Vaalburg W. In vivo delineation of 5-HT1A receptors in human brain with [18F]MPPF. J Nucl Med. 2000;41:1830–1835. [PubMed] [Google Scholar]
- Pierson ME, Andersson J, Nyberg S, McCarthy DJ, Finnema SJ, Varnas K, Takano A, Karlsson P, Gulyas B, Medd AM, Lee CM, Powell ME, Heys JR, Potts W, Seneca N, Mrzljak L, Farde L, Halldin C. [11C]AZ10419369: a selective 5-HT1B receptor radioligand suitable for positron emission tomography (PET). Characterization in the primate brain. Neuroimage. 2008;41:1075–1085. doi: 10.1016/j.neuroimage.2008.02.063. [DOI] [PubMed] [Google Scholar]
- Pike VW. Positron-emitting radioligands for studies in vivo—probes for human psychopharmacology. J Psychopharmacol. 1993;7:139–158. doi: 10.1177/026988119300700202. [DOI] [PubMed] [Google Scholar]
- Pinborg LH, Adams KH, Yndgaard S, Hasselbalch SG, Holm S, Kristiansen H, Paulson OB, Knudsen GM. [18F]altanserin binding to human 5HT2A receptors is unaltered after citalopram and pindolol challenge. J Cereb Blood Flow Metab. 2004;24:1037–1045. doi: 10.1097/01.WCB.0000126233.08565.E7. [DOI] [PubMed] [Google Scholar]
- Portas CM, Bjorvatn B, Fagerland S, Gronli J, Mundal V, Sorensen E, Ursin R. On-line detection of extracellular levels of serotonin in dorsal raphe nucleus and frontal cortex over the sleep/wake cycle in the freely moving rat. Neuroscience. 1998;83:807–814. doi: 10.1016/s0306-4522(97)00438-7. [DOI] [PubMed] [Google Scholar]
- Praschak-Rieder N, Hussey D, Wilson AA, Carella A, Lee M, Dunn E, Willeit M, Bagby RM, Houle S, Meyer JH. Tryptophan depletion and serotonin loss in selective serotonin reuptake inhibitor-treated depression: an [(18)F] MPPF positron emission tomography study. Biol Psychiatry. 2004;56:587–591. doi: 10.1016/j.biopsych.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Praschak-Rieder N, Wilson AA, Hussey D, Carella A, Wei C, Ginovart N, Schwarz MJ, Zach J, Houle S, Meyer JH. Effects of tryptophan depletion on the serotonin transporter in healthy humans. Biol Psychiatry. 2005;58:825–830. doi: 10.1016/j.biopsych.2005.04.038. [DOI] [PubMed] [Google Scholar]
- Quelch DR, Withey SL, Nutt DJ, Parker CA, Tyacke RJ. Species comparison of the binding properties for three D2/D3-DAR radioligands widely used in PET imaging. J Psychopharmacol. 2010;24 (suppl) [Google Scholar]
- Rabiner EA, Messa C, Sargent PA, Husted-Kjaer K, Montgomery A, Lawrence AD, Bench CJ, Gunn RN, Cowen P, Grasby PM. A database of [(11)C]WAY-100635 binding to 5-HT(1A) receptors in normal male volunteers: normative data and relationship to methodological, demographic, physiological, and behavioral variables. Neuroimage. 2002;15:620–632. doi: 10.1006/nimg.2001.0984. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Giovanetti E, Qian Y, Blakely RD. Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J Biol Chem. 1998;273:2458–2466. doi: 10.1074/jbc.273.4.2458. [DOI] [PubMed] [Google Scholar]
- Rbah L, Leviel V, Zimmer L. Displacement of the PET ligand 18F-MPPF by the electrically evoked serotonin release in the rat hippocampus. Synapse. 2003;49:239–245. doi: 10.1002/syn.10235. [DOI] [PubMed] [Google Scholar]
- Riad M, Watkins KC, Doucet E, Hamon M, Descarries L. Agonist-induced internalization of serotonin-1a receptors in the dorsal raphe nucleus (autoreceptors) but not hippocampus (heteroreceptors) J Neurosci. 2001;21:8378–8386. doi: 10.1523/JNEUROSCI.21-21-08378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riad M, Zimmer L, Rbah L, Watkins KC, Hamon M, Descarries L. Acute treatment with the antidepressant fluoxetine internalizes 5-HT1A autoreceptors and reduces the in vivo binding of the PET radioligand [18F]MPPF in the nucleus raphe dorsalis of rat. J Neurosci. 2004;24:5420–5426. doi: 10.1523/JNEUROSCI.0950-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice OV, Gatley SJ, Shen J, Huemmer CL, Rogoz R, DeJesus OT, Volkow ND, Gifford AN. Effects of endogenous neurotransmitters on the in vivo binding of dopamine and 5-HT radiotracers in mice. Neuropsychopharmacology. 2001;25:679–689. doi: 10.1016/S0893-133X(01)00287-1. [DOI] [PubMed] [Google Scholar]
- Rosel P, Arranz B, San L, Vallejo J, Crespo JM, Urretavizcaya M, Navarro MA. Altered 5-HT(2A) binding sites and second messenger inositol trisphosphate (IP(3)) levels in hippocampus but not in frontal cortex from depressed suicide victims. Psychiatry Res. 2000;99:173–181. doi: 10.1016/s0925-4927(00)00076-7. [DOI] [PubMed] [Google Scholar]
- Rosel P, Arranz B, Vallejo J, Oros M, Menchon JM, Alvarez P, Navarro MA. High affinity [3H]imipramine and [3H]paroxetine binding sites in suicide brains. J Neural Transm. 1997;104:921–929. doi: 10.1007/BF01285560. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH. Serotonin releasing agents. Neurochemical, therapeutic and adverse effects. Pharmacol Biochem Behav. 2002;71:825–836. doi: 10.1016/s0091-3057(01)00669-4. [DOI] [PubMed] [Google Scholar]
- Ruiz J, Gabilondo AM, Meana JJ, Garcia-Sevilla JA. Increased [3H] raclopride binding sites in postmortem brains from schizophrenic violent suicide victims. Psychopharmacology (Berl) 1992;109:410–414. doi: 10.1007/BF02247716. [DOI] [PubMed] [Google Scholar]
- Sandell J, Halldin C, Pike V, Chou Y, Varnas K, Hall H, Marchais S, Nowicki B, Wikstrom H, Swahn C, Farde L. New halogenated [11C]WAY analogues, [11C]6FPWAY and [11C]6BPWAY–radiosynthesis and assessment as radioligands for the study of 5-HT1A receptors in living monkey. Nucl Med Biol. 2001;28:177–185. doi: 10.1016/s0969-8051(00)00181-5. [DOI] [PubMed] [Google Scholar]
- Sari Y. Serotonin1B receptors: from protein to physiological function and behavior. Neurosci Biobehav Rev. 2004;28:565–582. doi: 10.1016/j.neubiorev.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc Natl Acad Sci USA. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DJ, Stohler CS, Koeppe RA, Zubieta JK. Time-course of change in [11C]carfentanil and [11C]raclopride binding potential after a nonpharmacological challenge. Synapse. 2007;61:707–714. doi: 10.1002/syn.20404. [DOI] [PubMed] [Google Scholar]
- Seeman P, Guan HC, Niznik HB. Endogenous dopamine lowers the dopamine D2 receptor density as measured by [3H]raclopride: implications for positron emission tomography of the human brain. Synapse. 1989;3:96–97. doi: 10.1002/syn.890030113. [DOI] [PubMed] [Google Scholar]
- Seeman P, Kapur S. Anesthetics inhibit high-affinity states of dopamine D2 and other G-linked receptors. Synapse. 2003;50:35–40. doi: 10.1002/syn.10221. [DOI] [PubMed] [Google Scholar]
- Sherry RL, Rauw G, McKenna KF, Paetsch PR, Coutts RT, Baker GB. Failure to detect amphetamine or 1-amino-3-phenylpropane in humans or rats receiving the MAO inhibitor tranylcypromine. J Affect Disord. 2000;61:23–29. doi: 10.1016/s0165-0327(99)00188-3. [DOI] [PubMed] [Google Scholar]
- Sibley DR, De Lean A, Creese I. Anterior pituitary dopamine receptors. Demonstration of interconvertible high and low affinity states of the D-2 dopamine receptor. J Biol Chem. 1982;257:6351–6361. [PubMed] [Google Scholar]
- Sibon I, Benkelfat C, Gravel P, Aznavour N, Costes N, Mzengeza S, Booij L, Baker G, Soucy JP, Zimmer L, Descarries L. Decreased [(18)F]MPPF binding potential in the dorsal raphe nucleus after a single oral dose of fluoxetine: a positron-emission tomography study in healthy volunteers. Biol Psychiatry. 2008;63:1135–1140. doi: 10.1016/j.biopsych.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Skaddan MB, Jewett DM, Sherman PS, Kilbourn MR. (R)-N-[11C]methyl-3-pyrrolidyl benzilate, a high-affinity reversible radioligand for PET studies of the muscarinic acetylcholine receptor. Synapse. 2002;45:31–37. doi: 10.1002/syn.10079. [DOI] [PubMed] [Google Scholar]
- Skinbjerg M, Liow JS, Seneca N, Hong J, Lu S, Thorsell A, Heilig M, Pike VW, Halldin C, Sibley DR, Innis RB. D2 dopamine receptor internalization prolongs the decrease of radioligand binding after amphetamine: a PET study in a receptor internalization-deficient mouse model. Neuroimage. 2010;50:1402–1407. doi: 10.1016/j.neuroimage.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleight AJ, Stam NJ, Mutel V, Vanderheyden PM. Radiolabelling of the human 5-HT2A receptor with an agonist, a partial agonist and an antagonist: effects on apparent agonist affinities. Biochem Pharmacol. 1996;51:71–76. doi: 10.1016/0006-2952(95)02122-1. [DOI] [PubMed] [Google Scholar]
- Staley JK, Van Dyck CH, Tan PZ, Al Tikriti M, Ramsby Q, Klump H, Ng C, Garg P, Soufer R, Baldwin RM, Innis RB. Comparison of [(18)F]altanserin and [(18)F]deuteroaltanserin for PET imaging of serotonin(2A) receptors in baboon brain: pharmacological studies. Nucl Med Biol. 2001;28:271–279. doi: 10.1016/s0969-8051(00)00212-2. [DOI] [PubMed] [Google Scholar]
- Stanley M, Virgilio J, Gershon S. Tritiated imipramine binding sites are decreased in the frontal cortex of suicides. Science. 1982;216:1337–1339. doi: 10.1126/science.7079769. [DOI] [PubMed] [Google Scholar]
- Talbot PS, Frankle WG, Hwang DR, Huang Y, Suckow RF, Slifstein M, Abi-Dargham A, Laruelle M. Effects of reduced endogenous 5-HT on the in vivo binding of the serotonin transporter radioligand 11C-DASB in healthy humans. Synapse. 2005;55:164–175. doi: 10.1002/syn.20105. [DOI] [PubMed] [Google Scholar]
- Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- Thomas DR, Atkinson PJ, Hastie PG, Roberts JC, Middlemiss DN, Price GW. [3H]-SB-269970 radiolabels 5-HT7 receptors in rodent, pig and primate brain tissues. Neuropharmacology. 2002;42:74–81. doi: 10.1016/s0028-3908(01)00151-4. [DOI] [PubMed] [Google Scholar]
- Tokugawa J, Ravasi L, Nakayama T, Lang L, Schmidt KC, Seidel J, Green MV, Sokoloff L, Eckelman WC. Distribution of the 5-HT(1A) receptor antagonist [(18)F]FPWAY in blood and brain of the rat with and without isoflurane anesthesia. Eur J Nucl Med Mol Imaging. 2007;34:259–266. doi: 10.1007/s00259-006-0228-x. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Tricklebank M, Middlemiss D.2004Hot topic: subtypes of the 5HT receptor Curr Drug Targets CNS Neurol Disord 31–90.14965240 [Google Scholar]
- Tsukada H, Nishiyama S, Fukumoto D, Ohba H, Sato K, Kakiuchi T. Effects of acute acetylcholinesterase inhibition on the cerebral cholinergic neuronal system and cognitive function: functional imaging of the conscious monkey brain using animal PET in combination with microdialysis. Synapse. 2004;52:1–10. doi: 10.1002/syn.10310. [DOI] [PubMed] [Google Scholar]
- Udo de Haes JI, Bosker FJ, Van Waarde A, Pruim J, Willemsen AT, Vaalburg W, Den Boer JA. 5-HT(1A) receptor imaging in the human brain: effect of tryptophan depletion and infusion on [(18)F]MPPF binding. Synapse. 2002;46:108–115. doi: 10.1002/syn.10134. [DOI] [PubMed] [Google Scholar]
- Udo de Haes JI, Cremers TI, Bosker FJ, Postema F, Tiemersma-Wegman TD, den Boer JA. Effect of increased serotonin levels on [18F]MPPF binding in rat brain: fenfluramine vs the combination of citalopram and ketanserin. Neuropsychopharmacology. 2005;30:1624–1631. doi: 10.1038/sj.npp.1300721. [DOI] [PubMed] [Google Scholar]
- Udo de Haes JI, Harada N, Elsinga PH, Maguire RP, Tsukada H. Effect of fenfluramine-induced increases in serotonin release on [18F]MPPF binding: a continuous infusion PET study in conscious monkeys. Synapse. 2006;59:18–26. doi: 10.1002/syn.20209. [DOI] [PubMed] [Google Scholar]
- Varnas K, Hall H, Bonaventure P, Sedvall G. Autoradiographic mapping of 5-HT(1B) and 5-HT(1D) receptors in the post mortem human brain using [(3)H]GR 125743. Brain Res. 2001;915:47–57. doi: 10.1016/s0006-8993(01)02823-2. [DOI] [PubMed] [Google Scholar]
- Varnas K, Nyberg S, Halldin C, Varrone A, Takano A, Karlsson P, Andersson J, McCarthy D, Smith M, Pierson ME, Soderstrom J, Farde L.2010Quantitative analysis of [(11)C]AZ10419369 binding to 5-HT(1B) receptors in human brain J Cereb Blood Flow Metab(in press) [DOI] [PMC free article] [PubMed]
- von Euler G, Mailleux P, Vanderhaeghen JJ, Fuxe K. Neurotensin reduces the affinity of dopamine D2 receptors in membranes from post mortem human caudate-putamen. Neurosci Lett. 1990;109:325–330. doi: 10.1016/0304-3940(90)90016-3. [DOI] [PubMed] [Google Scholar]
- Wainscott DB, Krushinski JH, Jr, Audia JE, Schaus JM, Zgombick JM, Lucaites VL, Nelson DL. [3H]LY334370, a novel radioligand for the 5-HT1F receptor. I. In vitro characterization of binding properties. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:169–177. doi: 10.1007/s00210-005-1035-9. [DOI] [PubMed] [Google Scholar]
- Willeit M, Ginovart N, Graff A, Rusjan P, Vitcu I, Houle S, Seeman P, Wilson AA, Kapur S. First human evidence of d-amphetamine induced displacement of a D2/3 agonist radioligand: a [11C]-(+)-PHNO positron emission tomography study. Neuropsychopharmacology. 2008;33:279–289. doi: 10.1038/sj.npp.1301400. [DOI] [PubMed] [Google Scholar]
- Williams WA, Shoaf SE, Hommer D, Rawlings R, Linnoila M. Effects of acute tryptophan depletion on plasma and cerebrospinal fluid tryptophan and 5-hydroxyindoleacetic acid in normal volunteers. J Neurochem. 1999;72:1641–1647. doi: 10.1046/j.1471-4159.1999.721641.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Onoe H, Tsukada H, Watanabe Y. Effects of increased endogenous serotonin on the in vivo binding of [11C]DASB to serotonin transporters in conscious monkey brain. Synapse. 2007;61:724–731. doi: 10.1002/syn.20422. [DOI] [PubMed] [Google Scholar]
- Yatham LN, Liddle PF, Shiah IS, Lam RW, Adam MJ, Zis AP, Ruth TJ. Effects of rapid tryptophan depletion on brain 5-HT(2) receptors: a PET study. Br J Psychiatry. 2001;178:448–453. doi: 10.1192/bjp.178.5.448. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Aronson JK, Blau K, Green AR, Grahame-Smith DG. Tranylcypromine (‘Parnate') overdose: measurement of tranylcypromine concentrations and MAO inhibitory activity and identification of amphetamines in plasma. Psychol Med. 1979;9:377–382. doi: 10.1017/s0033291700030890. [DOI] [PubMed] [Google Scholar]
- Zahniser NR, Doolen S. Chronic and acute regulation of Na+/Cl− -dependent neurotransmitter transporters: drugs, substrates, presynaptic receptors, and signaling systems. Pharmacol Ther. 2001;92:21–55. doi: 10.1016/s0163-7258(01)00158-9. [DOI] [PubMed] [Google Scholar]
- Zetterstrom T, Sharp T, Marsden CA, Ungerstedt U. In vivo measurement of dopamine and its metabolites by intracerebral dialysis: changes after d-amphetamine. J Neurochem. 1983;41:1769–1773. doi: 10.1111/j.1471-4159.1983.tb00893.x. [DOI] [PubMed] [Google Scholar]
- Zimmer L, Mauger G, Le Bars D, Bonmarchand G, Luxen A, Pujol JF. Effect of endogenous serotonin on the binding of the 5-hT1A PET ligand 18F-MPPF in the rat hippocampus: kinetic beta measurements combined with microdialysis. J Neurochem. 2002;80:278–286. doi: 10.1046/j.0022-3042.2001.00696.x. [DOI] [PubMed] [Google Scholar]
- Zimmer L, Rbah L, Giacomelli F, Le Bars D, Renaud B. A reduced extracellular serotonin level increases the 5-HT1A PET ligand 18F-MPPF binding in the rat hippocampus. J Nucl Med. 2003;44:1495–1501. [PubMed] [Google Scholar]
- Zimmer L, Riad M, Rbah L, Belkacem-Kahlouli A, Le Bars D, Renaud B, Descarries L. Toward brain imaging of serotonin 5-HT1A autoreceptor internalization. Neuroimage. 2004;22:1421–1426. doi: 10.1016/j.neuroimage.2004.03.020. [DOI] [PubMed] [Google Scholar]