

Abstract
The Ca2+-sensing receptor (CaR) regulates salt and water transport in the kidney as demonstrated by the association of gain of function CaR mutations with a Bartter syndrome-like, salt-wasting phenotype, but the precise mechanism for this effect is not fully established. We found previously that the CaR interacts with and inactivates an inwardly rectifying K+ channel, Kir4.1, which is expressed in the distal nephron that contributes to the basolateral K+ conductance, and in which loss of function mutations are associated with a complex phenotype that includes renal salt wasting. We now find that CaR inactivates Kir4.1 by reducing its cell surface expression. Mutant CaRs reduced Kir4.1 cell surface expression and current density in HEK-293 cells in proportion to their signaling activity. Mutant, activated Gαq reduced cell surface expression and current density of Kir4.1, and these effects were blocked by RGS4, a protein that blocks signaling via Gαi and Gαq. Other α subunits had insignificant effects. Knockdown of caveolin-1 blocked the effect of Gαq on Kir4.1, whereas knockdown of the clathrin heavy chain had no effect. CaR had no comparable effect on the renal outer medullary K+ channel, an apical membrane distal nephron K+ channel that is internalized by clathrin-coated vesicles. Co-immunoprecipitation studies showed that the CaR and Kir4.1 physically associate with caveolin-1 in HEK cells and in kidney extracts. Thus, the CaR decreases cell surface expression of Kir4.1 channels via a mechanism that involves Gαq and caveolin. These results provide a novel molecular basis for the inhibition of renal NaCl transport by the CaR.
Keywords: G Protein-coupled Receptors (GPCR), Heterotrimeric G Proteins, Kidney, Potassium Channels, Signal Transduction
Introduction
Sodium transport in the distal nephron determines body volume and blood pressure as demonstrated by the fact that mutations in genes expressed in this region of the kidney can cause either salt retention and high blood pressure or salt wasting and low blood pressure (1). A number of genetically distinct syndromes caused by abnormalities of transport proteins expressed in the distal nephron lead to phenotypes characterized by salt wasting. Bartter syndrome can result from loss of function mutations of the NaK2Cl co-transporter (NKCC2),2 the renal outer medullary K+ channel (ROMK or Kir1.1), or the ClC-Kb (basolateral Cl− channel). A variant also characterized by sensorineural deafness is caused by loss of function mutations in an accessory protein for ClC-Kb, barttin (Bartter syndrome neuraosensory deafness or BSND) (1–5). The hypocalciuric, hypomagnesemic variant of Bartter syndrome, Gitelman syndrome, is due to loss of function mutations of the thiazide-sensitive NaCl co-transporter (6). The recently described SeSAME (seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance) or EAST (epilepsy, ataxia, sensorineural deafness, and tubulopathy) syndrome is caused by loss of function mutations in Kir4.1 (KCNJ10), an inwardly rectifying K+ channel that is expressed in the basolateral membrane of the distal nephron (7–10). Presumably, reduced activity of Kir4.1 impairs recycling of K+ for the Na,K-ATPase reducing net transepithelial Na2+ transport. The CaR is an additional gene which can lead to a Bartter-like phenotype. However, in contrast to the other genes for which loss-of-function mutations lead to this phenotype, the CaR mutations are gain-of-function (11, 12). All of these transporters and CaR are present in the distal nephron, but precisely how the CaR reduces NaCl transport is not defined.
Activation of the CaR by hypercalcemia or mutations results in clinically significant salt and water wasting to the point of severe volume depletion and hypotension. Although CaR activates cellular signaling systems that can explain the inhibition of NKCC2 and ROMK, other receptors such as those for angiotensin II, endothelin, or α-adrenergic agonists stimulate the same signaling pathways without causing Na+, Cl−, and K+ wasting (13). This difference in effects on salt transport between the CaR and other receptors suggests that the CaR may act via distinct mechanisms to regulate electrolyte metabolism such as direct interaction with channels, pumps, or transporters.
To determine which factors result in the unique transport-regulatory properties of the CaR, we investigated proteins that interact with the CaR. We identified Kir4.1 as a protein that interacts with the CaR in yeast two-hybrid screens, heterologous expression systems, and in the distal convoluted tubule (DCT). We found that CaRWT inactivates Kir4.1, whereas the nonfunctional mutant CaRR795W has no effect. The activity of Kir4.1 is not affected by short term changes in Ca2+o, suggesting initially that inactivation of Kir4.1 is a relatively long term effect of CaRWT signaling that could involve processes such as protein trafficking (10). This mechanism has three notable features. The first is regulation of activity through control of cell surface expression via a mechanism dependent on Gαq and caveolin-1; the second is demonstration of physiologic regulation of Kir4.1; and the third feature is that the effect of the CaR on distal nephron NaCl transport appears to be attributable to physiologic regulation of a basolateral channel.
EXPERIMENTAL PROCEDURES
All chemicals were purchased from Sigma or Fisher Scientific unless specified otherwise. The monoclonal anti-CaR and anti-HA antibodies and HEK-293 cells that stably overexpress the wild-type CaR were described previously (10, 11). G418 sulfate and cell culture reagents were purchased from Invitrogen. The SuperSignal West Pico chemiluminescent substrate and BCA protein assay reagent were obtained from Pierce. Polyclonal anti-Myc (product is antibody A-14) and monoclonal anti-Myc (product no. 9E10) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal anti-Kir4.1 and anti-ROMK antibodies were supplied by Alomone Laboratories (Jerusalem, Israel).
Construction of Plasmids
To investigate the relationship between Kir4.1 and CaR without the limitation of specific antibodies, we made Myc- and HA-tagged constructs of the channel engineering epitope tags into the N terminus of the proteins. The cDNA coding for rat Kir4.1 was generously provided by Stephen Tucker at the University Laboratory of Physiology (University of Oxford) (28). The Kir4.1pBF construct was digested with EcoRI, blunt-ended, and then digested with KpnI. The pCruz-Myc or -HA constructs were digested with NotI, blunt-ended, and then digested with KpnI. The fragments were ligated into pCruz-Myc or pCruz-HA. All cDNA constructs were verified by direct sequencing. The ROMK pcDNA3.1 was generous gift from Wenhui Wang (New York Medical College, Valhalla, NY).
Transfection, Immunoblotting, and Immunoprecipitation
HEK-293 cells were transiently transfected with the CaRWT and CaR mutants (CaRI40F, CaRL125P, and CaRR795W), and the epitope-tagged channel constructs indicated (Myc-tagged Kir4.1-pCruz and HA-tagged Kir4.1-pCruz) or ROMK-pcDNA3.1 using the FuGENE 6 reagent and incubated at 37 °C for 24–48 h. For co-immunoprecipitation experiments, the medium was removed, and the cells were washed once with 1× PBS. The cells were lysed with 1× radioimmune precipitation assay buffer, and the lysates were centrifuged at 15,000 rpm for 1 h at 4 °C. The protein concentrations in the cell lysates were determined using the BCA protein assay reagent with BSA as a standard and then adjusted to the same concentration with buffer. The samples were subjected to 11% SDS-PAGE, processed for immunoblotting, and the levels of protein expression were determined. The cellular lysates were used for co-immunoprecipitation as described (16). Briefly, either a monoclonal anti-CaR antibody or a polyclonal anti-Kir4.1 antibody was separately loaded onto the Dynabead-protein A complex and slowly rotated for 2 h. The antibody-loaded Dynabead-protein A complex was rinsed twice, and the beads were mixed with the various cell lysates and rotated in the cold room overnight. The supernatants were discarded, and the Dynabead-protein A complex was washed. Loading buffer was added and vortexed vigorously. The tubes were placed in Dynal MPC to collect the sample buffer. The samples were heated at 55 °C for 20 min for channel proteins or boiled for 5 min for the CaR and then subjected to SDS-PAGE for immunoblotting using the antibodies indicated. For co-immunoprecipitation studies using rat kidney membrane extracts, rat kidneys were isolated, and the cortex and medulla were separated. The cortex and medulla were then cut into small pieces and homogenized in a buffer containing 20 mm HEPES, pH 8.0, 2 mm MgCl2, 1 mm EDTA, and protease inhibitors and centrifuged at 2000 rpm for 10 min. The supernatant was centrifuged at 13,500 rpm for 60 min to obtain crude membrane and cytosol fractions. The samples were used for immunoblotting to confirm the presence of the endogenous proteins. The crude membranes were extracted in 1× radioimmune precipitation assay buffer for 60 min and centrifuged at 13,500 rpm for 60 min. The resulting supernatants were used for immunoprecipitation using anti-CaR or Kir4.1 antibodies, and the immunoprecipitated samples were processed for immunoblotting using antibodies against CaR and Kir4.1 proteins.
Electrophysiology
HEK-293 cells were transiently transfected with plasmids containing the genes for study with a small amount of a plasmid-expressing EGFP (total amount of DNA transfected held constant by using empty vector) so that the transfected cells could be identified (14, 15). The extracellular medium (bath) contained 145 mm KCl, 2 mm MgCl2, 2 mm CaCl2, and 10 mm HEPES, pH 7.4, and the pipette contained 145 mm KCl, 2 mm EDTA, and 10 mm HEPES, pH 7.4. Capacitance and access resistance were monitored and compensated 75%, and the voltage protocol was a 0 mV holding potential and 400-ms steps from −100 to +100 mV in 20-mV increments (15).
Expression of siRNAs
The siRNA sequences were sense and antisense, respectively, for caveolin-1, 5′-CUAAACACCUCAACGAUGAUU-3′ and 5′-UCAUCGUUGAGGUGUUUAGUU-3′, and for the clathrin heavy chain, 5′-UCCAAUUCGAAGACCAAUU-3′ and 5′-AAUUGGUCUUCGAAUUGGA-3′ (16). The siRNAs (200 pmol) were transiently transfected into HEK-293 cells in 30-mm dishes with cDNAs as indicated using Polyfect and studied 2 days later. The effects of the siRNAs were documented by demonstrating reduced protein expression by Western blot.
Cell Surface Biotinylation
Cells were washed with PBS and incubated with sulfo-NHS-SS-biotin (biotin, 0.5 mg/ml) for 5 min on ice. The cells were washed three times with PBS, quenched with 100 mm glycine in PBS for 5 min, and dissolved in immunoprecipitation buffer (125 mm NaCl, 62.5 mm NaH2PO4, pH 7.2, and 0.625% lubrol, and protease inhibitors) and incubated overnight with avidin-agarose. The agarose-bound complexes were precipitated and size-fractionated by SDS-PAGE, and proteins were identified by immunoblotting (17).
RESULTS
Control of Kir4.1 Cell Surface Abundance by CaR
The activity of ion channels at the cell surface is a function of the number of channels and the open probability (Po). Many members of the Kir (inwardly-rectifying K+ channel) family of K+ channels are regulated by G protein-coupled receptors that alter the Po over short time frames through phosphatidylinositol bisphosphate metabolism or protein-protein interactions (18, 19). We found that co-expression of the CaRWT and Kir4.1 in Xenopus oocytes led to reduced whole cell currents, whereas the nonfunctional mutant CaRR795W had no effect (10). Inactivation of Kir4.1 by the CaR is not mediated by short term changes in receptor signaling, as changes in Ca2+ over the range 0.1–5 mm or addition of Gd+ (CaR agonist), had no effect on channel activity (10). Pursuing long term receptor effects on channel activity in this study, we investigated the ability of the CaRWT and CaR mutants with a range of signaling activities to affect Kir4.1 cell surface expression using cell surface biotinylation and current density with the whole cell patch technique in HEK-293 cells. The CaRWT (EC50 Ca, 3.2 mm) and three CaR mutants, CaRL125P (mutation associated with Bartter syndrome EC50 Ca, 1.4 mm), CaRI40F (EC50 Ca, 8.2 mm), and CaRR795W (nonfunctional mutant, EC50 Ca > 10 mm) ranged from constitutively active to inactive in normal cell culture conditions (11). As shown in Fig. 1A, the CaRWT reduces Kir4.1 cell surface expression to 52 ± 6.6% (S.E.) of the level seen with Kir4.1 alone, CaRL125P to 34 ± 5.9%, and CaRI40F to 62 ± 15%, whereas CaRR795W had no effect (94 ± 10%). To determine how cell surface expression of Kir4.1 relates to its activity, we measured whole cell current density in HEK-293 cells that transiently expressed Kir4.1 and the CaR mutants (Fig. 1B; current density (in pico Amps/pico Farads current capacitance): 1540 ± 89, 520 ± 82, 359 ± 101, 943 ± 124, and 1630 ± 189 for vector, CaRWT, CaRL125P, CaRI40F, and CaRR795W, respectively). The current density measurements have the same pattern as the cell surface determinations, showing that current density is reduced by the expressed CaR mutants in proportion to their level of signaling activity in a constant extracellular Ca2+ environment. CaRL125P has the least and the nonfunctional CaR mutant, CaRR795W has the most current density, similar to Kir4.1 alone. Despite the inability of the CaRR795W to signal, it co-immunoprecipitates with Kir4.1 when the two are co-expressed in HEK-293 cells, indicating that the interaction of the two proteins is not dependent on signaling activity of the CaR (Fig. 2A). The CaR and Kir4.1 co-immunoprecipitate reciprocally from rat kidney extracts, indicating that the CaR and Kir4.1 interact in vivo (Fig. 2B). These results demonstrate that the CaR reduces Kir4.1 cell surface expression and current density attributable to Kir4.1. The state of activation of the receptor, in this case determined by a range of mutations in the presence of constant Ca2+o, determines the magnitude of the response but does not affect the interaction of the receptor and the channel.
FIGURE 1.

Effect of CaR mutants on Kir4.1 cell surface expression (A) and current density (B and C). cDNAs coding for Kir4.1 and the CaR mutants were expressed transiently in HEK-293 cells. A, cell surface expression of Kir4.1 was measured using cell surface biotinylation. Ten μg of cell surface biotinylated protein was used for each lane in each blot, and Kir4.1 was identified using the Kir4.1 antibody. The top panel shows summary data (n = 7, mean ± S.E.). The bands corresponding to biotinylated Kir4.1 were quantitated using densitometry (Scion Image), and the values in each experiment were normalized for the level of expression of Kir4.1 alone. The middle panel Kir4.1 (Bio) shows a representative blot for biotinylated Kir4.1, and the bottom panel shows a corresponding cell extract blotted for Kir4.1 (Extr). B, top panel, summary data for whole cell current density ± S.E. (n = 8). All cells expressed Kir4.1 and CaR constructs as shown. C, the representative voltage-capacitance curves for Kir4.1 and CaR constructs are as shown. *, p < 0.05 compared with Kir4.1 alone (ANOVA). ** p < 0.05 between groups indicated (ANOVA).
FIGURE 2.

Co-immunoprecipitation of the CaRWT or CaRR795W with Kir4.1 (A), and reciprocal co-immunoprecipitation of the CaR and Kir4.1 from rat kidney extracts (B). A, HEK-293 cells were transiently transfected with either the CaRWT and Kir4.1 (lanes 1 and 2) or the CaRR795W (nonfunctional mutant) and Kir4.1 (lane 3) in the top and bottom two panels. The immunoprecipitation antibody (IP Ab) is shown above each panel. No indicates that agarose beads without an immunoprecipitating antibody were used. Extracts were prepared and incubated with agarose beads with or without immunoprecipitating antibody (lane 1) or the antibodies shown at the top of each panel. Immunoprecipitated proteins were identified using Western blotting (WB) and specific antisera as shown on the left. The panels on the right show expression of the target proteins in the cell lysate. B, rat kidney cortex extracts were prepared and incubated with the immunoprecipitating antibodies shown. In the first two panels, top and bottom, in lane 1, agarose beads without immunoprecipitating antibody were used. The right two lanes (lanes 2 and 3) are replicates, and the antibodies shown at the top of each panel were used for immunoprecipitation. The third and fourth panels (top and bottom) are controls using the Kir4.1 and Car immunoprecipitation antibodies or rabbit IgG as a negative control. The last (fifth) panels (top and bottom) show the CaR (top) and Kir4.1 (bottom) in the rat cortex lysate (Lys). Immunoprecipitated proteins were identified using Western blotting and specific antisera as shown on the left.
Signaling Pathways That Regulate Kir4.1 Cell Surface Expression and Current Density
The CaR can signal via members of the Gαi, Gαq, and Gα12/13 families of G protein α subunits (20, 21). To determine whether signaling via a G protein α subunit can explain the effects of the CaR on Kir4.1 cell surface expression and current density, and which G protein α subunit is responsible, we co-expressed constitutively active mutant forms of several G protein α subunits with Kir4.1 and measured Kir4.1 cell surface expression and current density in HEK-293 cells. The α subunits tested were Gαq (GαqQ209L), Gαi2 (GαiQ204L), Gα13 (Gα13Q226L), and Gαs (GαsQ227L). The CaR is not known to interact with Gαs, so we tested it as a negative control. Fig. 3A shows that GαqQ209L reproduces the effects of the CaR on Kir4.1, reducing cell surface expression measured by cell surface biotinylation (CaR, 35 ± 6.7%; Gαq, 37.1 ± 2.8% of Kir4.1 alone). Co-expression of Kir4.1 with the G protein αiQ204L, α13Q226L, or αsQ227L subunits did not reduce their cell surface expression (Fig. 3A). Similar results were obtained with measurements of current density where it was reduced by 52% by GαqQ209L (Kir4.1 (2,500 ± 400 pico Amps/pico Farads)) versus Kir4.1 and GαqQ204L (1,200 ± 200 pA/pF; p < 0.05) and not significantly affected by the other α subunits (Fig. 3B).
FIGURE 3.

G protein signaling and Kir4.1 cell surface expression and activity. cDNAs coding for Kir4.1 and the CaRWT or GTPase-deficient G protein α subunit mutants (Gαi2Q204L, Gα13Q226L, GαqQ209L, or GαsQ227L) were expressed transiently in HEK-293 cells. A, cell surface expression was measured using cell surface biotinylation. The top panel shows summary data (n = 4, mean ± S.E.). The bands corresponding to biotinylated Kir4.1 were quantitated using densitometry (Scion Image), and the values in each experiment were normalized for the level of expression of Kir4.1 alone. The middle panel Kir4.1 (Bio) shows a representative blot for biotinylated Kir4.1, and the bottom panel shows a representative cell extract blotted for Kir4.1 (Extr). All cells expressed Kir4.1 and the CaR or G protein α subunit constructs as shown. *, p < 0.05 compared with Kir4.1 alone, CaRWT and Gαq were not statistically different from each other (ANOVA). B, current density in HEK-293 cells expressing Kir4.1 and vector or the G protein α subunit constructs are shown (n = 8, ± S.E.). *, p < 0.05 compared with Kir4.1 (Vector) alone (ANOVA). C, the effect of RGS4 on CaR-mediated reduction in Kir4.1 whole cell current density is shown. cDNAs coding for Kir4.1 the CaRWT and RGS4 were expressed transiently in HEK-293 cells as indicated. Summary data for whole cell current density ± S.E. (n = 10). All cells expressed Kir4.1 and vector, and the CaRWT is as shown, and 0 to 1.5 μg of RGS4 cDNA as shown. *, p < 0.05 compared with CaR without RGS4 (ANOVA). D, cell surface expression was measured using cell surface biotinylation. The top panel shows summary data (n = 5, mean ± S.E.). The bands corresponding to biotinylated Kir4.1 were quantitated using densitometry (Scion Image), and the values in each experiment were normalized for the level of expression of Kir4.1 alone. The middle panel Kir4.1 (Bio) shows a representative blot for biotinylated Kir4.1, and the bottom panel shows a representative cell lysate blotted for Kir4.1 (Lys). *, p < 0.05 compared with Kir4.1 and CaRWT (ANOVA).
To verify that the CaR signals via Gαq to control Kir4.1, we co-expressed CaRWT with Kir4.1 and RGS4 (a regulator of G protein signaling protein). RGS4 accelerates the GTPase activity of members of the Gαi and Gαq families of G protein α subunits, and when co-expressed with them, prevents their activation. Because expression of Gαi2Q204L had no effect on Kir4.1 cell surface expression or channel density, an effect of RGS4 would have to occur through Gαq. In Fig. 3C, increasing amounts of RGS4 cDNA were co-expressed with Kir4.1 and the CaRWT, and whole cell current density was measured. From no RGS4 where the CaRWT reduced current density to ∼40% of the level found with Kir4.1 alone, increasing amounts of RGS4 blocked the effect of the CaRWT in a dose-dependent manner. Fig. 3D shows that the CaRWT reduces cell surface expression of Kir4.1 to ∼48.5 ± 6.8% of the level seen with Kir4.1 alone and that RGS4 blocks the effects of the CaRWT on Kir4.1 (88.7 ± 6.6% of Kir4.1 alone). These results show that the CaR acts via Gαq to reduce Kir4.1 cell surface expression and current density. Finally, to test for an effect of protein kinase C, a kinase downstream of Gαq, we treated HEK-293 cells expressing Kir4.1 alone or Kir4.1 with GαqQ204L with oleoyl-acetyl-glycerol (10 μm) a PKC activator. Oleoyl-acetyl-glycerol did not affect the current density of cells expressing Kir4.1 alone (Fig. 4) or alter the effect of GαqQ204L on Kir4.1. These results demonstrate that the CaR controls Kir4.1 cell surface expression and current density via a mechanism that requires Gαq but is independent of PKC.
FIGURE 4.
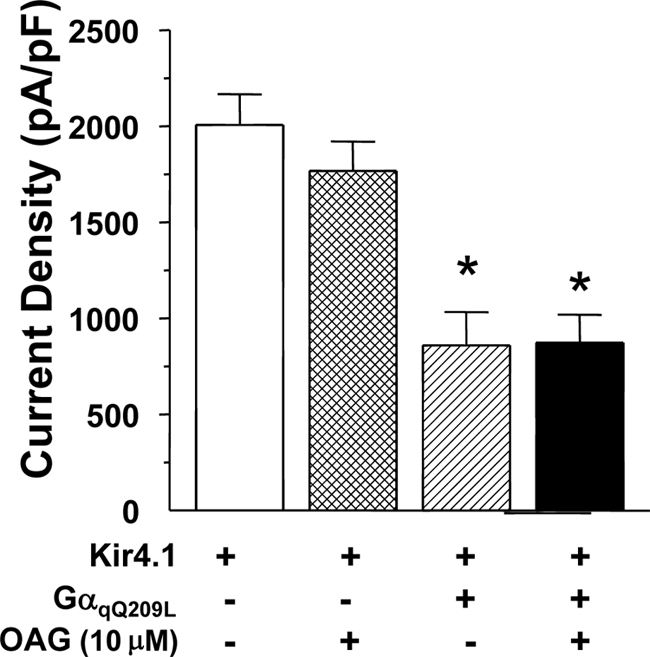
Effect of protein kinase C activation on Kir4.1 channel density. HEK-293 cells were transiently transfected with Kir4.1 and vector or GαqQ204L as shown and treated with oleoyl-acetyl-glycerol (OAG; 10 μm) for 1 h before rupture for current density measurements (n = 10, ± S.E.).
Specificity of CaR Effects on Kir4.1
To determine whether the effects of the CaR on Kir4.1 are selective, we compared the effects of the CaRWT on Kir4.1 and ROMK (Kir1.1), a K+ channel that is present in the apical membrane of the distal nephron and the trafficking of which involves clathrin-coated vesicles. Fig. 5A shows normalized current density and current-voltage (I-V) relationships for Kir4.1 and ROMK with and without the CaRWT. Fig. 5B shows cell surface expression of the two channels with or without the CaRWT. Expression of the CaR with ROMK does not affect its cell surface expression (98.7 ± 15% of control, S.E.), whereas the CaR has an effect on cell surface expression similar to that seen in other studies (56.6 ± 9.2% (S.E.) of control). The magnitude of current density and cell surface expression is similar for ROMK without and with the CaRWT, whereas CaRWT reduces Kir4.1 current density and cell surface expression. These results demonstrate that the CaR has differential effects on ROMK and Kir4.1 and that CaR selectively controls Kir4.1 cell surface expression.
FIGURE 5.

Selectivity of CaR effects on Kir4.1 for surface abundance and channel density. HEK-293 cells transiently transfected with cDNAs coding for the CaRWT or vector, and ROMK or Kir4.1 as shown. A, top panel (left), summary data for whole cell current density ± S.E. (n = 8); * p < 0.05 versus Kir4.1 alone (vector) (ANOVA). Currents were normalized to the maximum current for cells expressing Kir4.1 or ROMK without CaR at −100 mV. Top (right), representative current − capacitance curves for Kir4.1 or ROMK and CaRWT constructs. B, summary data and representative cell surface biotinylation studies for ROMK and Kir4.1 with and without the CaRWT. Cell surface biotinylation was performed as described in Fig. 1. The middle panel ROMK (or Kir4.1) shows a representative blot for biotinylated ROMK (Bio) or Kir4.1 (Bio), and the bottom panel shows a representative cell lysate blotted for ROMK or Kir4.1 (Lys). Bar graphs represent three separate experiments normalized to channel alone ± S.E.
Caveolin versus Clathrin-dependent Regulation of Kir4.1 Cell Surface Expression
We next examined whether the reduction in cell surface abundance of Kir4.1 by the CaR via Gαq occurs through a mechanism involving clathrin or caveolin-mediated trafficking. A caveolin-mediated mechanism is a good candidate because caveolin is expressed at high levels in the basolateral membrane of the DCT where the CaR and Kir4.1 are also expressed (22). We knocked down endogenous caveolin-1 or clathrin heavy chain using siRNA in HEK-293 cells co-expressing Kir4.1 and GαqQ209L, or vector(14). In both sets of studies, the GαqQ209L construct reduced current density by ∼50% in cells transfected with the control oligonucleotides (Fig. 6, A and B). The effect of GαqQ209L was blocked by siRNAs directed against caveolin-1 (Fig. 6A) but not clathrin heavy chain (Fig. 6B). In cells with siRNA directed against caveolin-1 (Fig. 6A), Kir4.1 current density appeared greater than in the absence of siRNA, suggesting that Kir4.1 may undergo baseline caveolin-1-dependent internalization. Knockdown of endogenous caveolin-1 and the clathrin heavy chain is demonstrated in the Western blot analysis (Fig. 6C). Thus, the CaR inactivates Kir4.1 via a signaling pathway that requires Gαq and caveolin leading to reduced cell surface expression.
FIGURE 6.

Caveolin versus clathrin-dependent cell surface expression of Kir4.1. Cells transiently expressing Kir4.1 alone or Kir4.1 and GαqQ209L were transfected with either a control (Cont.) oligonucleotide or siRNAs directed against caveolin-1 (A, n = 8) or the heavy chain of clathrin (B, n = 8) as indicated. Bars represent mean current density ± S.E. (*, p < 0.05 compared with Kir4.1 alone, ANOVA). Successful knockdown of caveolin-1 (A) and clathrin heavy chain (CHC) is shown by Western blot in C. D, co-immunoprecipitation of expressed Kir4.1 or the CaR with caveolin from HEK-293 cells. The top panels show the immunoprecipitating antibody (IP Ab) at the top, the Western blot antibody (WB Ab) is shown at the bottom right of each panel, and the expressed constructs are shown below the lanes of each panel. Bottom panels show control immunoprecipitations of caveolin-1 (Cav), Kir4.1, and the CaR. E, Co-immunoprecipitation of Kir4.1 or the CaR with caveolin-1 from kidney cortex extracts. The top panels show caveolin-1 co-immunoprecipitated with Kir4.1 (left) or the CaR (right), and the bottom panels show immunoprecipitation of Kir4.1 or the CaR with their respective antibodies. In the first lane in each panel, beads without immunoprecipitating antibody were used. This figure is representative of three separate experiments.
Physical Association of CaR and Kir4.1 with Caveolin
Caveolae are specialized membrane domains for formation of signaling complexes and for internalization of membrane proteins and other cargos (23). G protein-dependent signaling complexes, particularly those containing Gαq, are preferentially targeted to caveolae (24). The CaR and Kir4.1 physically associate (Fig. 2) (10). Caveolin-1 is a critical component of caveolae and mediates targeting of proteins to caveolae (23), so we investigated interactions among the CaR, Kir4.1, and caveolin-1. The CaR and Kir4.1 co-immunoprecipitate reciprocally with caveolin-1 when co-expressed in HEK-293 cells (Fig. 6D) and co-immunoprecipitate with caveolin-1 from rat kidney cortex extracts (Fig. 6E). These results indicate not only that the CaR and Kir4.1 interact with each other but that they also interact with caveolin in the kidney.
DISCUSSION
Hypercalcemia is well established as a cause of renal salt wasting, but the fact that it is receptor-mediated was not fully appreciated until the demonstration that activating mutations in the CaR lead to a Bartter-like salt-wasting syndrome (11, 12). The generally accepted mechanism by which the CaR reduces distal nephron NaCl reabsorption is inhibition of the activity of transporters in the TAL such as NKCC2 and ROMK by CaR-regulated second messengers including arachidonic acid metabolites, Ca2+i, and cAMP (13, 25).
We identify a novel mechanism for CaR-mediated renal salt wasting. This mechanism is characterized by long term CaR, Gαq, and caveolin-1 effects on Kir4.1 channel trafficking that result in reduced cell surface expression and activity of Kir4.1. In past studies using Xenopus oocytes, we found that the CaRWT inactivated Kir4.1 but that addition of CaR agonists or reducing Ca2+ levels over short time periods did not alter Kir4.1 currents, whereas CaRR795W had no effect (10). These results indicate that the effects of the CaR on Kir4.1 are distinct from the conventional effects of Gαq-coupled receptors that involve short term regulation of second messengers such as inositol 1,4,5-trisphosphate or phosphatidylinositol bisphosphate, kinases such as PKC, or protein-protein interactions that occur over seconds. Instead, they depend on protein trafficking that occurs over a time scale of a few to many minutes (10, 19).
Reduced Kir4.1 activity leads to reduced transepithelial NaCl transport, probably as a consequence of reduced recycling of K+ for the basolateral Na,K-ATPase. Evidence for this interpretation comes from the effects of human loss of function mutations in Kir4.1 and the phenotype of Kir4.1 knock-out mice (7–9). We show that the regulation of Kir4.1 by the CaR occurs at least partly via alterations of cell surface expression. The finding that the degree of reduction in cell surface expression corresponds to the nature and severity of the CaR mutation (most potent activating mutation, the mutation associated with Barter-like salt wasting, leads to maximum reduction in channel expression and currents, and the inactivating mutation has no effect on expression and currents) demonstrates that CaR effects are due to the level of CaR signaling and can thus explain the effects of hypercalcemia or CaR mutations on renal Na+ transport.
Our data show that the CaR reduces cell surface expression of Kir4.1 presumably by altering protein trafficking, a process that is slow compared with regulation of channel gating by second messengers or kinases. This mechanism appears to be appropriate because serum and extracellular Ca2+ concentrations change relatively slowly, for example following dietary ingestion of a significant amount of Ca2+ and possibly also Na+. Presumably, Ca2+ would be absorbed by the gastrointestinal tract over many minutes to hours. Even a sustained small rise in serum and interstitial Ca2+ would activate the CaR and lead to increased Ca2+ and Na+ excretion. The EC50 Ca is ∼1.0 mm, which means that under normal circumstances when ionized serum Ca2+ is on the order of 1.0 mm, the CaR is partially active, and Kir4.1 is in a partially active state so that it can be inactivated or activated by changes in Ca2+. Given the steep activation curve of the CaR in response to Ca2+ (Hill coefficient of 4), Kir4.1 should be sensitive to regulation over the relatively small variations in Ca2+ in the physiologic range. This relationship between Ca2+ and Na+ excretion could represent a mechanism to tie dietary intake of cations to their excretion.
The phenotypes of patients with activating CaR and inactivating Kir4.1 mutations are similar in that both are characterized by volume depletion with Na+, K+, Cl−, and Mg2+ wasting, but they differ in that patients with CaR mutations waste Ca2+, whereas patients with Kir4.1 mutations have low urine Ca2+ values (7, 8). Patients with Kir4.1 mutations also appear similar to those with Gitelman syndrome caused by loss of function mutations in the NaCl co-transporter localized to the apical membrane of the DCT. The differences between the phenotypes of patients with activating CaR and loss of function Kir4.1 mutations could be due to the fact that the CaR has effects beyond inactivating Kir4.1. The CaR reduces Ca2+ reabsorption in the TAL or the DCT by mechanisms that are independent of NaCl transport. Activation of the CaR in the TAL could reduce Ca2+ reabsorption by inhibiting paracellular transport (affecting NaCl transport to some degree) and reduce the activity of TRPV5 in the DCT (16, 26, 27).
The finding that expression of the Kir4.1 along the nephron varies with species and strain complicates assigning the effects of the CaR and Kir4.1 on NaCl transport to a specific nephron segment. Kir4.1 is expressed in the DCT, CNT, and at lower levels in the cortical collecting duct in all species studied (human, mouse, and rat), and in the CTAL of some mouse strains and humans, so depending on the species or strain, NaCl reabsorption would be reduced in the DCT, CNT, and cortical collecting duct, or possibly in the CTAL (8–10, 28). In all situations, Kir4.1 is important for the function of the DCT.
In a study using in vivo microperfused distal nephron segments from hypercalcemic rats that contained TAL as well as DCT and CNT, JNa was reduced (29). However, using microperfused mouse CTALs, two studies demonstrated effects of CaR agonists on cAMP or Ca2+i and Ca2+ transport, but no effect on NaCl transport (JNa), whereas one study (rat) found an effect on Cl− reabsorption associated with increased transepithelial voltage, an effect that could affect paracellular Cl− transport (26, 27, 30, 31). In these experiments, the basal activity of NKCC2 could have been low due to localization of the transporter in vesicles below the apical membrane, making further inhibition difficult to demonstrate (32).
A new explanation for Ca2+-mediated inhibition of distal nephron NaCl transport incorporates the relationship of the CaR to Kir4.1 with previous work. The CaR may not be effective in inhibiting NaCl reabsorption in nephron segments that lack Kir4.1. An electron microscopic study of the DCT in renal biopsy material from a patient with a loss of function mutation in Kir4.1 showed atrophy of this segment consistent with reduced NaCl transport (9). This result indicates that NaCl transport in the DCT is dependent on the function of Kir4.1 as well as the NaCl co-transporter and indicates that inactivation of Kir4.1 by the CaR is physiologically significant. The CaR could also regulate apical transporters (NKCC2 and ROMK) in a coordinate manner, although its inactivation of Kir4.1 should be sufficient to cause renal Na2+ loss based on the salt-wasting phenotypes of human loss of function Kir4.1 mutations and Kir4.1 knock-out mice (7–9).
The reason for the requirement of the CaR and Gαq signaling pathways that control Kir4.1 cell surface expression for caveolin-1 is not fully defined at this time. Caveolin is involved in both endocytosis and exocytosis, and we have not determined which, or whether both processes are important in reduction of cell surface expression of Kir4.1. Nevertheless, the involvement of caveolin has a number of implications. First, it may explain the selectivity of the CaR for Kir4.1 over ROMK. Internalization of ROMK is controlled via a mechanism that requires clathrin, so the CaR acting through caveolin-1 should have no effect on ROMK trafficking (14). Second, in at least two cell types, parathyroid cells and Saos-1 osteosarcoma cells, the CaR interacts with caveolin-1, suggesting that the CaR may normally function in caveolae (33, 34). In contrast to other G protein α subunits, Gαq is preferentially localized to caveolae and interacts with caveolin-1 in a manner that indicates that in addition to serving a scaffolding function, caveolin-1 may actively participate in signaling (24).
Control of caveolin-dependent endocytic pathways may be complex. The cell surface expression of TRPV5 is controlled by parathyroid hormone through caveolin (16). Oleoyl-acetyl-glycerol, presumably acting via PKC (often downstream of Gαq) increases surface expression of TRPV5, whereas it has no effect on basal or CaR-regulated cell surface expression of Kir4.1. These results indicate that the CaR-stimulated signaling pathway that controls Kir4.1 cell surface expression is different from that which controls TRPV5 and may contain novel components. Finally, along the nephron, staining for caveolin-1 is most intense on the basolateral membrane of the DCT where the CaR and Kir4.1 are both localized (10, 22, 35). Interestingly, caveolin-1−/− mice are hypercalciuric (35). Although the mechanism is not known, the effects are likely to be on the DCT, the TAL, or both, and could be attributable to abnormal CaR signaling.
CaR and Kir4.1 co-localize in the basolateral membrane of the DCT and the CNT, although expressed at lower levels in the CNT than in the DCT (7, 8, 10). In humans and some mouse stains, Kir4.1 is also expressed in the basolateral membrane of the CTAL along with the CaR (9). Inhibition of distal nephron NaCl transport via CaR-mediated inhibition of Kir4.1 is novel because it involves physiologic regulation of a basolateral channel. Although loss of function mutations of ClC-Kb and barttin (BSND) also reduce basolateral transport in the distal nephron, establishing the principle that basolateral transporters other than the Na,K-ATPase can affect transepithelial Na+ transport, physiologic regulation by this mechanism has not been reported.
This work was supported, in whole or in part, by National Institutes of Health Grants DK-59985 (to R. T. M.), DK-59530 (to C.-L. H.) and University of Texas Southwestern O'Brien Center grant P30DK079328. This was also supported by Veterans Affairs Merit Review grants (to C. H. and R. T. M.), the Leonard Rosenberg Research Foundation, and the Rammelkamp Center for Research and Education, Case Western Reserve University.
- NKCC2
- NaK2Cl− co-transporter
- aa
- amino acid
- CaR
- Ca2+-sensing receptor
- ANOVA
- analysis of variance
- CNT
- connecting tubule
- CTAL
- cortical thick ascending limb of Henle
- DCT
- distal convoluted tubule
- ROMK
- renal out medulla K+ channel
- TAL
- thick ascending limb of Henle.
REFERENCES
- 1. Lifton R. P., Gharavi A. G., Geller D. S. (2001) Cell 104, 545–556 [DOI] [PubMed] [Google Scholar]
- 2. Konrad M., Vollmer M., Lemmink H. H., van den Heuvel L. P., Jeck N., Vargas-Poussou R., Lakings A., Ruf R., Deschênes G., Antignac C., Guay-Woodford L., Knoers N. V., Seyberth H. W., Feldmann D., Hildebrandt F. (2000) JASN 11, 1449–1459 [DOI] [PubMed] [Google Scholar]
- 3. Simon D. B., Lifton R. P. (1998) Curr. Opin. Cell Biol. 10, 450–454 [DOI] [PubMed] [Google Scholar]
- 4. Ookata K., Tojo A., Suzuki Y., Nakamura N., Kimura K., Wilcox C. S., Hirose S. (2000) JASN 11, 1987–1994 [DOI] [PubMed] [Google Scholar]
- 5. Bonnardeaux A., Bichet D. G. (2000) in The Kidney (Brenner B. M. ed) pp. 1621–1678, W. B. Saunders, Philadelphia [Google Scholar]
- 6. Pearce S. H. (1998) Q. J. Med. 91, 5–12 [Google Scholar]
- 7. Scholl U. I., Choi M., Liu T., Ramaekers V. T., Häusler M. G., Grimmer J., Tobe S. W., Farhi A., Nelson-Williams C., Lifton R. P. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bockenhauer D., Feather S., Stanescu H. C., Bandulik S., Zdebik A. A., Reichold M., Tobin J., Lieberer E., Sterner C., Landoure G., Arora R., Sirimanna T., Thompson D., Cross J. H., van't Hoff W., Al Masri O., Tullus K., Yeung S., Anikster Y., Klootwijk E., Hubank M., Dillon M. J., Heitzmann D., Arcos-Burgos M., Knepper M. A., Dobbie A., Gahl W. A., Warth R., Sheridan E., Kleta R. (2009) NEJM 360, 1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reichold M., Zdebik A. A., Lieberer E., Rapedius M., Schmidt K., Bandulik S., Sterner C., Tegtmeier I., Penton D., Baukrowitz T., Hulton S. A., Witzgall R., Ben-Zeev B., Howie A. J., Kleta R., Bockenhauer D., Warth R. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 14490–14495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang C., Sindic A., Hill C. E., Hujer K. M., Chan K. W., Sassen M., Wu Z., Kurachi Y., Nielsen S., Romero M. F., Miller R. T. (2007) Am. J. Physiol. Renal Physiol. 292, F1073–1081 [DOI] [PubMed] [Google Scholar]
- 11. Vargas-Poussou R., Huang C., Hulin P., Houillier P., Jeunemaître X., Paillard M., Planelles G., Déchaux M., Miller R. T., Antignac C. (2002) JASN 13, 2259–2266 [DOI] [PubMed] [Google Scholar]
- 12. Watanabe S., Fukumoto S., Chang H., Takeuchi Y., Hasegawa Y., Okazaki R., Chikatsu N., Fujita T. (2002) Lancet 360, 692–694 [DOI] [PubMed] [Google Scholar]
- 13. Huang C., Miller R. T. (2010) Curr. Opin. Nephrol. Hypertens. 19, 106–112 [DOI] [PubMed] [Google Scholar]
- 14. He G., Wang H. R., Huang S. K., Huang C. L. (2007) J. Clin. Invest. 117, 1078–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lazrak A., Liu Z., Huang C. L. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cha S. K., Wu T., Huang C. L. (2008) Am. J. Physiol. Renal Physiol. 294, F1212–1221 [DOI] [PubMed] [Google Scholar]
- 17. Gottardi C. J., Dunbar L. A., Caplan M. J. (1995) Am. J. Physiol. Renal Physiol. 268, F285–295 [DOI] [PubMed] [Google Scholar]
- 18. Lopes C. M., Rohács T., Czirják G., Balla T., Enyedi P., Logothetis D. E. (2005) J. Physiol. 564, 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rohács T., Lopes C. M., Jin T., Ramdya P. P., Molnár Z., Logothetis D. E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang C., Miller R. T. (2007) Curr. Opin. Nephrol. Hypertens. 16, 437–443 [DOI] [PubMed] [Google Scholar]
- 21. Hofer A. M., Brown E. M. (2003) Nat. Rev. Mol. Cell Biol. 4, 530–538 [DOI] [PubMed] [Google Scholar]
- 22. Breton S., Lisanti M. P., Tyszkowski R., McLaughlin M., Brown D. (1998) J. Histochem. Cytochem. 46, 205–214 [DOI] [PubMed] [Google Scholar]
- 23. Lajoie P., Goetz J. G., Dennis J. W., Nabi I. R. (2009) J. Cell Biol. 185, 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oh P., Schnitzer J. E. (2001) Mol. Biol. Cell 12, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang W. H. (2006) Am. J. Physiol. Renal Physiol. 290, F14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desfleurs E., Wittner M., Simeone S., Pajaud S., Moine G., Rajerison R., Di Stefano A. (1998) Kidney Blood Press. Res. 21, 401–412 [DOI] [PubMed] [Google Scholar]
- 27. Motoyama H. I., Friedman P. A. (2002) Am. J. Physiol. Renal Physiol. 283, F399–406 [DOI] [PubMed] [Google Scholar]
- 28. Ito M., Inanobe A., Horio Y., Hibino H., Isomoto S., Ito H., Mori K., Tonosaki A., Tomoike H., Kurachi Y. (1996) FEBS Lett. 388, 11–15 [DOI] [PubMed] [Google Scholar]
- 29. Peterson L. N. (1990) Am. J. Physiol. Renal Physiol. 259, F122–129 [Google Scholar]
- 30. Friedman P. A. (1998) Annu. Rev. Physiol. 60, 179–197 [DOI] [PubMed] [Google Scholar]
- 31. De Jesus Ferreira M. C., Bailly C. (1998) Am. J. Physiol. Renal Physiol. 275, F198–203 [DOI] [PubMed] [Google Scholar]
- 32. Gamba G., Friedman P. A. (2009) Pflugers Arch. 458, 61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kifor O., Diaz R., Butters R., Kifor I., Brown E. M. (1998) J. Biol. Chem. 273, 21708–21713 [DOI] [PubMed] [Google Scholar]
- 34. Jung S. Y., Kwak J. O., Kim H. W., Kim D. S., Ryu S. D., Ko C. B., Cha S. H. (2005) Exp. Mol. Med. 37, 91–100 [DOI] [PubMed] [Google Scholar]
- 35. Cao G., Yang G., Timme T. L., Saika T., Truong L. D., Satoh T., Goltsov A., Park S. H., Men T., Kusaka N., Tian W., Ren C., Wang H., Kadmon D., Cai W. W., Chinault A. C., Boone T. B., Bradley A., Thompson T. C. (2003) Am. J. Path. 162, 1241–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]