

Abstract
A large family of membrane channel proteins selective for transport of water (aquaporins) or water plus glycerol (aquaglyceroporins) has been found in diverse life forms. Escherichia coli has two members of this family—a water channel, AqpZ, and a glycerol facilitator, GlpF. Despite having similar primary amino acid sequences and predicted structures, the oligomeric state and solute selectivity of AqpZ and GlpF are disputed. Here we report biochemical and functional characterizations of affinity-purified GlpF and compare it to AqpZ. Histidine-tagged (His-GlpF) and hemagglutinin-tagged (HA-GlpF) polypeptides encoded by a bicistronic construct were expressed in bacteria. HA-GlpF and His-GlpF appear to form oligomers during Ni-nitrilotriacetate affinity purification. Sucrose gradient sedimentation analyses showed that the oligomeric state of octyl glucoside-solubilized GlpF varies: low ionic strength favors subunit dissociation, whereas Mg2+ stabilizes tetrameric assembly. Reconstitution of affinity-purified GlpF into proteoliposomes increases glycerol permeability more than 100-fold and water permeability up to 10-fold compared with control liposomes. Glycerol and water permeability of GlpF both occur with low Arrhenius activation energies and are reversibly inhibited by HgCl2. Our studies demonstrate that, unlike AqpZ, a water-selective stable tetramer, purified GlpF exists in multiple oligomeric forms under nondenaturing conditions and is highly permeable to glycerol but less well permeated by water.
Living organisms can survive in hostile environments because their cell membranes are effective barriers to threats such as low pH and high levels of toxic solutes. Cell membranes must also be selectively permeable in order for cellular needs to be met in the form of water, specific ions, and nutrients. Cell membranes must also permit the release of unwanted substances and allow the cell to maintain its volume. In all living organisms, these physiological processes involve a multitude of membrane transport proteins each with distinct substrate specificities.
Among these transporters is the family of proteins sometimes referred to as “major intrinsic proteins.” Escherichia coli contains two members of this family: AqpZ, which is selectively permeated by water (1), and GlpF, the facilitator of glycerol transport (2). AqpZ and GlpF are representative of an early divergence, since water is essential for cellular hydration, and glycerol is necessary for cellular nutrition and osmotic balance. This dualism led to the functional division of mammalian homologs into two major groups: transporters selectively permeated by water (aquaporins) and transporters permeated by water and glycerol (aquaglyceroporins) (3).
The cloning of genes for AqpZ and GlpF yielded primary amino acid sequences that are clearly homologous (4–6). Moreover, cryoelectron microscopic analyses revealed the AqpZ and GlpF structures to be tetrameric with only subtle differences (7–10). The transport properties of these proteins are clearly distinct when expressed in Xenopus laevis oocytes (4, 11). AqpZ is freely permeated by water but not glycerol, whereas GlpF is known to be permeated by polyols such as glycerol (2), but is apparently not permeated by water (11).
The pore size may explain how a channel could be permeated by water, a smaller molecule, but not by glycerol. Nevertheless, it is not inherently obvious how a channel could be freely permeated by glycerol but not water. Specific sequence differences have been reported to determine a molecule's transport selectivity (12). Whereas AQP1 and other mammalian aquaporins are believed to be stable tetramers (13), GlpF has been reported to be a monomer in the membrane (14, 15). In contrast, cryoelectron microscopic analyses showed GlpF to be tetrameric (9, 10). To clarify these discrepancies, GlpF and AqpZ were expressed at high levels in E. coli, affinity-purified, and evaluated by velocity sedimentation and transport selectivity.
Materials and Methods
Materials.
n-Octyl β-d-glucopyranoside (OG) was obtained from Calbiochem. E. coli total lipid extract acetone/ether preparation was from Avanti Polar Lipids. Nickel nitrilotriacetate (NTA)-agarose, Ni-NTA-magnetic beads, and antibody to pentahistidine (BSA free) were from Qiagen. Purified murine monoclonal antibody to influenza virus hemagglutinin A epitope (HA) was from Covance (Richmond, CA). Complete protease inhibitor mixture was from Boehringer Mannheim. Restriction and DNA modifying enzymes were from New England Biolabs or Life Technologies. Other reagents were from Sigma or Aldrich.
Expression Plasmids and Bacterial Strains.
The GlpF open reading frame (ORF) was amplified from a Xenopus expression construct by PCR to create a 5′ MunI restriction site. The amplified fragment was cloned into a vector containing a histidine tag (His) upstream to the EcoRI insertion point (1). The resulting construct, pTrc10HisGlpF, contains the sequence of the commercially available pTrc99A plasmid (Pharmacia) with a NcoI–BamHI insertion coding for the His tag (H2N-MGHHHHHHHHHHSSGHIEGRHEL) preceding GlpF. Upstream to this sequence, the vector contains a Trc promoter inducible by isopropyl β-d-thiogalactoside (IPTG). The vector also encodes LacIq, the Trc repressor that ensures low transcriptional levels in the absence of inducer. The expression vector was transformed into the commercially available E. coli strain XL1B and was selected for by ampicillin resistance. HA-tagged recombinant protein was expressed from a similar plasmid where the N-terminal His tag was replaced by PCR with an HA tag (H2N-MAYPYDYPDYA). Coexpression of the His-tagged and HA-tagged forms was achieved by creating a bicistronic unit containing the two consecutive ORFs. A Shine–Dalgarno sequence was added upstream of HA-GlpF ORF, and the resulting fragment was cloned into pTrc10HisGlpF downstream of His-GlpF ORF.
Expression and Purification of Recombinant Proteins.
Luria broth cultures containing 50 μg/ml ampicillin were incubated for 13–16 hr at 37°C, diluted 100-fold into fresh broth, and propagated to a density of about 1.5 (OD at 600 nm). Expression of recombinant protein was induced by addition of 1 mM IPTG for 2 hr at 37°C before centrifugation (15 min at 2,000 × g). Harvested cells were resuspended in one 1/100 culture volume of ice-cold lysis buffer (100 mM K2HPO4/1 mM MgSO4/0.4 mg/ml lysozyme/0.1 mg/ml DNase I and protease inhibitors, pH 7.0) and subjected to three lysis cycles in a French press (125 × 106 Pa, at 4°C). Unbroken cells and debris were separated from the cell lysate by a 30-min centrifugation at 10,000 × g and discarded. Membrane fractions recovered from the supernatant by a 60-min centrifugation at 140,000 × g were resuspended to the original volume in solubilization buffer [3% OG in 100 mM K2HPO4/10% (vol/vol) glycerol/5 mM 2-mercaptoethanol/200 mM NaCl, pH 8.0] and incubated on ice for 1 hr. Insoluble material was pelleted by 45 min centrifugation at 140,000 × g. The soluble fraction was mixed with 1/50 vol of prewashed Ni-NTA-agarose beads (Qiagen) and incubated with agitation at 4°C for 1 hr. The beads were then packed in a plastic disposable column (Stratagene) and washed with 100 bead volumes of wash buffer (3% OG/100 mM K2HPO4/10% glycerol/5 mM 2-mercaptoethanol/200 mM NaCl/100 mM imidazole, pH 7.0) to remove nonspecifically bound material. Residual wash buffer was removed (2 min at 2,000 rpm in a bench-top microcentrifuge). Ni-NTA-agarose-bound material was eluted by incubation in 1 bed volume of elution buffer (3% OG/100 mM K2HPO4/10% glycerol/5 mM 2-mercaptoethanol/200 mM NaCl/1 M imidazole, pH 7.0) for 1 hr on ice. Analytical purifications were performed similarly except for the use of Ni-NTA-agarose magnetic beads (Qiagen). Typically, the first elution step of preparative purifications yielded pure protein at a concentration of 5–10 mg/ml, measured as described (16) with BSA as a standard. AqpZ was solubilized in 1.5% n-dodecyl β-d-maltoside and purified as described (1).
Sedimentation Analysis.
Velocity sedimentation analysis was used to determine the oligomeric structure of purified proteins or crude detergent extract of bacterial membranes expressing the recombinant protein. Detergent-solubilized material (2–10 μg of purified protein or membrane extract of 0.5 ml of culture in 200-μl sample volume) was layered on top of a continuous sucrose gradient (4 ml; 20 mM Tris⋅HCl/5 mM EDTA/3% OG/1 mM NaN3/5–20% sucrose, pH 8.0) and centrifuged at 140,000 × g for 18 hr. Up to 20 fractions were collected and analyzed by SDS/PAGE to determine the migration of the protein. Pure protein was detected by Coomassie brilliant blue staining of gels, whereas immunoblotting was used to detect recombinant proteins from membrane extracts. The sedimentation coefficient (s20,w) of each species was determined by interpolation of the relative migration vs. sedimentation coefficient linear function for the following standards: cytochrome c (1.8), carbonic anhydrase (2.9), BSA (4.3), β-amylase (8.9), and catalase (11.2).
Functional Reconstitution.
Purified protein was reconstituted into proteoliposomes. Briefly, a reconstitution mixture was prepared in a glass tube at room temperature by sequentially adding: 100 mM Mops–Na (pH 7.5), 1.25% (wt/vol) OG, purified protein (final concentration 100 μg/ml), and 10 mg/ml sonicated lipids. E. coli total lipid extract (acetone/ether preparation; Avanti Polar Lipids) was hydrated in 2 mM 2-mercaptoethanol to a final concentration of 50 mg/ml, incubated for 1 hr at room temperature, divided into aliquots, and frozen at −80°C. Before use, lipids were diluted into 500 μl in a borosilicate tube (16 × 125 mm) to a final concentration of 45 mg/ml in 100 mM Mops–Na (pH 7.5) and pulsed in a bath sonicator until a clear suspension was obtained. Lipids were always handled under nitrogen/argon atmosphere. The reconstitution mixture was loaded into Spectra/por 2.1 dialysis tubing (molecular weight cut-off 12,000–14,000, Spectrum Medical Industries) and dialyzed against 100 vol of assay buffer (50 mM Mops/150 mM N-methyl d-glucamine, adjusted to pH 7.5 with HCl/1 mM NaN3) for 24–72 hr at room temperature. Alternatively, the reconstitution mixture was injected into 25 vol of assay buffer under constant stirring to dilute the detergent. Liposomes were harvested by centrifugation (45 min at 140,000 × g) and resuspended into assay buffer. Protein content was measured as described (16) with BSA as a standard. The diameter of proteoliposomes obtained by dialysis (130 nm) was measured by light scattering.
Membrane Permeability Measurements.
The osmotic behavior of reconstituted proteoliposomes and control liposomes was analyzed by following the light scattering of the preparation in a stopped-flow apparatus (SF-2001; Kin Tek Instruments, University Park, PA) with a dead time of ≤1 ms. Water permeability was measured by rapidly mixing 100 μl of a liposomes suspension (1 μg of protein, 100 μg of lipids) in assay buffer (see above) with a similar volume of hyperosmolar solution (assay buffer with sucrose added as an osmolyte). The osmotic gradient (285 mosM) drives water efflux, and the consequent reduction in vesicle volume is measured as an increase in the intensity of scattered light (λ = 600 nm). The equation
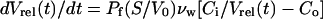 |
1 |
describes the change in volume as a function of membrane permeability (17). Vrel, the vesicular volume relative to the initial volume, is proportional to the intensity of scattered light (18) and is dimensionless. Pf is the osmotic water permeability, S/V0 is vesicle surface area to initial volume ratio, vw is the partial molar volume of water (18 cm3), Ci is the initial intravesicular osmolarity, and Co is the external osmolarity. The result of analytical integration of Eq. 1, which describes the time course of change in volume, cannot be easily fitted to experimental data. Thus, single-exponential time constants (k) were calculated by least-square fit of experimental data. A family of simulated curves was obtained by numerical integration of Eq. 1 and fitted to a single exponential. Pf was estimated by iterative comparison of the experimental time constants with the values obtained from the simulation by using MathCAD software.
To determine permeability to glycerol, liposomes were equilibrated in assay buffer supplemented with glycerol (≈570 mM glycerol). Liposome suspensions (100 μl, 1–2 μg of protein) were then rapidly mixed with a solution in which osmolality was compensated by a nonpermeant solute (sucrose). The external concentration of permeant solute is reduced by half (285 mM) without change in osmolality, driving the efflux of the permeant osmolyte and generating an outwardly oriented osmotic gradient. Water efflux causes a reduction in volume and an increase in the intensity of scattered light. Under our conditions it can be shown that the change in vesicle volume as a function of solute permeability (Psol) is described by the expression (17)
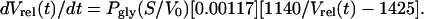 |
2 |
Experimental data were fitted to single-exponential equations, and the resulting time constants (k) were used to calculate Pgly as described above.
Results
Expression and Purification of GlpF.
Analysis of reconstituted, purified channel proteins may reveal biophysical properties that are not apparent when studied in whole cell membranes. We recently expressed the E. coli water channel as a 10-histidine-AqpZ chimeric polypeptide and functionally characterized the purified molecule (1). Here we have used the same system to study the E. coli glycerol facilitator as a 10-histidine-GlpF chimeric polypeptide. Transformed bacterial cultures were induced with IPTG, and membranes were solubilized in nondenaturing detergent (OG). Affinity purification with Ni-NTA columns yielded up to 2.5 mg of His-GlpF per liter of culture at a final concentration of 5–10 mg/ml. Coomassie-stained SDS/PAGE slabs showed a single band of ≈30 kDa (Fig. 1A).
Figure 1.

SDS/PAGE and velocity sedimentation analyses of His-GlpF and His-AqpZ. (A) Protein samples (≈1 μg) in 1% SDS and 140 mM 2-mercaptoethanol were analyzed with 10–20% acrylamide gradient gel slabs and stained with Coomassie brilliant blue (34). AqpZ samples were previously acidified with HCl and neutralized to disrupt the SDS-stable tetramer (1). (B) Membranes from transformed bacteria were solubilized in OG, layered over a 5–20% continuous sucrose gradient, and sedimented at 140,000 × g for 18 hr at 4°C. Twenty fractions were collected; top of gradient is on the left. Mobility of His-GlpF or His-AqpZ was determined by immunoblotting.
Sedimentation Analyses.
GlpF has been reported to exist in the membrane as a dispersion of single monomers (12, 15), whereas AQP1 and other members of the aquaporin family are stable homotetramers (13). When analyzed by velocity sedimentation through sucrose density gradients, AqpZ migrates as a tetrameric protein of 5.7 S (1). Bacterial cultures transformed with the His-GlpF construct were induced with IPTG, and membranes were solubilized and directly analyzed by velocity sedimentation through 5–20% sucrose density gradients. Anti-His immunoblots of the profiles revealed a major band at 30 kDa in fractions of lesser density (Fig. 1B, lanes 6 and 7), whereas a ladder of SDS-stable higher molecular weight oligomers appeared in fractions of intermediate and greater density (lanes 8–12). This profile was distinct from that achieved with membranes containing His-AqpZ, which formed a single SDS-stable band in fractions of greater density (lanes 10–13).
To further evaluate the GlpF monomer vs. tetramer paradox, a GlpF chimeric molecule was tagged with an N-terminal epitope from hemagglutinin A (HA). This construct was cloned into the same expression vector by itself or as a second ORF in a bicistronic artificial operon. When expressed alone, HA-GlpF did not adsorb to Ni-NTA affinity columns (not shown). When His-GlpF and HA-GlpF were cotranslated from the bicistronic construct in the same bacterial cultures, both polypeptides eluted from the Ni-NTA affinity column (Fig. 2A). These data indicate that a significant part of solubilized GlpF exists as multisubunit oligomers. Detergent-extracted membranes were incubated at 4°C for up to 5 hr without decreased copurification of HA-GlpF, indicating that the multimeric species is stable under the conditions used for solubilization (not shown). When His-GlpF and HA-GlpF were expressed in separate bacterial cultures, and OG-solubilized membranes were mixed before Ni-NTA affinity chromatography, only His-GlpF eluted from the column (Fig. 2B). Thus, although His-GlpF/HA-GlpF heterooligomers formed readily when the two polypeptides were cotranslated in the same bacterial cultures, no exchange between independently expressed oligomers in solution was observed.
Figure 2.

Interaction between His-GlpF and HA-GlpF. Membranes from bacteria expressing His-GlpF, HA-GlpF, or both were extracted in 3% OG and incubated for 1 hr at 4°C. The solutions were then mixed with Ni-NTA magnetic beads and eluted with imidazole. (Note that only His-GlpF or proteins associated with His-GlpF will adsorb to Ni-NTA beads.) The eluates were analyzed by SDS/PAGE gels stained with Coomassie brilliant blue (CBB) or probed with monoclonal antibodies to His (α-His) or HA (α-HA). (A) Membranes from a bacterial culture coexpressing His-GlpF and HA-GlpF from a bicistronic operon were adsorbed to Ni-NTA and eluted. (B) Membranes from a bacterial culture expressing His-GlpF and from a second culture expressing HA-GlpF were mixed before adsorption to Ni-NTA and elution.
Velocity sedimentation analyses of Ni-NTA affinity-purified His-GlpF/HA-GlpF proteins were undertaken with a series of protein standards to reveal approximate sedimentation coefficients (Fig. 3A). When the 5–20% sucrose gradients contained 3% OG and 20 mM Tris but no additional salt, the profile contained ≈30-kDa bands in fractions from top to bottom, whereas inclusion of 300 mM NaCl in the gradients shifted the profile toward denser fractions at the bottom (Fig. 3B). When 300 mM MgCl2 was included in the gradient instead of NaCl, the major peak was even better defined in denser fractions presumed to contain tetramers (lanes 10 and 11). Substitution of 0.1% SDS for 3% OG completely shifted the peak to fractions of lesser density (lanes 6–8) presumed to contain monomers and possibly dimers. Thus, the equilibrium between oligomeric forms of affinity-purified GlpF may be altered by the ionic strength of the gradient.
Figure 3.

Velocity sedimentation analysis of purified His-GlpF. OG-solubilized, affinity-purified His-GlpF was sedimented at 140,000 × g, for 18 hr at 20°C on a continuous gradient containing 5–20% sucrose. The gradient contained 20 mM Tris⋅HCl (pH 8.0), 5 mM EDTA, and 3% OG. (A) The sedimentation coefficient of GlpF was determined by comparison with multiple water-soluble standards. (B) Mobility of purified GlpF was determined in gradients also containing no additional salt, 300 mM NaCl, or 300 mM MgCl2, or in 0.1% SDS. Twenty fractions were collected; top of gradient is on the left. Mobility of His-GlpF was determined by immunoblotting. (C) Fractions 6 and 11 from a gradient containing OG and 300 mM NaCl were analyzed by immunoblotting with anti-His or anti-HA monoclonal antibodies before (a) and after (b) a second affinity purification using Ni-NTA-agarose magnetic beads.
His-GlpF and HA-GlpF polypeptides were both present when gradient fractions 6 and 11 were evaluated by immunoblotting (Fig. 3C, lanes a). Thus, the HA-GlpF in fraction 6 must have originated from His-GlpF/HA-GlpF heterooligomers that disassociated during sedimentation. Attempts to repurify the protein from these fractions over Ni-NTA columns revealed that His-GlpF and HA-GlpF were both present in fraction 11 (Fig. 3C, lanes b). Thus, whereas Ni-NTA affinity-purified His-GlpF and His-GlpF/HA-GlpF are predominantly oligomeric when first eluted, dissociation of the oligomer occurs during sedimentation.
Membrane Permeability Measurements.
The functional division of aquaporins and aquaglyceroporins implies that the latter are permeated by both water and glycerol (3). When expressed in X. laevis oocytes, GlpF exhibited large glycerol transport activity and negligible water permeability (11). Thus, GlpF is often regarded as a glycerol-specific transporter. We sought to resolve this discrepancy by evaluating water permeability and glycerol permeability in reconstituted proteoliposomes containing Ni-NTA affinity-purified proteins, GlpF (His-GlpF/HA-GlpF), AqpZ (His-AqpZ), or GlpF + AqpZ with direct comparison to liposomes containing no reconstituted protein (Fig. 4 and Table 1).
Figure 4.

Functional reconstitution of glycerol transport activity in GlpF and AqpZ + GlpF proteoliposomes. GlpF (100 μg), AqpZ + GlpF (100 μg each), or no protein was mixed with pure phospholipid, and proteoliposomes were formed and equilibrated with glycerol (855 mosM). A concentration gradient for glycerol was then imposed by rapidly mixing 100 μl of the equilibrated proteoliposome suspension (1 μg of protein) with an equal volume of isoosmotic solution containing sucrose as a nonpermeant osmolyte in a stopped-flow apparatus. Release of glycerol from the proteoliposomes creates an osmotic gradient producing water efflux, and vesicle shrinkage was measured by light scattering. The average kinetics of 5–10 measurements were normalized and fitted to a characteristic single-order exponential equation dependent on the time course of glycerol efflux. Notice the different time scales in plots.
Table 1.
Permeabilities of reconstituted proteoliposomes
| Permeability | Conditions | Liposomes | GlpF | AqpZ | GlpF + AqpZ |
|---|---|---|---|---|---|
| Glycerol | |||||
| kgly, s−1 | Control at 5°C | 0.019 ± 0.006 | 7.8 ± 0.7 | 0.011 ± 0.005 | 38 ± 1 |
| Control at 20°C | 0.17 ± 0.02 | 18 ± 1 | 0.14 ± 0.02 | 54 ± 7 | |
| HgCl2 at 20°C* | 0.11 ± 0.02 | 0.28 ± 0.01 | 0.14 ± 0.02 | 16 ± 1† | |
| Pgly, cm/s at 5°C | 6.2 × 10−8 | 2.5 × 10−5 | 3.6 × 10−8 | 1.2 × 10−4 | |
| Ea, kcal/mol | 27 ± 2 | 9.6 ± 1.5 | 25 ± 2 | 4.9 ± 1.5 | |
| Water | |||||
| kw, s−1 | Control at 5°C | 2.4 ± 0.2 | 23 ± 3 | 154 ± 30 | |
| Control at 20°C | 13.5 ± 0.2 | 41 ± 1 | 158 ± 18 | ||
| HgCl2 at 20°C* | 2.5 ± 0.2 | 4.8 ± 0.1† | 98 ± 6 | ||
| HgCl2 + DTT at 20°C* | ND | 32 ± 5† | ND | ||
| Pf, cm/s at 5°C | 5.1 × 10−4 | 4.9 × 10−3 | 3.3 × 10−2 | ||
| Ea, kcal/mol | 16 | 7 | 3 |
Proteoliposomes were preincubated with 100 μM HgCl2 for 30 min at 37°C. Reversal of HgCl2 inhibition was achieved by incubation with 1 mM DTT at 37°C for 30 min. ND, not determined.
This value corresponds to 30 μM HgCl2.
Glycerol permeability was measured with proteoliposomes and liposomes previously loaded with reconstitution buffer containing glycerol. These preparations were then rapidly mixed with isotonic reconstitution buffer containing sucrose as the nonpermeant osmolyte (in place of glycerol), and light scattering was measured. With this system, glycerol released from the proteoliposomes creates a small outwardly oriented osmotic gradient which is dissipated by efflux of water. Control liposomes exhibited very low glycerol permeability, and AqpZ proteoliposomes were virtually identical (Fig. 4, Table 1). In contrast, GlpF proteoliposomes exhibited a 400-fold increase in glycerol permeability, and proteoliposomes reconstituted with GlpF + AqpZ exhibited even faster kinetics, presumably because of fast release of glycerol through GlpF and faster release of water through AqpZ. GlpF-containing proteoliposomes displayed only a 3-fold increase in urea permeability at 5°C as compared with control liposomes (not shown). Arrhenius activation energy of glycerol transport was high for control liposomes, low for GlpF proteoliposomes, and even lower for GlpF + AqpZ proteoliposomes (Table 1). The glycerol permeability of GlpF proteoliposomes was significantly inhibited by 0.1 mM HgCl2 (Table 1). As expected, GlpF proteoliposomes are exceedingly well permeated by glycerol, but water permeation is rate-limiting during this measurement.
The osmotic water permeability of these same liposome and proteoliposome preparations was measured. The preparations were rapidly mixed with hyperosmolar reconstitution buffer containing sucrose as the nonpermeant osmolyte, and light scattering was measured (Fig. 5 and Table 1). At 5°C, control liposomes exhibited low water permeability, GlpF proteoliposomes exhibited 10-fold faster water permeability, and AqpZ proteoliposomes exhibited 60-fold faster water permeability (Fig. 5 and Table 1). Arrhenius activation energies of these processes were high for control liposomes, low for GlpF proteoliposomes, and even lower for AqpZ proteoliposomes (Table 1). The osmotic water permeability of GlpF proteoliposomes was significantly inhibited by 30 μM HgCl2 (Table 1). Incubation with 1 mM DTT fully reversed the mercurial inhibition. Thus GlpF is clearly permeated by water, although it is significantly less permeable to water than AqpZ. The presence of up to 100 mM glycerol did not affect the kinetics of water transport (not shown).
Figure 5.

Functional reconstitution of water transport activity in GlpF and AqpZ proteoliposomes. GlpF, AqpZ, or no protein was mixed with pure phospholipid and proteoliposomes were formed. A 2-fold osmotic gradient was imposed by rapidly mixing 100 μl of proteoliposome suspension (1 μg of protein) with an equal volume of hyperosmotic solution containing sucrose as a nonpermeant osmolyte in a stopped-flow apparatus. Water efflux causes vesicle shrinkage, which was measured by light scattering. The average kinetics of 5–10 measurements was normalized and fitted to a characteristic single-order exponential equation dependent on the time course of water efflux. Notice the different time scales in plots.
Discussion
The physiological needs for water and glycerol are unrelated, and the chemical compositions of these two liquids are distinct, so it was surprising that water and glycerol channel proteins turned out to be closely related. These proteins have recently drawn large scientific interest and, in the past 5 years, several hundred manuscripts have been published describing the biology of water channel proteins and their cousins the glycerol transporters (reviewed in refs. 19 and 20). Mammalian homologs are conveniently grouped into those permeable to water, aquaporins, and those permeable to water and glycerol, aquaglyceroporins (reviewed in ref. 3). Members of this protein superfamily are found throughout nature, and virtually all microorganisms have at least one member. E. coli has two, AqpZ and GlpF, but despite their high degree of sequence similarity, recent publications have noted major differences in oligomeric structure (1, 15), which may be linked to substrate specificity (12, 15).
When analyzed by velocity sedimentation, AQP1 and other aquaporins were predicted to be homotetramers (13). Velocity sedimentation of detergent-solubilized membrane proteins is technically difficult, because the protein standards are freely soluble in water (21). Nevertheless, ultrastructural studies confirmed that AQP1 is tetrameric in the plasma membranes of transfected cells (22) and in reconstituted membrane crystals (23, 24). AqpZ forms particularly stable tetramers which remain noncovalently associated even in SDS (1). It was therefore surprising that GlpF was reported to be a monomer when analyzed by velocity sedimentation in nondenaturing detergent (14) and by freeze–fracture electron microscopy of oocyte membranes (15). These studies initially seemed incompatible with reports describing the tetrameric organization of purified GlpF reconstituted into membrane crystals (9, 10).
Our studies confirm the findings of both groups of investigators. Sedimentation analysis of GlpF solubilized into OG directly from bacterial membranes shows the protein is predominantly disassociated in the less dense fractions (Fig. 1), whereas GlpF behaves as a multisubunit oligomer when studied by selective adsorption and elution from Ni-NTA (Fig. 2). When velocity sedimentation was undertaken with affinity-purified GlpF in gradients of different compositions, it became clear that the GlpF exists in multiple oligomeric states (Fig. 3). Unlike AqpZ, GlpF is almost entirely disassociated by SDS (Fig. 3B), whereas sedimentation of GlpF in 20 mM Tris without added salt or with 300 mM NaCl resulted in a smear in fractions ranging from monomeric to tetrameric masses. Only sedimentation in 300 mM MgCl2 produced a better-defined peak of tetrameric mass. Other workers using mass spectrometry and chemical crosslinking have concluded that GlpF forms oligomers when urea is present (25). It remains to be established if the same will happen when glycerol is present. Taken together, these studies indicate that: (i) GlpF exists in multiple oligomeric states; (ii) tetramers are stabilized while in the membrane and during affinity purification; (iii) the affinity of the GlpF monomer–dimer–tetramer associations is much lower than for AqpZ. In this way, GlpF resembles other integral membrane proteins such as the anion exchanger (AE1/band 3) which exists as an equilibrium of monomers, dimers, and tetramers, although it can be stabilized in specific oligomeric forms (26).
The mechanisms for selective water or glycerol permeation have also evoked significant interest (reviewed in ref. 27). When expressed in Xenopus laevis oocytes, GlpF was shown to exhibit high levels of glycerol transport but negligible water transport (11). This apparently caused the third mammalian homolog (AQP3) to be misidentified as transporter of glycerol but not water (28), although it is now clear that mammalian homologs AQP3 (29, 30), as well as AQP7 and AQP9, are permeated by water and glycerol, hence “aquaglyceroporins” (3). Biophysical measurements of purified GlpF reconstituted into proteoliposomes provide much better precision than studies in oocytes (Figs. 4 and 5). This permitted the demonstration that GlpF has significant water permeability, albeit significantly lower than AqpZ (Table 1). In addition, this system demonstrated the extremely low permeability of control liposomes to glycerol, which is increased several hundredfold in GlpF proteoliposomes (Table 1).
The selectivity for water or glycerol permeation has been proposed to be determined by two aromatic residues, tyrosine and tryptophan, near the top of the sixth transmembrane domain (12). In a study of the insect water channel protein AQPcic, tyrosine and tryptophan (YW, the sequence in AQPcic) were changed to proline and leucine (PL, the corresponding sequence in GlpF), and the protein lost its water permeability but gained glycerol permeability when studied in oocytes (12). We developed the reciprocal mutants in AqpZ (FW to PL) and in GlpF (PL to FW). The mutant proteins were expressed in E. coli and purified by Ni-NTA chromatography; however, neither mutant exhibited detectable water or glycerol permeability (data not shown). Thus the two-residue selectivity hypothesis does not bear general relevance for all members of this protein superfamily.
While our studies were nearing completion, the atomic structures were reported for human red cell AQP1 by cryoelectron microscopy (31) and GlpF by x-ray crystallography (32). The structures are very closely related and have been referred to as “fraternal twins” (33). The fourfold axis of symmetry of GlpF contains two magnesium ions complexed with tryptophan-42 and glutamate-43 near the tops of the second-bilayer spanning α-helices (32). This structure may explain the better definition of GlpF as a tetramer in MgCl2 (Fig. 3B). Nevertheless, the tryptophan and glutamate residues are not conserved in AQP1 or other members of this protein superfamily, so the general significance in tetramer formation is yet unclear. The atomic model of AQP1 provides insight into how water, but not protons, can pass through the pore because of the isolation of a single water molecule at the pore center by two highly conserved asparagine residues in the two NPA motifs (31). In addition, the structure of GlpF demonstrates the importance of two bulky and hydrophobic pore-lining residues (tryptophan-48 and phenylalanine-200) that provide a hydrophobic wedge over which the carbon backbone of glycerol is oriented (32). While the atomic structure of GlpF explains why glycerol can permeate the channel, it does not explain how water can be prohibited from permeating the pore, and the x-ray structure revealed the presence of water molecules interspersed between the glycerol molecules (32). Faced with the greater molarity of water in biological solutions (55 M), it is clear that glycerol transporters cannot completely block water movements through the pore. These findings appear to be well reconciled with our biophysical studies.
Acknowledgments
We thank Peter C. Maloney and Giuseppe Calamita for assistance. We also thank Peter Pedersen, Suresh Ambudkar, and Alan Finkelstein for critical suggestions. This work was supported in part by the National Institutes of Health.
Abbreviations
- OG
n-octyl β-d-glucopyranoside
- IPTG
isopropyl β-d-thiogalactoside
- NTA
nitrilotriacetate
- HA
hemagglutinin A epitope
References
- 1.Borgnia M J, Kozono D, Calamita G, Maloney P C, Agre P. J Mol Biol. 1999;291:1169–1179. doi: 10.1006/jmbi.1999.3032. [DOI] [PubMed] [Google Scholar]
- 2.Heller K B, Lin E C C, Wilson T H. J Bacteriol. 1980;144:274–278. doi: 10.1128/jb.144.1.274-278.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agre P, Bonhivers M, Borgnia M J. J Biol Chem. 1998;273:14659–14662. doi: 10.1074/jbc.273.24.14659. [DOI] [PubMed] [Google Scholar]
- 4.Calamita G, Bishai W R, Preston G M, Guggino W B, Agre P. J Biol Chem. 1995;270:29063–29066. doi: 10.1074/jbc.270.49.29063. [DOI] [PubMed] [Google Scholar]
- 5.Muramatsu S, Mizuno T. Nucleic Acids Res. 1989;17:4378. [PMC free article] [PubMed] [Google Scholar]
- 6.Sweet G, Gandor C, Voegele R, Wittekindt N, Beuerle J, Truniger V, Lin E C C, Boos W. J Bacteriol. 1990;172:424–430. doi: 10.1128/jb.172.1.424-430.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ringler P, Borgnia M J, Stahlberg H, Maloney P C, Agre P, Engel A. J Mol Biol. 1999;291:1181–1190. doi: 10.1006/jmbi.1999.3031. [DOI] [PubMed] [Google Scholar]
- 8.Scheuring S, Ringler P, Borgnia M, Stahlberg H, Muller D J, Agre P, Engel A. EMBO J. 1999;18:4981–4987. doi: 10.1093/emboj/18.18.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun T, Philippsen A, Borgnia M J, Agre P, Kuhlbrandt W, Engel A, Stahlberg H. EMBO Rep. 2000;1:183–189. doi: 10.1093/embo-reports/kvd022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stahlberg, H., Braun, T., de Groot, B. L., Philippsen, A., Borgnia, M. J., Agre, P., Kühlbrandt, W. & Engel, A. (2001) J. Struct. Biol., in press. [DOI] [PubMed]
- 11.Maurel C, Reizer J, Schroeder J I, Chrispeels M J, Saier M H J. J Biol Chem. 1994;269:11869–11872. [PubMed] [Google Scholar]
- 12.Lagree V, Froger A, Deschamps S, Hubert J F, Delamarche C, Bonnec G, Thomas D, Gouranton J, Pellerin I. J Biol Chem. 1999;274:6817–6819. doi: 10.1074/jbc.274.11.6817. [DOI] [PubMed] [Google Scholar]
- 13.Smith B L, Agre P. J Biol Chem. 1991;266:6407–6415. [PubMed] [Google Scholar]
- 14.Lagree V, Froger A, Deschamps S, Pellerin I, Delamarche C, Bonnec G, Gouranton J, Thomas D, Hubert J F. J Biol Chem. 1998;273:33949–33953. doi: 10.1074/jbc.273.51.33949. [DOI] [PubMed] [Google Scholar]
- 15.Bron P, Lagree V, Froger A, Rolland J P, Hubert J F, Delamarche C, Deschamps S, Pellerin I, Thomas D, Haase W. J Struct Biol. 1999;128:287–296. doi: 10.1006/jsbi.1999.4196. [DOI] [PubMed] [Google Scholar]
- 16.Schaffner W, Weissmann C. Anal Biochem. 1973;56:502–514. doi: 10.1016/0003-2697(73)90217-0. [DOI] [PubMed] [Google Scholar]
- 17.Zeidel M L, Ambudkar S V, Smith B L, Agre P. Biochemistry. 1992;31:7436–7440. doi: 10.1021/bi00148a002. [DOI] [PubMed] [Google Scholar]
- 18.Illsley N P, Verkman A S. J Membr Biol. 1986;94:267–278. doi: 10.1007/BF01869722. [DOI] [PubMed] [Google Scholar]
- 19.Borgnia M, Nielsen S, Engel A, Agre P. Annu Rev Biochem. 1999;68:425–458. doi: 10.1146/annurev.biochem.68.1.425. [DOI] [PubMed] [Google Scholar]
- 20.Calamita G. Mol Microbiol. 2000;37:254–262. doi: 10.1046/j.1365-2958.2000.02016.x. [DOI] [PubMed] [Google Scholar]
- 21.Sadler J E. J Biol Chem. 1979;254:4443. [PubMed] [Google Scholar]
- 22.Verbavatz J M, Brown D, Sabolic I, Valenti G, Ausiello D A, van Hoek A N, Ma T, Verkman A S. J Cell Biol. 1993;123:605–618. doi: 10.1083/jcb.123.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walz T, Smith B L, Agre P, Engel A. EMBO J. 1994;13:2985–2993. doi: 10.1002/j.1460-2075.1994.tb06597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitra A K, Yeager M, van Hoek A N, Wiener M C, Verkman A S. Biochemistry. 1994;33:12735–12740. doi: 10.1021/bi00209a001. [DOI] [PubMed] [Google Scholar]
- 25.Manley D M, McComb M E, Perreault H, Donald L J, Duckworth H W, O'Neil J D. Biochemistry. 2000;39:12303–12311. doi: 10.1021/bi000703t. [DOI] [PubMed] [Google Scholar]
- 26.Van Dort H M, Moriyama R, Low P S. J Biol Chem. 1998;273:14819–14826. doi: 10.1074/jbc.273.24.14819. [DOI] [PubMed] [Google Scholar]
- 27.Heymann J B, Engel A. J Mol Biol. 2000;295:1039–1053. doi: 10.1006/jmbi.1999.3413. [DOI] [PubMed] [Google Scholar]
- 28.Ma T, Frigeri A, Hasegawa H, Verkman A S. J Biol Chem. 1994;265:21845–21849. [PubMed] [Google Scholar]
- 29.Echevarria M, Windhager E E, Tate S S, Frindt G. Proc Natl Acad Sci USA. 1994;91:10997–11001. doi: 10.1073/pnas.91.23.10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishibashi K, Sasaki S, Fushimi K, Uchida S, Kuwahara M, Saito H, Furukawa T, Nakajima K, Yamaguchi Y, Gojobori T. Proc Natl Acad Sci USA. 1994;91:6269–6273. doi: 10.1073/pnas.91.14.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann J B, Engel A, Fujiyoshi Y. Nature (London) 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 32.Fu D, Libson A, Miercke L J, Weitzman C, Nollert P, Krucinski J, Stroud R M. Science. 2000;290:481–486. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- 33.Unger V M. Nat Struct Biol. 2000;7:1082–1084. doi: 10.1038/81914. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]