

Abstract
The discovery of methods suitable for the conversion in vitro of native proteins into amyloid fibrils has shed light on the molecular basis of amyloidosis and has provided fundamental tools for drug discovery. We have studied the capacity of a small library of tetracycline analogues to modulate the formation or destructuration of β2-microglobulin fibrils. The inhibition of fibrillogenesis of the wild type protein was first established in the presence of 20% trifluoroethanol and confirmed under a more physiologic environment including heparin and collagen. The latter conditions were also used to study the highly amyloidogenic variant, P32G. The NMR analysis showed that doxycycline inhibits β2-microglobulin self-association and stabilizes the native-like species through fast exchange interactions involving specific regions of the protein. Cell viability assays demonstrated that the drug abolishes the natural cytotoxic activity of soluble β2-microglobulin, further strengthening a possible in vivo therapeutic exploitation of this drug. Doxycycline can disassemble preformed fibrils, but the IC50 is 5-fold higher than that necessary for the inhibition of fibrillogenesis. Fibril destructuration is a dynamic and time-dependent process characterized by the early formation of cytotoxic protein aggregates that, in a few hours, convert into non-toxic insoluble material. The efficacy of doxycycline as a drug against dialysis-related amyloidosis would benefit from the ability of the drug to accumulate just in the skeletal system where amyloid is formed. In these tissues, the doxycycline concentration reaches values several folds higher than those resulting in inhibition of amyloidogenesis and amyloid destructuration in vitro.
Keywords: Amyloid, Cell Death, Protein-Drug Interactions, Protein Folding, Protein Stability, Inhibition of Fibrillogenesis, Tetracyclines
Introduction
Amyloidosis associated with long term hemodialysis results from the deposition of full-length β2-microglobulin (β2-m)2 and its N-terminal truncated species ΔN6β2m in target tissues (1, 2). Although all peripheral organs (but not the brain) can be potentially affected (3), the muscle-skeletal tissues are the preferential target always involved in this type of amyloidosis. Despite significant progress achieved in the hemodialysis techniques, including the increased biocompatibility and the active removal of circulating β2-m, the onset of this amyloidosis can be delayed but not avoided in dialysis-related amyloidosis patients (4). New therapeutic approaches, targeting the process of protein aggregation and promoting fibril solubilization (5), are under investigation for the treatment of different types of amyloid diseases. Up until now, different classes of structurally unrelated compounds have been investigated for their ability to interfere with protein self-aggregation and to weaken the intermolecular interactions that stabilize the fibrillar structure of the aggregates (6). Over 10 years ago, iododoxorubicin was serendipitously discovered as the prototype of a class of compounds able to inhibit protein aggregation (7), but this compound was subsequently abandoned for its toxicity. The resemblance of the polycyclic conjugated structure of tetracyclines with iododoxorubicin prompted a group studying the pathologic aggregation of the prion protein to test the properties of those antibiotics in the propagation of prion-related diseases (8). The successful results thereof stimulated further challenges. So far it has been shown that tetracyclines are able to antagonize the infectivity of the PrPsc aggregates (9), to inhibit in vitro the aggregation of amyloidogenic variants of transthyretin (10), the amyloid-β peptide fibrillogenesis (11, 12), and amylin fibrillogenesis both in vitro (13) and in vivo (14). The generic anti-aggregation property of tetracyclines has been confirmed in the fibrillogenesis of myoglobin (15), a protein that, although not amyloidogenic in humans, represents a very informative model of the mechanism of conversion of globular proteins into fibrillar assemblies.
Based on these and other results obtained in vitro and in animal models, at least three clinical trials have been undertaken to assess possible clinical benefits of tetracyclines in the treatment of the prion3 amyloid-β peptide (16), and transthyretin4 amyloidosis. In this study, we sought to evaluate the effect of tetracyclines in modulating, in vitro, the formation and the stability of β2-m fibrils. It is worth noting that tetracyclines selectively accumulate in the skeletal system (17), where they maintain biological properties such as metalloprotease inhibition (18). Therefore, the local concentration of bioactive tetracyclines, at the site of amyloid formation, should largely exceed that needed to interfere with stability and growth of the β2-m amyloid deposits that accumulate preferentially onto bone joints and cartilage. We show here that doxycycline and a group of analogues inhibit amyloid aggregation and induce amyloid fibril disassembly with the initial appearance of toxic oligomers that subsequently reorganize into harmless assemblies.
EXPERIMENTAL PROCEDURES
The 13 tetracycline congeners, reported in supplemental Fig. 1 in the hydrochloride form, were obtained from Sigma-Aldrich. Their purity was always above 95%. Drugs were dissolved in deionized water at 3 mm (stock solutions) and kept at −20 °C for no more than 2 months. Under these conditions, they were stable as determined by reverse phase HPLC. Aliquots of drugs were added to 50 or 70 mm phosphate buffer at pH 7.4 or 6.7, containing 100 mm NaCl, in the presence or absence of 20% (v/v) trifluoroethanol (TFE) or in pure water and immediately used to minimize their oxidation.
In Vitro Fibrillogenesis of β2-m and Effect of Doxycycline
Expression and purification of recombinant β2-m species were carried out as reported previously (19). In addition to wild type protein that makes fibrils under non-physiologic conditions, we have also investigated the P32G variant that can make fibrils in more physiological conditions (20), at neutral pH, and in the absence of any organic solvent. Recombinant wild type β2-m (100 μm) was incubated at 37 °C in 50 mm phosphate buffer, 100 mm NaCl, pH 7.4, containing 20% (v/v) TFE and preformed β2-m fibrils seeds at 2.5 μg/ml (21). Recombinant β2-m presenting the P32G mutation, at a concentration of 40 μm, was incubated at 37 °C under agitation at 250 rpm in 25 mm sodium phosphate buffer, pH 7.0, in the presence of 100 μg/ml heparin and preformed β2-m fibrils seeds at a concentration of 2.5 μg/ml. Quantification of amyloid fibril formation was performed using thioflavin T (ThT) as described previously (22). β2-m fibrillogenesis was carried out in the presence of different concentrations of tetracyclines (50, 100, 200, 300 μm) to evaluate their effect on the growth of fibrils. Dose-response fluorescence data were fitted by non-linear regression using GraphPad Prism 5.03 (GraphPad Software, San Diego, CA), and IC50 was determined from the best fitting of three experiments using a standard Hill slope dose-response curve
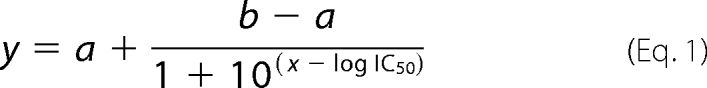 |
where a and b are the bottom and top plateau, respectively. 30 μl of the fibrillogenesis mixture with 300 μm different tetracycline analogues were centrifuged after 96 h of incubation at 10,000 × g for 15 min. The supernatant and protein pellets were analyzed by SDS-PAGE under reducing conditions (23). Pellets were resuspended in 3 μl of PBS buffer to be analyzed. Quantification of bands within each lane was carried out using the Quantity One software (Bio-Rad), and the percentages of β2-m in both supernatant and pellet were determined as compared with control β2-m. Destructuration of β2-m fibrils by doxycycline was evaluated by ThT assay and by electron microscopy by incubating ex vivo and synthetic β2-m fibrils in the presence of 300 μm doxycycline for 12 days.
NMR Experiments
The interaction between β2-m and doxycycline was analyzed by NMR spectroscopy under three different solvent conditions: (a) 70 mm sodium phosphate buffer, 100 mm NaCl, pH 6.7; (b) 70 mm sodium phosphate buffer, 100 mm NaCl, 18% (v/v) TFE, pH 6.7; (c) pure water, pH 5.5–6.6. A few microliters of 100 mm stock solution of doxycycline in pure water were incrementally added to the 0.125–0.7 mm β2-m samples to reach protein:doxycycline molar ratios of 1:0.3, 1:0.6, 1:1, 1:1.5, 1:2, 1:2.6, 1:3, and 1:6. All the NMR samples contained 5% D2O for frequency lock purpose. NMR experiments were recorded on a Bruker AVANCE spectrometer operating at 500 MHz (1H) and equipped with x-y-z-gradient accessory. Besides one-dimensional spectra, conventional proton homonuclear two-dimensional total correlation spectroscopy (24) and NOESY (25) were recorded to assign doxycycline. Two-dimensional [1H-15N] HSQC (26) experiments were acquired with 128–256 increments collected in t1 with 2,048 data points and 16–32 scans/FID in t2, over a spectral width of 14 and 33.5 ppm in proton and nitrogen dimension, respectively. Data were processed with Topspin (Bruker Biospin) and analyzed with Sparky. The chemical shift variations of the [1H-15N] HSQC spectra were calculated according Mulder et al. (27) using the formula
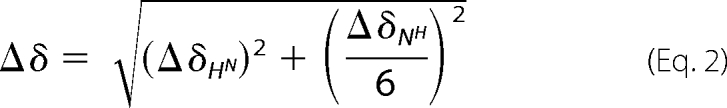 |
with the hydrogen (first term) and nitrogen (second term) Δδ values expressed in ppm. The chemical shift values of β2-m are deposited at the Biological Magnetic Resonance Bank (BMRB) (accession number 15521). Diffusion coefficients were measured by using the convection-compensated two-dimensional double stimulated echo-bipolar pulse (DSTE-BPP) sequence (28) to collect matrices of 2,048 (t2) by 80 points (t1). The z axis gradient strength was varied linearly from 2 to 95% of its maximum value (61.1 G/cm). Water suppression was achieved with the addition, in the specific sequence, of a sculpting module (29) or using a flip-back pulse in the HSQC experiments (30).
Inhibition of MTT Reduction
Human SH-SY5Y neuroblastoma cells were obtained from ATCC (Manassas, VA) and cultured in 1:1 Ham's F-10:DMEM medium supplemented with 10% fetal calf serum, 3.0 mm glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin, in a 5.0% CO2 humidified atmosphere at 37 °C. Cell viability was assessed by the MTT reduction inhibition assay. The cells were plated on 96-well plates at a density of 6,000 cells/well in 200 μl of fresh medium. After 72 h, the cells were exposed for 24 h to β2-m previously resolubilized in PBS in the absence or in the presence of different concentrations of drug at 37 °C for 1 h or with vehicle for control. The cells were also exposed to preformed fibrils previously treated at 37 °C with different concentrations of doxycycline for different lengths of time. At the end of the incubation, the cell culture medium was removed, and the cells were incubated for 2.0 h with 100 μl of a 0.5 mg/ml MTT solution in DMEM without phenol red. After incubation, cell lysis solution (100 μl/well: 20% SDS, 50% N,N-dimethylformamide) was added to the samples that were incubated overnight at 37 °C in a humidified incubator. Blue formazan absorbance was measured at 570 nm using an automatic plate reader (Bio-Rad).
Atomic Force Microscopy
Aggregation of β2-m in the presence of fibrillar collagen and heparin was performed as reported previously (31), in the absence and in the presence of 360 μm doxycycline. For atomic force microscopy (AFM) inspection, a collagen aliquot was extracted from the protein solution, deposited onto freshly cleaved mica, gently washed with buffer, and dried under a nitrogen stream. β2-m fibrils formed in the presence of TFE as detailed above were employed to test the effects of exposure to doxycycline on fibril structure. A 10-μl aliquot of β2-m fibrils treated with doxycycline was incubated on freshly cleaved mica for 5 min, rinsed with Milli-Q water to remove the materials not adhering to the substrate, and then dried under mild vacuum. Tapping mode AFM images were acquired in air using a Dimension 3100 scanning probe microscope equipped with a “G” scanning head (maximum scan size 100 μm) and driven by a Nanoscope IIIa controller and a MultiMode scanning probe microscope equipped with an “E” scanning head (maximum scan size 10 μm) and driven by a Nanoscope IV controller (Digital Instruments-Veeco, Santa Barbara, CA). Single beam uncoated silicon cantilevers (type OMCL-AC160TS, Olympus, Tokyo, Japan) were used. The drive frequency was between 320 and 340 kHz, and the scan rate was between 0.5 and 1.5 Hz. Aggregate sizes were measured from the height in cross-section of the topographic AFM images; due to the drying procedure applied, the measured sizes are reduced with respect to fully hydrated conditions.
RESULTS
Titration of the Inhibition of β2-m Fibrillogenesis by Different Tetracyclines
We initially tested the concentration-dependent ability of 13 tetracycline analogues (supplemental Fig. 1) to inhibit fibrillogenesis of wild type β2-m in vitro. According to the ThT assay proposed by Yamamoto et al. (21), we found that only seven analogues were able to inhibit β2-m fibril growth at a drug concentration limit <300 μm. For these seven compounds, we determined the IC50 activity value that corresponds to the molar concentration of the drug at which the specific fluorescence of the ThT-amyloid fibril complex is reduced by 50% (Fig. 1). The IC50 values for these seven compounds ranged from 47 ± 5 μm for doxycycline and 49 ± 9 μm for 4-epi-oxytetracycline (the most effective compounds) to 136 ± 9 μm for anhydrochlortetracycline and rolitetracycline. The inhibition of fibrillogenesis by tetracyclines is consistent with the inhibition of protein self-aggregation. The inhibition of aggregation was confirmed by quantifying the soluble fraction of β2-m after centrifugation. Electrophoresis of the supernatant and the precipitate (Fig. 2) showed that both doxycycline and 4-epi-oxytetracycline exhibit the highest effect of solubilization of β2-m fibrils. Other analogues, such as minocycline or 4-epianhydrotetracycline, left most of the protein in the precipitate. The ability of doxycycline to inhibit β2-m fibrillogenesis was confirmed using the P32G variant that can elongate fibril seeds in phosphate buffer at pH 7.0 (20). Fig. 3 reports the dose-response effect of doxycycline on fibril formation by this variant from which we derived an IC50 of 95 ± 22 μm.
FIGURE 1.

Inhibition of wild type β2-m fibrillogenesis by seven tetracycline congeners was followed by thioflavin fluorescence assay. Experiments were carried out in triplicate at different molar drug concentrations (50, 100, 200, 300 μm). Data were normalized and fitted using a standard Hill slope dose-response curve. Means ± S.D. of three experiments are shown for each compound. The structure of doxycycline, the most effective compound, is also reported.
FIGURE 2.

a, SDS-PAGE of the supernatant and protein pellet obtained after centrifugation of the fibrillogenesis mixtures containing 100 μm β2-m and 300 μm of each tetracycline congener. Samples were centrifuged after 48 h of incubation. Alongside the molecular mass standards, supernatant (s) and pellet (p) are shown for 4-epi-oxytetracycline (lane 1), anhydrochlortetracycline (lane 2), minocycline (lane 3), demeclocycline (lane 4), doxycycline (lane 5), and 4-epianhydrotetracycline (lane 6), respectively. Treated and non-treated β2-m are shown as controls. b, the percentage of protein in both supernatant and pellet of samples with and without tetracyclines. Proteins within each lane were quantified by densitometric analysis, and the percentage was determined as compared with control β2-m samples.
FIGURE 3.

Doxycycline dose-dependent inhibition of fibrillogenesis of the P32G β2-m variant. Experiments were carried out in triplicate in the same conditions reported in the legend for Fig. 1. Error bars indicate means ± S.D.
Doxycycline Inhibits β2-m Fibrillogenesis in the Presence of Collagen
Inhibitors of protein/peptide fibrillogenesis of potential pharmacological interest should be tested in assays mimicking the in vivo environment. For β2-m, we have recently set up a method of in vitro fibrillogenesis that generates β2-m fibrils under conditions reproducing a physiological milieu in the presence of two ubiquitous constituents of amyloid fibrils: collagen and heparin (31, 32). Although the method exhibits some variability in the time of onset of fibril growth, probably because of the complexity of the aggregation conditions, AFM analysis is extremely sensitive and specific for monitoring fibril growth on the surface of collagen fibers. Therefore, we used these conditions to investigate the effect of doxycycline on β2-m fibrillization onto collagen. We compared the AFM pattern of a β2-m sample aggregated in the absence of doxycycline (Fig. 4a) or in the presence of 360 μm doxycycline (Fig. 4b) after 3 months of incubation. In the absence of doxycycline, large bundles of fibrils (black arrows), intertwined or running parallel to each other, were present in close proximity to collagen fibers (white arrows). The latter exhibit a much larger size and appear completely covered by a homogeneous layer of aggregated proteins that masks, at least partially, their typical banding pattern. At variance, in the presence of doxycycline, a completely different pattern was observed (Fig. 4b). Strikingly, in this case, β2-m fibrils are totally missing, whereas both collagen fibers and background appear covered by tightly packed globular material (Fig. 4b, inset) that masks, even more effectively, the banding pattern of collagen fibers. Having assessed the inhibitory effect of doxycycline, we tested whether fibril growth was inhibited even at lower (50–200 μm) doxycycline concentrations. After 2 months of aggregation, fibrils were repeatedly observed in the sample incubated with 50 μm doxycycline, whereas at 100 and 200 μm doxycycline, the fibrils were absent (results not shown).
FIGURE 4.

Tapping mode atomic force microscopy image (amplitude data) of a β2-m sample incubated for 3 months at 37 °C in the presence of fibrillar collagen and heparin, in the absence (a) and in the presence (b) of 360 μm doxycycline. Scan size was 2.8 μm. Amplitude data are shown to better visualize the sample surface, with relatively thin amyloid fibrils (a, black arrows) lying on the surface of large corrugations corresponding to the collagen fibers (white arrows). The inset shows a topographic image (height data) of the texture of the background of b (corresponding to a flat, homogeneous portion of the sample) at a higher magnification; scan size 460 nm, Z range 20 nm.
Effect of Tetracyclines on Preformed β2-m Fibrils
The tetracyclines previously found to display the best anti-fibrillogenic activity with soluble β2-m were tested for their ability to solubilize preformed β2-m fibrils. The specific results obtained with doxycycline are reported in Fig. 5a. Fibrils at a concentration of 100 μm (based on monomeric β2-m) were exposed to increasing concentrations of doxycycline, and the ThT fluorescence was measured after 12 h of incubation. The IC50 required for fibril decomposition was much higher (5-fold) than that necessary to inhibit fibril growth. The material incubated with doxycycline at the concentration inducing 100% inhibition of ThT fluorescence was further analyzed by electron microscopy that confirmed the complete loss of the fibrillar structure (Fig. 5, b and c). Despite the destructuration of the ordered fibrillar assembly, the material coming from doxycycline-treated fibrils was still insoluble in physiologic buffer, suggesting that the fibrillar protein does not recover a native or native-like soluble structure upon incubation with doxycycline.
FIGURE 5.

Solubilization of preformed β2-m fibrils. a, comparative analysis of the doxycycline effect on fibrillogenesis (filled circles) and on fibril destructuration (filled triangles). Error bars indicate means ± S.D. b and c, electron microscopic images of ex vivo fibrils taken before (b) and after (c) 48 h of incubation with doxycycline.
Tetracycline-β2-m Interaction at Molecular Level
To provide information on the drug effects at the atomic level, the interaction between β2-m (0.12–0.7 mm) and doxycycline was analyzed by NMR spectroscopy under different solvent conditions, namely aqueous 70 mm phosphate buffer, 100 mm NaCl (pH 6.7), the same saline buffer containing 18% TFE (v/v), and pure water. These environments, although non-physiological, were selected to monitor the effect of doxycycline under conditions supporting (saline buffer, and much less effectively, pure water) or challenging (TFE) the conformational stability of the protein in solution. As in most biophysical investigations, however, physiologically distant conditions enable highlighting trends and drawing conclusions that help in interpreting functional biology. In particular, the saline buffer containing 18% TFE, although preventing excessive protein precipitation, is a fibrillogenic condition in the presence of nucleating fibril seeds (33) that is expected to approach very closely the partial unfolding and spreading over conformational ensembles also including fibril-competent species. Despite being far from the physiological fibrillogenic conditions, a TFE content around 20% in aqueous suspensions of fibril nuclei (seeds) has been repeatedly shown to result in the growth of fibrils that exhibit the same properties as the natural ones (21), making such a condition an established method of in vitro fibrillogenesis of wild type β2-m (34). At the doxycycline concentrations that were used for NMR experiments (0.12–3.0 mm), as imposed by the required protein concentrations and ratios, the best solubility was observed in pure water.
NMR in Aqueous Phosphate
In aqueous phosphate and NaCl, with or without TFE, doxycycline addition to protein solution was always accompanied by visible clouding and partial precipitation. When doxycycline was added to the β2-m solution in the absence of the organic co-solvent but in the presence of phosphate buffer and NaCl, we found very minor changes in the 1H-15N HSQC spectrum of the protein recorded at 37 °C. The chemical shifts of the protein amide correlation, along with the other 1H resonances, remained constant up to a nominal protein:doxycycline stoichiometry of ∼1:6. Minor chemical shift changes could be detected only for the 15N-1H correlation of the Phe-22 residue, whereas a number of other backbone amide cross-peaks exhibited distinct and progressive amplitude changes (supplemental Fig. 2). The corresponding residues map to the A-B loop, the B and C strands together with the intervening loop, the C′ strand and the following C′-D loop, the D-E loop, the F strand, and the C-terminal segment. Invariably, under any of the investigated solvent conditions, the doxycycline resonances shifted from the frequency observed without the protein and showed only minor line width increase (supplemental Fig. 3) due to a weak interaction leading to fast exchange. The observed doxycycline shifts were rather limited, and in phosphate buffer, with or without TFE, the most detectable effects occurred for the C4-N,N dimethyl group, the adjacent location (C4-H), and the aromatic hydrogens (data not shown), whereas in pure water, substantial shifts were unequivocally observed on the same side also for H4′, H5′, H6, and CH3(C6) (supplemental Fig. 3). The weakness of the fast exchange interaction was confirmed by the difficulty to detect direct NOEs between the protein and the doxycycline ligand, except a few saturation transfer difference correlations (35) (data not shown) that arise, by definition, from spin diffusion and are therefore nonspecific.
NMR Results in Pure Water
The NMR spectrum of the isolated protein (0.12–0.59 mm) at 25 °C in the absence of saline buffers revealed increased line widths, suggesting that the lack of an ionic environment increases the average extent of β2-m association. Upon titration with doxycycline at a titrant:protein ratio up to 3:1, several amide resonances of the 15N-1H HSQC spectrum showed small shifts and/or intensity changes (supplemental Figs. 4 and 5), namely at the N-terminal tail, the A-B, B-C, D-E, and F-G loops, the E strand, and the C-terminal tail. In addition, the HSQC spectra also revealed a general line sharpening that prompted us to run diffusion-ordered spectroscopy measurements to estimate the translational diffusion coefficient (D) of the protein and hence its effective molecular weight in solution. We calculated an apparent molecular mass of 20.2 kDa for β2-m in pure water, i.e. about twice the actual value (11.9 kDa), suggesting the occurrence of fast association equilibria with an average distribution around the dimeric adduct. Upon the addition of doxycycline, the diffusion-ordered spectroscopy spectra showed an increased D value of β2-m (Fig. 6a), consistent with a decrease of the protein apparent molecular mass to 12 kDa at 1:3 stoichiometric ratio with respect to doxycycline. Again, the whole NMR experimental pattern points to a fast exchange interaction between the tetracycline and the folded β2-m molecule. This interaction effectively stabilizes the protein and shifts its dynamic association equilibria in aqueous solvent toward the monomeric native state. The analysis of the chemical shift and intensity changes throughout doxycycline titration experiments (with and without saline buffer, supplemental Figs. 2 and 5), along with simultaneous diffusion-ordered spectroscopy controls and similar analysis as a function of the protein concentration in the absence of doxycycline (supplemental Fig. 6), enabled us to identify and distinguish unequivocally the protein sites involved in ligand interaction and in protein-protein association in pure water. In particular, doxycycline binds with the highest affinity to the region comprising the end of strand A, the initial part of strand B, and the central residues of the intervening loop (supplemental Fig. 7). A second doxycycline binding region occurs at the N-terminal stretch (fragments 2–4) and the facing B-C and F-G loops (supplemental Figs. 6b and 7). The protein-protein interaction surface comprises the N-terminal segment and the neighbor B-C and D-E loops (supplemental Fig. 6), in agreement with previous molecular dynamics evidence (36).
FIGURE 6.

a, two-dimensional diffusion-ordered spectroscopy map for β2-m in pure water in the presence of doxycycline at 1:0 molar ratio (black), at 1:1 molar ratio (blue), and at 1:3 molar ratio (red). The diffusion coefficients were estimated in the limit of a globular isotropic assumption for aqueous β2-m, using the trimeric EMILIN1 gC1q D value as a calibrator for a 51.2-kDa isotropic globule (41). b, β2-m interaction sites with doxycycline in pure water. These locations were obtained from the 15N-1H HSQC chemical shift (Δδ) and intensity change analysis upon doxycycline titration. The compensated Δδ values were calculated as described under “Experimental Procedures” after Mulder et al. (27). The color code is: yellow, the residues with Δδ ≤ Δδ + σ, where Δδ is the mean chemical shift change and σ is the chemical shift difference standard deviation; orange, residues with Δδ + σ < Δδ ≤ Δδ + 2σ; red, residues with Δδ > Δδ Δδ + 2σ.
NMR Analysis in Water/TFE
The scenario changed drastically when β2-m was titrated with doxycycline in the presence of 18% (v/v) TFE that approaches a highly fibrillogenic condition (33). We deliberately avoided reaching 20% (v/v) TFE, a condition that impairs NMR observation; rather, we chose to deal with a native structure destabilization close to the fibrillogenic transition limit (33), while retaining at 25 °C a soluble native-like species, as inferred from the chemical shift dispersion pattern very closely related to that observed in the absence of the organic co-solvent (supplemental Fig. 8). In the absence of doxycycline, an increase of the temperature to 37 °C resulted in massive protein precipitation, whereas the NMR pattern for most of the protein fraction still in solution was consistent with a largely unfolded species, with a native-like species being visible only at very low contour levels (Fig. 7a). Increasing the temperature to 37 °C in the presence of doxycycline (1.96 mm), at a nominal 3:1 ratio with respect to β2-m, resulted again in substantial protein precipitation, but in this case, the 1H-15N HSQC map revealed the equilibrium of the largely unfolded species and the native-like form at higher contour level (Fig. 7b). Therefore, the net effect of the tetracycline on the thermal destabilization of β2-m in 18% TFE is to shift the native/unfolded equilibrium toward the native-like species, although the largely unfolded species still predominates. The result was also confirmed by CD analysis (supplemental Fig. 9). Interestingly, the effect was also observed when the tetracycline was added after the denaturing exposure at 37 °C, revealing a surprising reversibility (in 18% TFE) of the doxycycline/β2-m system (supplemental Fig. 10). Whatever the timing of ligand addition, the NMR parameters of β2-m in aqueous TFE, i.e. 1H-15N peak chemical shifts and amplitudes, did not show substantial changes, nor did the NMR parameters of the doxycycline itself. Therefore, the observed difference in the distribution pattern of β2-m species in aqueous TFE (Fig. 7, a and b) should be attributed to a weak, transient, preferential interaction of the doxycycline with the native-like β2-m species. A quantitative assessment using pairs of resolved cross-peaks in the spectra of Fig. 7, a and b, led us to estimate a doxycycline-induced gain in ΔGunfolding of 0.9 kJ/mol. This very limited stabilization is not surprising in 18% aqueous TFE at 37 °C, i.e. a condition of substantial destabilization of the native protein (ΔGunfolding ∼−4 kJ/mol).
FIGURE 7.

a and b, 1H-15N HSQC spectra of β2-m 0.65 mm in TFE 18% (phosphate 70 mm, NaCl 100 mm) at 37 °C recorded without (a) or with (b) doxycycline (1.96 mm). The spectra were obtained using the same acquisition conditions from two samples prepared using the same β2-m mother solution, with a 10.0-μl addition of 0.100 m doxycycline for b. The experimental data were treated with the same processing parameters and at the same absolute scaling (TOPSPIN, Bruker). The reported maps were drawn starting at the same lowest contour level and with the same level numbers and spacing.
Doxycycline Modifies the Cytotoxicity of Soluble and Fibrillar β2-m
A fundamental issue regarding the mechanism of tissue damage caused by the amyloid deposits consists in defining the cytotoxic role of prefibrillar oligomeric aggregates. We recently reported that soluble β2-m is cytotoxic if oligomers are present (3), thus confirming and enriching previous reports on β2-m cytotoxicity (37). As some tetracyclines are apparently capable of decomposing fibrils into smaller aggregates, we studied the cytotoxicity of native and fibrillar β2-m exposed to doxycycline. Cytotoxicity was quantified by the MTT and lactate dehydrogenase release assays, widely used cell viability tests. We confirmed that the native β2-m is cytotoxic for human neuroblastoma SH-SY5Y cells when oligomers are not eliminated from the solution, whereas the fibrils are not toxic even at 20 μm (monomer concentration), a value exceeding by 4-fold the IC50 of soluble β2-m cytotoxicity (3). Fig. 8a shows that doxycycline, at a concentration equimolar to β2-m, abrogated the cytotoxicity of native soluble β2-m. On the contrary, and unexpectedly, fibril exposure to doxycycline triggered a cytotoxicity that was absent in untreated fibrils (Fig. 8b). The rescue of fibril cytotoxicity was directly related to the drug concentration. The results reported in Fig. 8b refer to an experiment in which β2-m fibrils were incubated for 72 h with doxycycline before their inclusion in the cell culture medium. However, to check whether a longer fibril incubation with doxycycline could modify cytotoxicity, we also analyzed β2-m fibrils incubated with the tetracycline for 2, 3, 4, 7, and 15 days. Fig. 8c shows that the cytotoxicity of this material reached a maximal activity after 3 days of incubation and then progressively declined to eventually disappear after 7 days. It is noteworthy that the cells incubated with β2-m fibrils not treated with doxycycline showed a surprisingly greater viability, as compared with control cells incubated only with vehicle. This aspect will be further investigated as it might suggest a positive role of the fibrillar net on cell growth. To this end, we notice that the presence of non-toxic fibrils in the cell medium can somehow promote cell adhesion to the support, as is known in the case of the curli amyloid fibrils favoring bacterial attachment to the substrate (38). Consistently, although ThT decreases along a monophasic curve, the cytotoxicity trend reflects a multiphasic process. Such a different dependence of ThT fluorescence and cytotoxicity on the time of fibril incubation with doxycycline (Fig. 8d) suggests that remodeling of the aggregated protein goes far beyond the modifications detected by the ThT assay that reaches a plateau after 48 h of fibril incubation with the drug.
FIGURE 8.

Effects of doxycycline on cytotoxicity of β2-m. a, β2-m (100 μm) was resolubilized in the absence or in the presence of different concentrations of doxycycline at 37 °C for 1.0 h. 10-μl aliquots of the samples were diluted in 90 μl of cell medium and added to wells coated with SH-SY5Y cells whose viability is reported as a function of the initial doxycycline concentration. b and c, preformed β2-m fibrils (100 μm, monomer concentration) were treated with doxycycline (Dox) for 72 h at the indicated concentrations (b) or at a fixed concentration (300 μm) for the indicated time lengths (c) and then added to the culture medium to estimate toxicity. Cell viability was measured by the MTT assay. The data are expressed as the percentage with respect to the control values obtained in three independent experiments, each value being the average of six trials. d, effect of the time of exposure to doxycycline of preformed β2-m fibrils on cell viability and ThT fluorescence quenching (same experiment as c). In all panels, error bars indicate means ± S.D.
Fig. 9 shows the relation between aggregate toxicity and the evolution of fibril morphology imaged by AFM as a function of the incubation time in the presence of doxycycline. Fig. 9a shows β2-m fibrils immediately after drug addition. Exposure to doxycycline gives rise to a partial fibril disaggregation into smaller units, as shown in Fig. 9b, acquired after 24 h of incubation. An extensive release of such oligomeric units took place in the subsequent hours. After 72 h of incubation with doxycycline (Fig. 9c), the accumulation of oligomers reached its maximum level; this result correlated very well with the maximum cytotoxicity found in the same conditions. The size distribution of these oligomers is rather broad, with height values ranging from less than 1 nm to ∼10 nm. At longer incubation times, oligomers are reassembled into more complex structures. In particular, after 7 days, the sample mainly consisted of ∼1-nm-high aggregates of variable length (Fig. 9d). Again, this result correlates well with the decrease of cytotoxicity found in the same conditions.
FIGURE 9.

Tapping mode AFM images (amplitude data) showing the evolution of the morphology of preformed β2-m fibrils as a function of the incubation time with doxycycline (300 μm). a, immediately after drug addition. b, at 24 h, when partial fibril dissolution into smaller units becomes visible. c, at 72 h, corresponding to maximum oligomer release and minimum cell viability, with initial rearrangement of the oligomers into more complex structures. d, at 7 days, when the sample is mostly aggregated into structures with uniform height (1 nm). Scan size: 2 μm (a, c, and d) and 1 μm (b).
DISCUSSION
The inhibition of the fibrillogenesis of wild type β2-m by tetracyclines displays the features of a dose-response reaction, and the effective concentration is graded from 47 and 49 μm of the more effective compounds (doxycycline and 4-epi-oxytetracycline, respectively) to 136 μm (anhydrochlortetracycline-rolitetracycline) and to >300 μm as measured for minocycline. The poor activity of minocycline was quite surprising because, among the tetracyclines, this drug was the most active against the aggregation of another amyloidogenic protein, PrPsc (9). This finding represents unequivocal evidence that in amyloidogenesis inhibition, a specific structure-function relation does exist between the single amyloidogenic proteins and each tetracycline species. On the basis of the first screening, we decided to focus our investigation on the anti-amyloidogenic effects of doxycycline because a vast pharmacological and toxicological experience already exists for this drug. The interference of doxycycline with β2-m aggregation was characterized by NMR spectroscopy that confirms the global persistence of the connectivities of the native protein even when it was exposed to conditions that favor unfolding and can trigger fibrillogenesis by seeding.
The anti-aggregating effect of doxycycline was confirmed by NMR analysis for its ability to rescue soluble native-like β2-m from the aggregates grown in ∼20% TFE and to decrease β2-m association in pure water. The stronger β2-m self-association in pure water reflects the effect of exposed hydrophobicity; such an effect is attenuated by the presence of salts that assist molecule solvation. The strong activity of doxycycline and 4-epi-oxytetracycline, in comparison with other tetracyclines, highlights the relevance of the C5 oxydryl. Despite the highly dynamic character of the interaction, the contact surfaces in the β2-m/tetracycline adduct have been successfully identified by specific NMR observations. Based on the current evidence, it is possible to establish that the N,N-dimethyl group of doxycycline, and probably, to a larger extent, the adjacent locations (C4′, C5, C5′, and C6), are involved in the transient adduct with the protein, whereas the protein appears to employ preferentially the region at the end of strand A and the initial part of strand B together with a part of the AB loop area. An additional lower affinity site for doxycycline binding appears to overlap the region that β2-m transiently uses for dynamic oligomerization, i.e. the N-terminal end with the facing B-C, D-E, and F-G loops. The structural details of this behavior will be discussed in a forthcoming study. Here, however, two aspects of the doxycycline-β2-m interaction are worth stressing, namely (i) the efficiency of the transient contact between doxycycline and the target protein, reaching a proper turnover rate because of the relative lability of the interaction, and (ii) the preferential doxycycline affinity for the natively folded conformer of β2-m that also accounts for the results obtained in the presence of TFE. The ability of doxycycline to inhibit the amyloidogenesis of β2-m was validated not only in 20% TFE but also in physiologic-like conditions, by using the variant P32G or in the presence of pro-amyloidogenic cofactors, i.e. collagen and heparin.
In particular, the effect of the drug in conditions mimicking the environment in which wild type β2-m makes fibrils in vivo has been qualitatively confirmed in tests carried out in the presence of collagen and heparin, when fibril growth is intimately attached to the collagen surface (31). We are aware that the concentrations of doxycycline required for the abrogation of fibrillogenesis in all the conditions we assayed largely exceed that reached in vivo by the drug in classical therapeutic regimes, i.e. typically 1–5 μm. However, tetracyclines are highly concentrated (up to 2,000-fold over the bulk tissue value) in the skeletal system (17), the elective target of this amyloidosis. Therefore, the successful abrogation in vitro of amyloid formation, at the drug concentration we used, supports the possibility that doxycycline can also be effective in vivo. It is worthy of note that upon prolonged exposure to the drug, the structure of β2-m fibrils is deeply remodeled, as shown by the loss of the extended polymeric pattern in the electron microscopy images and, consistently, by the remarkable reduction of ThT reactivity, although the protein remains irreversibly insoluble.
Dilemmas regarding the identification of the cytotoxic entity of amyloid proteins apply to dialysis-related amyloidosis similarly to the other systemic and localized amyloidoses. We have recently established that in the cell culture, oligomeric β2-m is cytotoxic, thus confirming previous data on the biological activity of soluble β2-m that is cytotoxic for certain cells (3) and mitogenic for other strains such as the osteoclasts (39). Because doxycycline deeply interferes with β2-m aggregation, we evaluated the effect of the drug on the biological activity of soluble and fibrillar β2-m. At equimolar concentrations, the presence of doxycycline fully abrogates the toxicity of soluble β2-m oligomers. This is consistent with the NMR data highlighting the suppression induced by doxycycline of the dynamic β2-m association in solution. On the other hand, the effect of the drug on the biological activity of mature β2-m fibrils is significantly more complex. The fibrils, per se, are not cytotoxic; however, fibril exposure to doxycycline elicits a transient cytotoxic activity that reaches the maximum effect after 3 days of incubation and vanishes after 7 days of incubation. The inspection by AFM of the evolution of the morphology of β2-m fibrils incubated with doxycycline at time intervals corresponding to those at which cytotoxicity was measured allowed us to identify the assemblies responsible for toxicity. In fact, in the toxic specimens, we imaged globular oligomeric aggregates that were substantially absent in the fibrillar samples not exposed to the drug. The same oligomers were also absent in the fibrillar samples incubated with doxycycline for 7 days, i.e. a time lapse over which the presence of the drug results in extensive structural reorganization. As it has been recently demonstrated for another amyloidogenic protein (40), it is likely that cytotoxicity results from the exposure of hydrophobic groups for β2-m oligomers as well; such exposure is very likely to drive the rearrangement of oligomers into more stable structures with reduced toxicity.
The dissection of the molecular effects of the interaction between doxycycline and the amyloidogenic protein β2-m has therefore confirmed and better clarified the intimate mechanism by which tetracyclines counteract the tendency of amyloidogenic proteins to acquire an ordered fibrillar structure and, in their prefibrillar state, a specific toxic structure. Such information could be rapidly translated into a new therapeutic opportunity for this amyloidosis. In our view, a selected population of patients like those under prolonged hemodialysis at high risk of amyloid deposition or with early amyloid deposits are the natural candidates for this therapy. The onset of dialysis-related amyloidosis can be easily recognized by the occurrence of carpal tunnel syndrome. Therefore, these patients could be treated before entering a stage of the disease characterized by irreversible tissue damage and in which the excessive amount of amyloid could defeat the therapeutic efficacy of the drug. Furthermore, as the deposition of this amyloid takes place in a tissue environment where tetracyclines naturally reach the highest possible concentration, we suggest that the in vivo validation of the doxycycline therapy in dialysis-related amyloidosis could represent an extraordinary proof of principle of the effectiveness of the class of anti-amyloid compounds acting by inhibiting amyloid fibril growth.
Acknowledgment
We thank Ruggero Tenni for providing natural collagen fibers.
This work was supported by grants from the Italian Ministry of University, Research and Instruction (MIUR) (projects FIRB and PRIN), the European Union (EU) (EURAMY Project LSHM-CT-2005-037525), and the Fondazione Cariplo (2007.5151 and 2009-2543).
This article was selected as a Paper of the Week.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–10.
F. Tagliavini, personal communication.
M. Sarajva, personal communication.
- β2-m
- β2-microglobulin
- ThT
- thioflavin T
- TFE
- trifluoroethanol
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- AFM
- atomic force microscopy
- HSQC
- heteronuclear single quantum coherence.
REFERENCES
- 1. Gejyo F., Yamada T., Odani S., Nakagawa Y., Arakawa M., Kunitomo T., Kataoka H., Suzuki M., Hirasawa Y., Shirahama T., Cohen A. S., Schmid K. (1985) Biochem. Biophys. Res. Commun. 129, 701–706 [DOI] [PubMed] [Google Scholar]
- 2. Giorgetti S., Stoppini M., Tennent G. A., Relini A., Marchese L., Raimondi S., Monti M., Marini S., Østergaard O., Heegaard N. H., Pucci P., Esposito G., Merlini G., Bellotti V. (2007) Protein Sci. 16, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giorgetti S., Raimondi S., Cassinelli S., Bucciantini M., Stefani M., Gregorini G., Albonico G., Moratti R., Montagna G., Stoppini M., Bellotti V. (2009) Nephrol. Dial. Transplant. 24, 1176–1181 [DOI] [PubMed] [Google Scholar]
- 4. Yamamoto S., Kazama J. J., Maruyama H., Nishi S., Narita I., Gejyo F. (2008) Clin. Nephrol. 70, 496–502 [DOI] [PubMed] [Google Scholar]
- 5. Bellotti V., Nuvolone M., Giorgetti S., Obici L., Palladini G., Russo P., Lavatelli F., Perfetti V., Merlini G. (2007) Ann. Med. 39, 200–207 [DOI] [PubMed] [Google Scholar]
- 6. De Lorenzi E., Giorgetti S., Grossi S., Merlini G., Caccialanza G., Bellotti V. (2004) Curr. Med. Chem. 11, 1065–1084 [DOI] [PubMed] [Google Scholar]
- 7. Merlini G., Ascari E., Amboldi N., Bellotti V., Arbustini E., Perfetti V., Ferrari M., Zorzoli I., Marinone M. G., Garini P., Diegoli M., Trizio D., Ballinari D. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 2959–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tagliavini F., Forloni G., Colombo L., Rossi G., Girola L., Canciani B., Angeretti N., Giampaolo L., Peressini E., Awan T., De Gioia L., Ragg E., Bugiani O., Salmona M. (2000) J. Mol. Biol. 300, 1309–1322 [DOI] [PubMed] [Google Scholar]
- 9. De Luigi A., Colombo L., Diomede L., Capobianco R., Mangieri M., Miccolo C., Limido L., Forloni G., Tagliavini F., Salmona M. (2008) PLoS ONE 3, e1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cardoso I., Saraiva M. J. (2006) FASEB J. 20, 234–239 [DOI] [PubMed] [Google Scholar]
- 11. Forloni G., Colombo L., Girola L., Tagliavini F., Salmona M. (2001) FEBS Lett. 487, 404–407 [DOI] [PubMed] [Google Scholar]
- 12. Diomede L., Cassata G., Fiordaliso F., Salio M., Ami D., Natalello A., Doglia S. M., De Luigi A., Salmona M. (2010) Neurobiol. Dis. 40, 424–431 [DOI] [PubMed] [Google Scholar]
- 13. Aitken J. F., Loomes K. M., Konarkowska B., Cooper G. J. (2003) Biochem. J. 374, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aitken J. F., Loomes K. M., Scott D. W., Reddy S., Phillips A. R., Prijic G., Fernando C., Zhang S., Broadhurst R., L'Huillier P., Cooper G. J. (2010) Diabetes 59, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malmo C., Vilasi S., Iannuzzi C., Tacchi S., Cametti C., Irace G., Sirangelo I. (2006) FASEB J. 20, 346–347 [DOI] [PubMed] [Google Scholar]
- 16. Loeb M. B., Molloy D. W., Smieja M., Standish T., Goldsmith C. H., Mahony J., Smith S., Borrie M., Decoteau E., Davidson W., McDougall A., Gnarpe J., O'donnell M., Chernesky M. (2004) J. Am. Geriatr. Soc. 52, 381–387 [DOI] [PubMed] [Google Scholar]
- 17. Agwuh K. N., MacGowan A. (2006) J. Antimicrob. Chemother. 58, 256–265 [DOI] [PubMed] [Google Scholar]
- 18. Saikali Z., Singh G. (2003) Anticancer Drugs 14, 773–778 [DOI] [PubMed] [Google Scholar]
- 19. Esposito G., Michelutti R., Verdone G., Viglino P., Hernández H., Robinson C. V., Amoresano A., Dal Piaz F., Monti M., Pucci P., Mangione P., Stoppini M., Merlini G., Ferri G., Bellotti V. (2000) Protein Sci. 9, 831–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jahn T. R., Parker M. J., Homans S. W., Radford S. E. (2006) Nat. Struct. Mol. Biol. 13, 195–201 [DOI] [PubMed] [Google Scholar]
- 21. Yamamoto S., Yamaguchi I., Hasegawa K., Tsutsumi S., Goto Y., Gejyo F., Naiki H. (2004) J. Am. Soc. Nephrol. 15, 126–133 [DOI] [PubMed] [Google Scholar]
- 22. Esposito G., Ricagno S., Corazza A., Rennella E., Gümral D., Mimmi M. C., Betto E., Pucillo C. E., Fogolari F., Viglino P., Raimondi S., Giorgetti S., Bolognesi B., Merlini G., Stoppini M., Bolognesi M., Bellotti V. (2008) J. Mol. Biol. 378, 887–897 [DOI] [PubMed] [Google Scholar]
- 23. Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 24. Braunschweiler L., Ernst R. R. (1983) J. Magn. Reson. 53, 521–528 [Google Scholar]
- 25. Jeener J., Meier B. H., Bachmann P., Ernst R. R. (1979) J. Chem. Phys. 71, 286–292 [Google Scholar]
- 26. Bodenhausen G., Ruben D. J. (1980) Chem. Phys. Lett. 69, 185–189 [Google Scholar]
- 27. Mulder F. A., Schipper D., Bott R., Boelens R. (1999) J. Mol. Biol. 292, 111–123 [DOI] [PubMed] [Google Scholar]
- 28. Jerschow A., Müller N. (1998) J. Magn. Reson. 132, 13–18 [DOI] [PubMed] [Google Scholar]
- 29. Hwang T. L., Shaka A. J. (1995) J. Magn. Reson. Ser. A. 112, 275–279 [Google Scholar]
- 30. Grzesiek S., Bax A. (1993) J. Am. Chem. Soc. 115, 12593–12594 [Google Scholar]
- 31. Relini A., Canale C., De Stefano S., Rolandi R., Giorgetti S., Stoppini M., Rossi A., Fogolari F., Corazza A., Esposito G., Gliozzi A., Bellotti V. (2006) J. Biol. Chem. 281, 16521–16529 [DOI] [PubMed] [Google Scholar]
- 32. Relini A., De Stefano S., Torrassa S., Cavalleri O., Rolandi R., Gliozzi A., Giorgetti S., Raimondi S., Marchese L., Verga L., Rossi A., Stoppini M., Bellotti V. (2008) J. Biol. Chem. 283, 4912–4920 [DOI] [PubMed] [Google Scholar]
- 33. Yamaguchi K., Naiki H., Goto Y. (2006) J. Mol. Biol. 363, 279–288 [DOI] [PubMed] [Google Scholar]
- 34. Yamaguchi K., Takahashi S., Kawai T., Naiki H., Goto Y. (2005) J. Mol. Biol. 352, 952–960 [DOI] [PubMed] [Google Scholar]
- 35. Mayer M., Meyer B. (1999) Angew. Chem. Int. Ed. Engl. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 36. Fogolari F., Corazza A., Viglino P., Zuccato P., Pieri L., Faccioli P., Bellotti V., Esposito G. (2007) Biophys. J. 92, 1673–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gordon J., Wu C. H., Rastegar M., Safa A. R. (2003) Int. J. Cancer 103, 316–327 [DOI] [PubMed] [Google Scholar]
- 38. Barnhart M. M., Chapman M. R. (2006) Annu. Rev. Microbiol. 60, 131–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Menaa C., Esser E., Sprague S. M. (2008) Kidney Int. 73, 1275–1281 [DOI] [PubMed] [Google Scholar]
- 40. Campioni S., Mannini B., Zampagni M., Pensalfini A., Parrini C., Evangelisti E., Relini A., Stefani M., Dobson C. M., Cecchi C., Chiti F. (2010) Nat. Chem. Biol. 6, 140–147 [DOI] [PubMed] [Google Scholar]
- 41. Verdone G., Corazza A., Colebrooke S. A., Cicero D., Eliseo T., Boyd J., Doliana R., Fogolari F., Viglino P., Colombatti A., Campbell I. D., Esposito G. (2009) J. Biomol. NMR 43, 79–96 [DOI] [PubMed] [Google Scholar]