

Abstract
ATP binding cassette transporter G1 (ABCG1) mediates the transport of cellular cholesterol to HDL, and it plays a key role in maintaining macrophage cholesterol homeostasis. During inflammation, HDL undergoes substantial remodeling, acquiring lipid changes and serum amyloid A (SAA) as a major apolipoprotein. In the current study, we investigated whether remodeling of HDL that occurs during acute inflammation impacts ABCG1-dependent efflux. Our data indicate that lipid free SAA acts similarly to apolipoprotein A-I (apoA-I) in mediating sequential efflux from ABCA1 and ABCG1. Compared with normal mouse HDL, acute phase (AP) mouse HDL containing SAA exhibited a modest but significant 17% increase in ABCG1-dependent efflux. Interestingly, AP HDL isolated from mice lacking SAA (SAAKO mice) was even more effective in promoting ABCG1 efflux. Hydrolysis with Group IIA secretory phospholipase A2 (sPLA2-IIA) significantly reduced the ability of AP HDL from SAAKO mice to serve as a substrate for ABCG1-mediated cholesterol transfer, indicating that phospholipid (PL) enrichment, and not the presence of SAA, is responsible for alterations in efflux. AP human HDL, which is not PL-enriched, was somewhat less effective in mediating ABCG1-dependent efflux compared with normal human HDL. Our data indicate that inflammatory remodeling of HDL impacts ABCG1-dependent efflux independent of SAA.
Keywords: high density lipoprotein, serum amyloid A, reverse cholesterol transport, macrophage, ATP binding cassette AI
Macrophages possess a number of mechanisms to regulate the balance between cholesterol uptake/synthesis and export. Of major importance are transport mechanisms that promote the efflux of excess cholesterol to extracellular acceptors, i.e., macrophage reverse cholesterol transport (RCT). The removal of excess cholesterol is critical in the vessel wall, where macrophage uptake of lipoprotein-derived lipid can lead to a pathological cholesterol load in the absence of sufficient removal systems. On the basis of studies in mice, two members of the ATP binding cassette (ABC) superfamily of transmembrane transporters, ABCA1 and ABCG1, play critical roles in preventing cholesterol accumulation in macrophages. In mice, combined deficiency of ABCA1 and ABCG1 in macrophages leads to impaired cellular cholesterol efflux in vitro and a massive increase in macrophage lipid accumulation in vivo (1–3). However, the role of ABCG1 in cholesterol efflux in human monocyte-derived macrophages has recently been questioned (4). Accumulating evidence suggests that ABCA1 and ABCG1 act through distinct, yet synergistic, mechanisms to promote macrophage RCT. Whereas lipid-poor apolipoproteins serve as extracellular acceptors for ABCA1-mediated phospholipid (PL) and cholesterol efflux, ABCG1 appears to promote efflux by redistributing intracellular cholesterol to plasma membrane domains accessible for removal by HDL, but not lipid-poor apolipoprotein A-I (apoA-I) (5). ABCA1 and ABCG1 may act sequentially to mediate efflux, such that nascent HDL generated through the lipidation of lipid-poor/free apoA-I by ABCA1 in turn serves as a substrate for cellular cholesterol export through ABCG1 (6, 7). Studies to measure macrophage RCT in vivo confirm that ABCA1 and ABCG1 have an additive effect on macrophage RCT in mice (8).
An important issue to be addressed is how the cooperative interaction between ABCA1 and ABCG1 functions during acute inflammation. In this condition, serum amyloid A (SAA) is a major acute phase (AP) protein highly induced in the liver (9). SAA is also induced by inflammatory stimuli in peripheral cells expressing ABCA1 and ABCG1, such as macrophages and adipocytes (9). Plasma SAA concentrations can increase up to 1000-fold during an AP response, with peak concentrations exceeding 1 mg/ml. Approximately 95% of AP SAA in the plasma is found associated with HDL, where it composes the major apolipoprotein (10). In addition, inflammatory HDL undergoes significant changes in lipid composition, with triglycerides tending to increase (11). Further, during inflammation there is concomitant induction (∼100-fold) of Group IIA secretory phospholipase A2 (sPLA2-IIA) in the liver, which leads to selective hydrolysis of HDL PL that alters the particle's structure and promotes its catabolism (12, 13).
Lipid-poor SAA has been shown to promote ABCA1-dependent cholesterol efflux similar to apoA-I (14–17). In this study, we investigated the extent to which SAA and AP HDL promote ABCG1-dependent efflux. Our data show that SAA acts analogously to apoA-I in effecting sequential efflux from ABCA1 and ABCG1. With respect to compositional changes in HDL that occur during inflammation, alterations in PL content and not the presence of SAA impact the ability of AP HDL to promote ABCG1-dependent efflux.
MATERIALS AND METHODS
Human subjects
Blood was collected from healthy volunteers for isolation of normal (N) HDL and from patients undergoing cardiac surgery using a membrane oxygenator (coronary artery bypass, valve replacement), 24 h post-operatively for isolation of AP HDL. Blood was collected only from patients who underwent successful uncomplicated surgery and who gave informed consent. The study was approved by the University of Kentucky Medical Institutional Review Board.
Animals
C57BL/6 mice were obtained from Jackson Laboratories. Mice lacking SAA1.1 and SAA 2.1 were generated by targeted deletion of both mouse acute phase SAA genes SAA1 and SAA2 (InGenious Targeting Laboratory Inc., Stony Brook, NY) using embryonic stem cells derived from C57BL/6×129 SVEV mice (18). The targeting vector contained a neo cassette that replaced ∼10.1 kb of SAA1 and SAA2, including exon 2 of both oppositely orientated genes. SAA null (SAAKO) mice and littermate controls [wild-type (WT)] were maintained in a pathogen-free facility under equal light-dark cycles with free access to water and food. To elicit an AP response, 12-16 week-old mice were injected intra-peritoneally with 6 µg lipopolysaccharide (LPS) (Escherichia coli 0111:B4, Sigma Chemical Co.) per gram of body weight. After 24 h the mice were humanely euthanized, and plasma was collected for preparation of HDL. All procedures were carried out in accordance with PHS policy and approved by the Veterans Administration Medical Center Institutional Animal Care and Use Committee (Assurance number A3506-01).
Cell culture
THP-1 monocytes originally obtained from the American Type Culture Collection were grown in RPMI supplemented with 10% heat-inactivated FBS, 1% penicillin, 1% streptomycin, and 2 mM glutamine. The cells were seeded into 35 mM culture wells (4 × 106 cells/well) and differentiated into macrophages by incubating them for 24 h in media supplemented with 50 ng/ml phorbol 12-myristate 13-acetate (PMA; Sigma). THP-1 macrophages were loaded with cholesterol by incubating the cells for 48 h with RPMI supplemented with 10% lipoprotein deficient serum, 100 µg/ml acetylated LDL, and 50 ng/ml PMA (6).
Baby hamster kidney (BHK) cells stably transfected with human ABCG1 cDNA under the control of the mifepristone-inducible GeneSwitch system were grown in DMEM supplemented with 10% FBS, 1% penicillin, 1% streptomycin, and 2 mM glutamine and kept under selection with 100 µg/ml hygromycin (Invitrogen) and 250 µg/ml Zeocin™ (Invitrogen) (5, 7). ABCG1 was induced by incubating cells in DMEM containing 0.2% fatty acid-free BSA and 10 nM mifepristone (Invitrogen). Control cells received no mifepristone.
Lipoproteins
Mouse HDL (d = 1.063-1.21 g/ml), human HDL (d = 1.063-1.21 g/ml) and human LDL (d = 1.019-1.063 g/ml) were isolated from mouse plasma (N or AP) or human plasma (N or AP) by density gradient ultracentrifugation, dialyzed against 150 mmol/l NaCl, 0.01% EDTA, sterile filtered, and stored under argon gas at 4°C (10). Protein concentrations were determined by the method of Lowry et al. (19).
Nascent HDL preparations
THP-conditioned acceptor particles (nascent HDL) were generated according to a published protocol (6). Briefly, lipid-loaded THP-1 macrophages were incubated for 24 h in RPMI containing 0.2% fatty acid-free-BSA and either 10 µg/ml human apoA-I (Biodesign) or 20 µg/ml SAA (corresponding to human SAA1α except for the presence of an N-terminal methionine and substitution of asparagine for aspartic acid at position 60 and arginine for histidine at position 71; Biovision). The media was harvested and centrifuged (1500 g for three minutes) to remove detached cells, and the concentration of nascent HDL particles was determined prior to use in efflux experiments by quantitative immunoblotting using purified apoA-I and SAA as standards.
Cholesterol efflux experiments
Cellular cholesterol efflux experiments in BHK cells were carried out essentially as described (20). Cells (∼70% confluent) in 12-well plates were labeled with 0.2 μCi/ml [3H]cholesterol (35-50 Ci/mmol, Amersham Biosciences) in complete DMEM medium for 48 h. Cells were then washed three times with PBS containing 1 mg/ml BSA (PBS-BSA) and equilibrated overnight in DMEM containing 0.2% fatty acid-free BSA (DMEM-BSA). Cells were incubated overnight in media containing 10 nM mifepristone to induce ABCG1 expression. Control cells received no mifepristone. Following two additional washes with PBS-BSA, cells were incubated for 5 h at 37°C in DMEM-BSA with or without HDL, hydrolyzed HDL, or lipid-free or THP-1-conditioned apoA-I or SAA, as indicated. Following incubation, the medium was collected and centrifuged to remove detached cells. Adherent cells were washed at 4°C twice with PBS-BSA and twice with PBS. Radioactivity in the media was measured directly in a Packard β liquid scintillation counter. Cellular lipid was extracted with hexane/isopropyl alcohol (3:2 v/v) for 30 min at room temperature and counted for radioactivity. Efflux of cellular [3H] cholesterol to media was expressed as the percentage of total radioactivity in media and cells. ABCG1-specific values were calculated as the difference between the efflux values in mifepristone-treated and control cells.
HDL hydrolysis
Mouse HDL (0.6-1.0 mg/ml HDL protein) was hydrolyzed by human recombinant sPLA2-IIA in Tris-buffered saline (pH 7.4) containing 10 mg/ml fatty-acid free BSA and 2 mM CaCl2. Lipolysis was terminated after 24 h incubations at 37°C by the addition of EDTA (final concentration 20 mM). Mock hydrolyzed HDL was generated under the same conditions but omitting sPLA2-IIA. Hydrolysis reactions were carried out as reported in the literature (21–23) and approximated physiological conditions. The extent of PL hydrolysis was assessed by measuring the amount of free fatty acids released (Wako Chemicals).
Gradient gel electrophoresis and Western blots
Aliquots containing 25-50 ng lipid free apoA-I or SAA, or apoA-I or SAA in conditioned media from THP-1 macrophages, were separated by nondenaturing gradient gel electrophoresis (GGE). Electrophoresis was carried out in 4-20% polyacrylamide gels for 3.5 h at 200 V at 4°C, and the samples were then transferred to PVDF membranes (100 min at 100 V at 4°C) and immunoblotted using either anti-human apoA-I (Calbiochem) or anti-human SAA (Behring, Germany) antibodies, as indicated. Bound antibodies were detected by enhanced chemiluminescence (GE Healthcare, NJ). To assess the effect of PL hydrolysis on the size distribution of mouse HDL, mock-hydrolyzed and sPLA2-hydrolyzed HDL (1 μg HDL protein) were separated by GGE as described above and immunoblotted using an anti-mouse apoA-I antibody (Biodesign International).
Statistical analyses
Data are expressed as mean ± SEM. After testing for normalcy and equal variance, data was analyzed for statistical significance as indicated in the figure legends. Significance was set at ★ = <0.05; ★★ = <0.01; and ★★★ = <0.001.
RESULTS
THP-1 macrophages convert lipid-free SAA to nascent HDL particles
Previous studies determined that lipid-poor SAA stimulates ABCA1-dependent cholesterol efflux (14–17). In the current study, we investigated whether SAA is lipidated through the action of ABCA1 to generate nascent HDL particles in a manner analogous to ABCA1-mediated lipidation of apoA-I (6). THP-1 macrophages were treated with PMA and cholesterol loaded to upregulate ABCA1 expression, and then incubated with 10 µg/ml lipid-free apoA-I or SAA for 24 h. The media from the cells were subjected to nondenaturing GGE followed by immunoblotting to assess the extent of lipidation of the THP-1-conditioned apoA-I and SAA (Fig. 1A, B). Lipid-free apoA-I migrated as a predominant band below the smallest size standard, whereas lipid-free SAA migrated as two distinct bands on the nondenaturing gel, possibly due to its propensity to aggregate. After THP-1 conditioning, a portion of apoA-I migrated as larger-sized particles, indicating formation of nascent HDL. Thus, our results were analogous to a previous report that ∼15% of lipid-free apoA-I converts to nascent HDL when incubated with THP-1 macrophages under similar conditions (24). In contrast to apoA-I, virtually all of the SAA migrated as larger-sized HDL particles after incubation with THP-1 macrophages (Fig. 1B). The size distribution of THP-1-conditioned apoA-I and SAA was distinct.
Fig. 1.

Nascent HDL particles generated by THP-1 macrophages. Equal amounts of lipid-poor apoA-I and apoA-I incubated for 24 h with cholesterol-loaded THP-1 macrophages were separated by nondenaturing GGE and immunoblotted using anti-human apoA-I (A). Equal amounts of lipid-poor SAA and SAA incubated for 24 h with cholesterol-loaded THP-1 macrophages were separated by nondenaturing GGE and immunoblotted using anti-human SAA (B). Exposures to ECL reagent were chosen to optimize the visualization of nascent HDL particles. apoA-I, apolipoprotein A-I; GGE, gradient gel electrophoresis; SAA, serum amyloid A.
THP-1-conditioned SAA stimulates ABCG1-mediated cholesterol efflux
Using the ABCG1-inducible BHK in vitro model system (5, 7), we next assessed the ability of lipid-poor and THP-1-conditioned SAA to serve as acceptors for ABCG1-independent and ABCG1-dependent cholesterol efflux. Efflux to lipid-poor and THP-1-conditioned apoA-I was measured for comparison (Fig. 2). In BHK cells without ABCG1 induction, cholesterol efflux to lipid-poor apoA-I was negligible. Lipidation of apoA-I by THP-I conditioning resulted in significantly increased ABCG1-independent efflux. Lipid-poor SAA was more efficient than apoA-I in mediating ABCG1-independent efflux, similar to what has been reported for untransfected HeLa cells (16) and HepG2 cells (17). However, lipidation of SAA had no effect on ABCG1-independent efflux. As expected, lipid-poor apoA-I was not an effective acceptor for ABCG1-dependent cholesterol efflux, which was defined as the difference in efflux by BHK cells induced to express ABCG1 (Total) and control BHK cells (ABCG1-independent) (5, 6). When compared with lipid-poor apoA-I, ABCG1-dependent efflux to THP-1-conditioned apoA-I was significantly increased (6.5-fold), confirming an earlier report that THP-1 macrophages convert lipid-poor apoA-I to nascent HDLs that are sufficiently lipidated to serve as substrates for ABCG1-mediated cholesterol export (6). Interestingly, THP-1 macrophages had a similar effect on lipid-poor SAA, such that ABCG1-dependent efflux was 9.5-fold higher for THP-1-modified SAA compared with lipid-poor SAA. This enhanced ABCG1-dependant efflux suggests that the increased size of THP-1-conditioned SAA (Fig. 1B) is likely due to lipidation rather than extensive aggregation. Taken together, our results suggest that ABCA1 and ABCG1 can act sequentially to mediate cellular cholesterol export to SAA.
Fig. 2.

ABCG1-mediated cholesterol efflux to nascent HDL particles. Transfected BHK cells were labeled with [3H] cholesterol and then induced to express ABCG1 as described in “Materials and Methods.” Cellular cholesterol efflux stimulated by 5 h incubations with either lipid-poor apoA-I or SAA or conditioned media from THP-1 cells incubated with apoA-I or SAA (10 µg/ml apolipoprotein) was determined. ABCG1-dependent efflux represents the difference between BHK cells treated with mifepristone (Total) and untreated cells (ABCG1-independent). Values are the mean ± SEM of triplicate determinations. Total and ABCG1-independent efflux to lipid-poor apoA-I and SAA, or THP-1-conditioned apoA-I and SAA data were analyzed using one way ANOVA with Tukey-adjusted pairwise comparisons. Different lower case letters identify different means among apoA-I groups; different capital letters identify different means among SAA groups (P ≤ 0.004). In addition, the ABCG1-dependent component was compared across groups in posthoc tests. ★★★ indicates P < 0.001. Similar results were obtained in two additional experiments. ABCG1, ATP binding cassette transporter G1; apoA-I, apolipoprotein A-I; BHK, baby hamster kidney; SAA, serum amyloid A.
SAA present on mouse AP HDL is not responsible for an enhanced capacity of AP HDL to stimulate ABCG1-mediated efflux
During an AP response, the majority (∼95%) of SAA in plasma is associated with HDL (10). Thus, it was of interest to determine whether SAA-containing AP HDL differed in its ability to serve as an acceptor for ABCG1-dependent efflux compared with N HDL. Accordingly, HDL was isolated from plasma of N mice (N WT HDL) and mice 24 h after injection with LPS (AP WT HDL). Whereas SAA was not detectable in N WT HDL, it comprised approximately 40% of the apolipoprotein associated with AP WT HDL (data not shown). SAA-bearing AP HDL exhibited a modest but significant reduced capacity to elicit ABCG1-independent cholesterol efflux compared with N WT HDL. However, this same HDL elicited significantly more ABCG1-dependent efflux compared with N WT HDL (Fig. 3, white and black bars).
Fig. 3.

ABCG1-mediated cholesterol efflux to N WT HDL, AP WT HDL, and AP SAAKO HDL. Transfected BHK cells were labeled with [3H] cholesterol and then induced to express ABCG1 as described in “Materials and Methods.” Cellular cholesterol efflux stimulated by 5 h incubations with 25 µg/ml N WT HDL (white bars), AP WT HDL (black bars), and AP SAAKO HDL (gray bars) was measured. ABCG1-dependent efflux represents the difference between BHK cells treated with mifepristone (Total) and untreated cells (ABCG1-independent). The data shown are representative of five experiments with five separate preparations of ligands, each performed in triplicate. Total and ABCG1-independent efflux were analyzed using one way ANOVA with Tukey-adjusted pairwise comparisons. Different letters correspond to different means (P < 0.001, except a versus b where P < 0.05). In addition, the ABCG1-dependent component was compared across groups in posthoc tests. ★★★ indicates P < 0.001. ABCG1, ATP binding cassette transporter G1; AP, acute phase; BHK, baby hamster kidney; N, normal; SAAKO, mice with targeted deletion of SAA1.1 and SAA2.1; WT, wild-type.
To dissect out the role of SAA versus other modifications of AP WT HDL in mediating enhanced ABCG1-dependent efflux, we isolated N and AP HDL from mice lacking the two major AP SAA isoforms, SAA1.1 and SAA2.1, and assessed their ability to stimulate efflux through ABCG1. In efflux assays using five separate HDL preparations, there was no significant difference in ABCG1-dependent efflux stimulated by N WT HDL (2.06 ± 0.13% of total label) and N SAAKO HDL (1.95 ± 0.20% of total label; data not shown). This result was not surprising, given that HDLs isolated from untreated WT and SAAKO mice do not differ in their lipid or apolipoprotein content (18). However, AP SAAKO HDL was even more effective in mediating ABCG1-dependent cholesterol efflux compared with AP WT HDL (Fig. 3, gray bars). In efflux assays using five separate N and AP HDL preparations from WT and SAAKO mice, incubations with SAA-containing AP WT HDL resulted in a ∼17% increase in ABCG1-dependent efflux compared with the corresponding N HDL, whereas ABCG1-dependent efflux to AP SAAKO HDL was increased more than 2-fold (data not shown). Thus, it appears that the presence of SAA per se does not account for the enhanced capacity of mouse AP HDL to serve as an acceptor for ABCG1-mediated efflux and that other modifications of HDL that occur during inflammation can significantly influence cellular cholesterol efflux by ABCG1.
Phospholipid depletion of HDL reduces ABCG1-mediated efflux
We recently reported that AP WT HDL has decreased protein and increased PL content compared with N WT HDL, and these differences are even more pronounced in AP SAAKO HDL that lacks SAA (18). We therefore speculated that alterations in the surface composition of N and AP HDL may account for differences in ABCG1-dependent efflux. To investigate whether increased PL content of AP HDL enhances its ability to accept cellular cholesterol through ABCG1, N WT HDL (0.92 nmol PL/µg HDL protein), AP WT HDL (0.97 nmol PL/µg HDL protein), and AP SAAKO HDL (1.18 nmol PL/µg HDL protein) were incubated in the presence or absence of sPLA2-IIA and then tested for their ability to stimulate ABCG1-dependent efflux (Fig. 4A–C). PL hydrolysis was assessed by measuring the release of FFA (data not shown) and was calculated to result in approximately 30-50% loss of PL from HDL particles. PL hydrolysis was also confirmed by size reduction in sPLA2-treated HDLs (Fig. 4A–C, inserts). For each of the three HDL preparations, hydrolysis by sPLA2 significantly reduced ABCG1-mediated cellular cholesterol efflux, indicating that decreasing the PL content of HDL irrespective of the presence of SAA has a negative effect on ABCG1-mediated efflux.
Fig. 4.
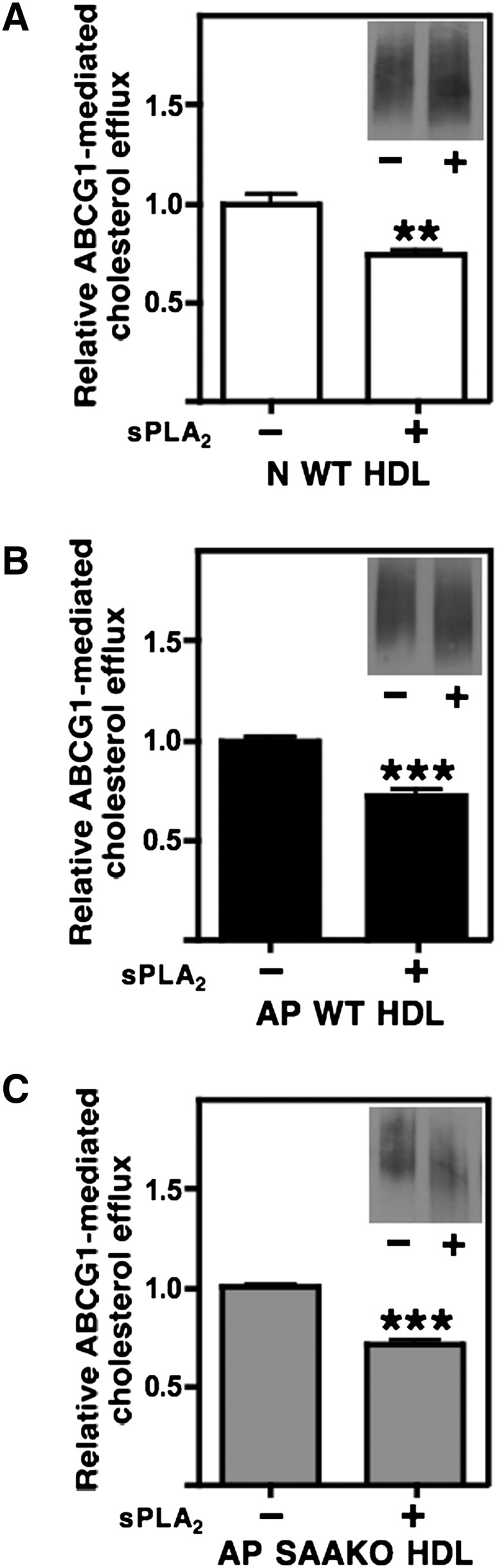
ABCG1-mediated cholesterol efflux to sPLA2-hydrolyzed N WT HDL, AP WT HDL, and AP SAAKO HDL. N WT HDL (A), AP WT HDL (B), and AP SAAKO HDL (C) were hydrolyzed overnight in the absence or presence of sPLA2-IIA (2 µg/mg HDL), as indicated. Transfected BHK cells were labeled with [3H] cholesterol, induced to express ABCG1, and then incubated for 5 h with the indicated HDLs (25 µg/ml). ABCG1-mediated efflux was calculated as the difference between ABCG1-expressing cells and control cells that were not induced to express ABCG1. Values represent the mean ± SEM of triplicate determinations. Inserts: Aliquots of mock-hydrolyzed (−) and sPLA2-hydrolyzed (+) HDL were separated by nondenaturing GGE and immunoblotted with anti- apoA-I to show the size reduction of hydrolyzed HDLs. Significance between mock- and sPLA2-hydrolyzed samples was determined by Student t-test. ABCG1, ATP binding cassette transporter G1; AP, acute phase; BHK, baby hamster kidney; N, normal; SAAKO, mice with targeted deletion of SAA1.1 and SAA2.1; sPLA2-IIA, Group IIA secretory phospholipase A2; WT, wild-type.
To substantiate this finding, AP WT HDL was incubated with increasing concentrations of sPLA2-IIA and then assessed in ABCG1-dependent efflux assays. As expected, incubations with increasing concentrations of sPLA2-IIA resulted in more extensive PL hydrolysis, as assessed by the release of FFA (Fig. 5A) and the generation of progressively smaller HDLs (data not shown). In BHK cells lacking ABCG1, PL hydrolysis did not alter the ability of AP WT HDL to serve as an acceptor for cholesterol efflux (Fig. 5B). However, for cells induced to express ABCG1, hydrolysis by sPLA2 decreased efflux in a dose-dependent manner, indicating that ABCG1-mediated efflux is reduced as PL content is decreased.
Fig. 5.

ABCG1-mediated cholesterol efflux to sPLA2-hydrolyzed AP WT HDL. AP WT HDL was incubated overnight with the indicated concentration of sPLA2-IIA. PL hydrolysis was monitored by measuring the release of FFA (A). Transfected BHK cells were radiolabeled with [3H] cholesterol and then treated with (black bars) or without (white bars) mifepristone to induce ABCG1. Cellular cholesterol efflux stimulated by 5 h incubations with 25 µg/ml AP HDL hydrolyzed with the indicated amount of sPLA2-IIA was measured; values represent the mean ± SEM of triplicate determinations (B). Significance between mock- and sPLA2-hydrolyzed samples was determined by one way ANOVA followed by Bonferroni's posttest. ABCG1, ATP binding cassette transporter G1; AP, acute phase; BHK, baby hamster kidney; sPLA2-IIA, Group IIA secretory phospholipase A2; WT, wild-type.
AP human HDL is not a better acceptor for ABCG1-dependent efflux than normal human HDL
In humans, the AP response is characterized by a marked increase in plasma sPLA2-IIA activity that does not occur in C57BL/6 mice due to a frame shift mutation in the mouse sPLA2-IIA gene (25). Thus, it was of interest to determine if AP human HDL is enriched in PL similar to AP WT HDL and if AP human HDL is altered in its ability to serve as an acceptor for ABCG1-mediated cholesterol efflux. For these studies, HDL was isolated from the plasma of normal volunteers (N hu HDL) and from patients 24 h after cardiac surgery (AP hu HDL). Despite marked enrichment with SAA (Fig. 6A), the protein and lipid content of AP hu HDL was not significantly altered compared with N hu HDL (Table 1). As previously reported, AP hu HDL migrated as slightly larger particles compared with N hu HDL when subjected to nondenaturing GGE (Fig. 6B). This finding is likely due to the enrichment of the HDL surface with SAA (10). Compared with N hu HDL, AP hu HDL evoked a modest but significant decrease in ABCG1-dependent cholesterol efflux (Fig. 6C). Taken together, our data indicate that the impact of acute inflammation on ABCG1-dependent efflux to mouse and human HDL is different and may be related to the effect of the AP response on the PL content of the respective HDLs.
Fig. 6.

ABCG1-mediated cholesterol efflux to N and AP human HDL. HDL (d = 1.063-1.21 g/ml) was isolated from the plasma of normal subjects (N) or from patients 24 h post cardiac surgery (AP). Visualization was by Coomassie staining of HDL subjected to SDS-PAGE (A) and GGE (B). Transfected BHK cells were labeled with [3H] cholesterol and then induced to express ABCG1 as described in “Materials and Methods.” Cellular cholesterol efflux stimulated by 5 h incubations with 25 µg/ml N and AP human HDL was measured. ABCG1-mediated efflux was calculated as the difference between ABCG1-expressing cells and control cells that were not induced to express ABCG1 (C). Values (mean ± SEM) were obtained from eight experiments performed with three preparations of human N and AP HDL and are expressed relative to the corresponding N hu HDL. Significance was determined by Student t-test. ABCG1, ATP binding cassette transporter G1; AP, acute phase; BHK, baby hamster kidney; GGE, gradient gel electrophoresis; N, normal.
TABLE 1.
Composition of human HDL
| Component | N | AP |
|---|---|---|
| % | % | |
| Prot | 49.5 ± 0.77 | 51.3 ± 1.13 |
| FC | 4.4 ± 0.24 | 3.9 ± 0.50 |
| CE | 13.4 ± 0.90 | 10.8 ± 0.73 |
| TG | 2.4 ± 0.93 | 4.2 ± 0.74 |
| PL | 30.4 ± 1.54 | 29.8 ± 1.88 |
HDL was isolated from blood of healthy volunteers (N) and cardiac surgery patients 24 h post-operatively (AP). Protein (prot) concentration was determined by the method of Lowry. Total cholesterol, free cholesterol (FC), triglycerides (TG), and phospholipids (PL) were determined enzymatically using commercial kits (WAKO chemicals, Richmond, VA). Cholesterol ester (CE) was calculated as the difference between total cholesterol and FC. Values represent the mean ± SEM of three HDL preparations.
DISCUSSION
It has long been recognized that plasma HDL concentrations are inversely related to the risk of atherosclerotic cardiovascular disease. The protective effect of HDL is largely attributed to its key role in RCT, whereby excess cholesterol from peripheral cells is transported back to the liver for excretion in the bile. During acute inflammation, HDL undergoes substantial changes in lipid and apolipoprotein composition that could alter its ability to carry out individual steps in the RCT pathway. In this study, we focused on the impact of inflammation on ABCG1-dependent cellular cholesterol efflux. We utilized a well-established in vitro system that provides inducible expression of ABCG1 and thus allows for direct measurements of ABCG1-dependent cellular cholesterol efflux (5, 7, 26). We determined that, similar to apoA-I, lipid-poor SAA supports the cooperative interaction of ABCA1 and ABCG1 to produce nascent HDL. We also showed that inflammatory remodeling of mouse HDL modestly enhances ABCG1-dependent efflux and that the presence of SAA on inflammatory HDL is not required for this effect. Our data indicate that PL enrichment of HDL, such as what occurs during an AP response in mice, has a positive effect on ABCG1-dependent efflux. This is in agreement with an earlier report that ABCG1-dependent efflux correlates with the PL content of acceptor particles (6).
We showed that lipid-free SAA is highly effective in stimulating ABCA1-dependent efflux, consistent with previous reports (14–17, 27, 28). This finding is not unexpected, given that nascent HDL generated through ABCA1 appears to require the presence of amphophilic α-helices as a key conformation in the acceptor polypeptide, rather than a specific amino acid sequence (27, 28). The predicted secondary structure of human SAA1 indicates two amphophilic α-helical segments (29). Like apoA-I, lipid-poor SAA has been shown to stabilize ABCA1 protein, providing additional evidence that SAA interacts with ABCA1 in a manner similar to apoA-I (15). Based on their migration on nondenaturing gels, the interaction of ABCA1 with lipid-poor SAA appears to generate nascent HDLs with a size distribution that is distinctly larger compared with nascent HDL generated by apoA-I (Fig. 1). Consistent with our analysis, Abe-Dohmae et al. (14) determined by size-exclusion chromatography that apoA-I-containing nascent HDL elutes in two distinct peaks, both of which are smaller than nascent HDL generated by SAA. They also identified distinct differences in the migration of the two HDLs when separated by agarose gel electrophoresis, reflecting the respective isoelectric point values of apoA-I and SAA. Thus, nascent HDLs generated by apoA-I and SAA clearly possess different physiochemical properties.
The majority of SAA in plasma is found associated with HDL, predominantly in the denser HDL3 subfraction (10). The process by which SAA-containing HDL is formed during acute inflammation is not entirely clear. One proposed mechanism is that lipid-free SAA secreted by hepatocytes associates with existing spherical HDL particles through a remodeling process that may involve the displacement of apoA-I (10, 30, 31). However, induction of an AP response in apoA-I-deficient mice leads to the formation of large, spherical HDL particles in which >90% of the protein is SAA, suggesting that SAA is capable of sequestering lipid to form HDL in the absence of apoA-I and other apolipoproteins (30, 31). On the other hand, adenoviral vector-mediated expression of SAA in apoA-I-deficient mice in the absence of inflammation results in circulating SAA that is mostly in a lipid-poor form, suggesting that components of the AP response are required for the biogenesis of SAA-rich HDL in the absence of apoA-I (32). Studies in ABCA1-deficient mice demonstrate that the formation of SAA-containing HDL is dependent on ABCA1 (15), consistent with several reports, including the current study, that incubating cells with exogenous SAA results in robust cholesterol release in an ABCA1-dependent manner (14–16). In this study, we show for the first time that nascent HDL generated by SAA serves as an efficient substrate for ABCG1-mediated cellular cholesterol efflux. The extent to which ABCG1 contributes to the biogenesis of SAA-containing HDLs in vivo requires further study. What also is not entirely clear is whether ABCG1 plays a major role in cholesterol efflux in humans. Although extensive evidence indicates that ABCA1 and ABCG1 act in an additive manner to promote RCT in mice (1–3), the importance of this interaction in humans is likely more complex and less well studied. LXR-stimulated cholesterol efflux in human cholesterol-loaded macrophages (THP-1 cells or peripheral blood monocytes) was reported to be independent of ABCG1 (4). Others, however, have shown that in type II diabetic patients, reduced expression of ABCG1 is associated with increased cholesterol accumulation in macrophages (33). The precise relevance of the interaction between ABCA1 and ABCG1 in humans merits further investigation given the known species differences in the regulation of key genes involved in cholesterol homeostasis (34–36).
The impact of acute inflammation on macrophage RCT in vivo has been investigated in mice (37, 38). For these studies, 3H-cholesterol-labeled J774 macrophages or primary mouse macrophages were administered into the peritoneal cavity of normal and LPS-injected mice, and the movement of 3H-cholesterol from these cells into plasma, liver, and feces was monitored. The results of both studies indicated that acute inflammation impairs macrophage-to-feces RCT. A likely contributing factor to this reduction in RCT was decreased hepatic expression of ABCG5 and ABCG8, major transport proteins mediating biliary cholesterol secretion (37, 38). Whether SAA per se contributes to decreased RCT during the AP response was addressed by Annema et al. (37), who determined that adenovirus overexpression of mouse SAA (but not human SAA) results in a significant reduction in fecal excretion of the macrophage-derived 3H-cholesterol tracer. However, there was no evidence that the rate of movement of 3H-cholesterol from macrophages to the plasma or the liver was impaired as a result of SAA overexpression. Taken together, in vivo studies suggest that the integrated effect of inflammation is to retard macrophage-to-feces RCT, although the impact of individual components of the AP response on specific steps in the RCT pathway has not been completely delineated. Our data indicate that reduced macrophage-to-feces RCT during acute inflammation in mice is not likely due to a detrimental effect on ABCG1-dependent efflux.
Studies focusing on the ability of AP-HDL to promote macrophage cholesterol efflux in vitro have been carried out. In one study, N and AP human HDL promoted cholesterol efflux from THP-1 cells in a similar manner, but enrichment of the N HDL with SAA ex vivo reduced cellular cholesterol efflux by 30% (39). McGillicuddy et al. (38) reported that inflammatory remodeling of mouse HDL impairs its capacity to serve as an acceptor for macrophage cholesterol efflux. They also reported that HDL isolated from humans subjected to experimental endotoxemia was less effective in stimulating cholesterol efflux from cholesterol-loaded J774 macrophages compared with normal human HDL. Similarly, Annema et al. (37) concluded that efflux from cholesterol-loaded THP-1 macrophages to plasma or HDL isolated from sepsis patients was markedly reduced compared with healthy controls. On the other hand, on the basis of their studies in J774 macrophages, Kisilevsky et al. concluded that SAA on AP HDL promotes macrophage efflux by mobilizing intracellular cholesterol stores, thereby facilitating its transport out of cells (40).Thus, available data regarding the effect of inflammatory remodeling and/or SAA enrichment of HDL on macrophage cholesterol efflux is somewhat conflicting and likely influenced by the cell system utilized and the source of the HDL ligand. To date, the impact of inflammation on ABCG1-dependent efflux has not been specifically addressed.
In the current study, we determined that inflammatory remodeling of mouse HDL, particularly PL enrichment, had an enhancing effect on ABCG1 efflux that was more pronounced in mice lacking SAA (Fig. 3). In humans, inflammatory remodeling of HDL appeared to negatively impact efflux through ABCG1, as HDL isolated from patients undergoing an AP response due to cardiac surgery was modestly deficient in its ability to stimulate ABCG1-dependent efflux compared with HDL from healthy controls (Fig. 6). We recently reported that acute inflammation in WT mice results in a modest increase in HDL PL content, and AP HDL from SAA DKO mice is even more PL-enriched (18). In contrast, compositional analyses indicated that the AP response in humans is not associated with PL enrichment of HDL (Table 1), possibly due to the induction of sPLA2-IIA that occurs during the AP response in humans but not C57BL/6 mice. Thus, our data suggests that increasing the PL content of HDL has a positive effect on ABCG1-dependent efflux. This conclusion is substantiated by our finding that sPLA2-IIA hydrolysis of normal and AP mouse HDL significantly decreased efflux through ABCG1 (Figs. 4 and 5). However, the difference in ABCG1-dependent efflux between normal and AP HDLs may not be entirely attributable to differences in PL content. For example, sPLA2 hydrolysis of AP WT HDL resulted in a 31% decrease in PL content, and this was associated with a 30% decrease in efflux (Fig. 4B). A similar 33% difference in ABCG1-dependent efflux was observed between N WT HDL and AP WT HDL (Fig. 3), despite only a 5% difference in PL content between these two HDLs. Studies to compare macrophage RCT in WT and human sPLA2-IIA transgenic mice have demonstrated a decreased rate of appearance of 3H-cholesterol in plasma of transgenic mice, indicating that sPLA2 may decrease macrophage cholesterol efflux during inflammation (37). These results appear to contradict the work of Sankaranarayanan et al. (26), who reported that HDL3 incubated with multilamellar vesicles to enrich the particles with PL were not altered in their ability to serve as substrates for ABCG1-dependent efflux. The reason for the discrepant results is not clear. It is possible that enriching HDL with PL ex vivo generates a particle whose structure is not equivalent to HDLs that are remodeled during acute inflammation in vivo.
In summary, our data indicate that the extensive remodeling of HDL that occurs during acute inflammation has modest effects on ABCG1-dependent efflux. Interestingly, inflammatory remodeling of mouse HDL increases ABCG1-dependent efflux, whereas efflux stimulated by human HDL appears to decrease during inflammation. We provide evidence that this differential effect is likely due to differences in the PL content of AP HDL from the respective species. The presence of SAA on AP HDL does not appear to alter the capacity of the particle to serve as a substrate for ABCG1. The capacity of lipid-poor SAA to promote sequential efflux from ABCA1 and ABCG1 may be important for cholesterol flux out of atherosclerotic lesions, where each of these three factors is present.
Acknowledgments
The authors thank Vicky Noffsinger and Darrell Robertson for excellent technical assistance.
Footnotes
Abbreviations:
- ABC
- ATP binding cassette transporter
- AP
- acute phase
- apoA-I
- apolipoprotein A-I
- BHK
- baby hamster kidney
- GGE
- gradient gel electrophoresis
- LPS
- lipopolysaccharide
- N
- normal
- PL
- phospholipid
- PMA
- phorbol 12-myristate 13-acetate
- RCT
- reverse cholesterol transport
- SAA
- serum amyloid A
- SAAKO
- mice with targeted deletion of SAA1.1 and SAA2.1
- sPLA2-IIA
- Group IIA secretory phospholipase A2
- WT
- wild-type
This work was supported by National Institutes of Health Grant P01 HL-086670. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Studies were supported with resources and facilities provided by the Lexington, KY, Veterans Affairs Medical Center.
REFERENCES
- 1.Out R., Hoekstra M., Habets K., Meurs I., de Waard V., Hildebrand R. B., Wang Y., Chimini G., Kuiper J., Van Berkel T. J., et al. 2008. Combined deletion of macrophage ABCA1 and ABCG1 leads to massive lipid accumulation in tissue macrophages and distinct atherosclerosis at relatively low plasma cholesterol levels. Arterioscler. Thromb. Vasc. Biol. 28: 258–264. [DOI] [PubMed] [Google Scholar]
- 2.Out R., Jessup W., Le Goff W., Hoekstra M., Gelissen I. C., Zhao Y., Kritharides L., Chimini G., Kuiper J., Chapman M. J., et al. 2008. Coexistence of foam cells and hypocholesterolemia in mice lacking the ABC transporters A1 and G1. Circ. Res. 102: 113–120. [DOI] [PubMed] [Google Scholar]
- 3.Yvan-Charvet L., Ranalletta M., Wang N., Han S., Terasaka N., Li R., Welch C., Tall A. R. 2007. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Invest. 117: 3900–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larrede S., Quinn C. M., Jessup W., Frisdal E., Olivier M., Hsieh V., Kim M. J., Van Eck M., Couvert P., Carrie A., et al. 2009. Stimulation of cholesterol efflux by LXR agonists in cholesterol-loaded human macrophages is ABCA1-dependent but ABCG1-independent. Arterioscler. Thromb. Vasc. Biol. 29: 1930–1936. [DOI] [PubMed] [Google Scholar]
- 5.Vaughan A. M., Oram J. F. 2005. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 280: 30150–30157. [DOI] [PubMed] [Google Scholar]
- 6.Gelissen I. C., Harris M., Rye K. A., Quinn C., Brown A. J., Kockx M., Cartland S., Packianathan M., Kritharides L., Jessup W. 2006. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler. Thromb. Vasc. Biol. 26: 534–540. [DOI] [PubMed] [Google Scholar]
- 7.Vaughan A. M., Oram J. F. 2006. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J. Lipid Res. 47: 2433–2443. [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Collins H. L., Ranalletta M., Fuki I. V., Billheimer J. T., Rothblat G. H., Tall A. R., Rader D. J. 2007. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 117: 2216–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhlar C. M., Whitehead A. S. 1999. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 265: 501–523. [DOI] [PubMed] [Google Scholar]
- 10.Coetzee G. A., Strachan A. F., van der Westhuyzen D. R., Hoppe H. C., Jeenah M. S., de Beer F. C. 1986. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 261: 9644–9651. [PubMed] [Google Scholar]
- 11.Khovidhunkit W., Kim M. S., Memon R. A., Shigenaga J. K., Moser A. H., Feingold K. R., Grunfeld C. 2004. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J. Lipid Res. 45: 1169–1196. [DOI] [PubMed] [Google Scholar]
- 12.de Beer F. C., de Beer M. C., van der Westhuyzen D. R., Castellani L. W., Lusis A. J., Swanson M. E., Grass D. S. 1997. Secretory non-pancreatic phospholipase A2: influence on lipoprotein metabolism. J. Lipid Res. 38: 2232–2239. [PubMed] [Google Scholar]
- 13.Pruzanski W., Vadas P., Browning J. 1993. Secretory non-pancreatic group II phospholipase A2: role in physiologic and inflammatory processes. J. Lipid Mediat. 8: 161–167. [PubMed] [Google Scholar]
- 14.Abe-Dohmae S., Kato K. H., Kumon Y., Hu W., Ishigami H., Iwamoto N., Okazaki M., Wu C. A., Tsujita M., Ueda K., et al. 2006. Serum amyloid A generates high density lipoprotein with cellular lipid in an ABCA1- or ABCA7-dependent manner. J. Lipid Res. 47: 1542–1550. [DOI] [PubMed] [Google Scholar]
- 15.Hu W., Abe-Dohmae S., Tsujita M., Iwamoto N., Ogikubo O., Otsuka T., Kumon Y., Yokoyama S. 2008. Biogenesis of HDL by SAA is dependent on ABCA1 in the liver in vivo. J. Lipid Res. 49: 386–393. [DOI] [PubMed] [Google Scholar]
- 16.Stonik J. A., Remaley A. T., Demosky S. J., Neufeld E. B., Bocharov A., Brewer H. B. 2004. Serum amyloid A promotes ABCA1-dependent and ABCA1-independent lipid efflux from cells. Biochem. Biophys. Res. Commun. 321: 936–941. [DOI] [PubMed] [Google Scholar]
- 17.van der Westhuyzen D. R., Cai L., de Beer M. C., de Beer F. C. 2005. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J. Biol. Chem. 280: 35890–35895. [DOI] [PubMed] [Google Scholar]
- 18.De Beer M. C., Webb N. R., Wroblewski J. M., Noffsinger V. P., Rateri D. L., Ji A., Van Der Westhuyzen D. R., De Beer F. C. 2010. Impact of serum amyloid A on high density lipoprotein composition and levels. J. Lipid Res. 51: 3117–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193: 265–275. [PubMed] [Google Scholar]
- 20.Francis G. A., Knoop R. H., Oram J. F. 1995. Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier disease. J. Clin. Invest. 96: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rye K. A., Hime N. J., Barter P. J. 1995. The influence of cholesteryl ester transfer protein on the composition, size, and structure of spherical, reconstituted high density lipoproteins. J. Biol. Chem. 270: 189–196. [DOI] [PubMed] [Google Scholar]
- 22.Rye K. A., Hime N. J., Barter P. J. 1997. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J. Biol. Chem. 272: 3953–3960. [DOI] [PubMed] [Google Scholar]
- 23.Rye K. A., Wee K., Curtiss L. K., Bonnet D. J., Barter P. J. 2003. Apolipoprotein A-II inhibits high density lipoprotein remodeling and lipid-poor apolipoprotein A-I formation. J. Biol. Chem. 278: 22530–22536. [DOI] [PubMed] [Google Scholar]
- 24.Bielicki J. K., McCall M. R., Forte T. M. 1999. Apolipoprotein A-I promotes cholesterol release and apolipoprotein E recruitment from THP-1 macrophage-like foam cells. J. Lipid Res. 40: 85–92. [PubMed] [Google Scholar]
- 25.Kennedy B. P., Payette P., Mudgett J., Vadas P., Pruzanski W., Kwan M., Tang C., Rancourt D. E., Cromlish W. A. 1995. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J. Biol. Chem. 270: 22378–22385. [DOI] [PubMed] [Google Scholar]
- 26.Sankaranarayanan S., Oram J. F., Asztalos B. F., Vaughan A. M., Lund-Katz S., Adorni M. P., Phillips M. C., Rothblat G. H. 2009. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J. Lipid Res. 50: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Remaley A. T., Stonik J. A., Demosky S. J., Neufeld E. B., Bocharov A. V., Vishnyakova T. G., Eggerman T. L., Patterson A. P., Duverger N. J., Santamarina-Fojo S., et al. 2001. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 280: 818–823. [DOI] [PubMed] [Google Scholar]
- 28.Remaley A. T., Thomas F., Stonik J. A., Demosky S. J., Bark S. E., Neufeld E. B., Bocharov A. V., Vishnyakova T. G., Patterson A. P., Eggerman T. L., et al. 2003. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J. Lipid Res. 44: 828–836. [DOI] [PubMed] [Google Scholar]
- 29.Turnell W., Sarra R., Glover I. D., Baum J. O., Caspi D., Baltz M. L., Pepys M. B. 1986. Secondary structure prediction of human SAA1. Presumptive identification of calcium and lipid binding sites. Mol. Biol. Med. 3: 387–407. [PubMed] [Google Scholar]
- 30.Cabana V. G., Reardon C. A., Wei B., Lukens J. R., Getz G. S. 1999. SAA-only HDL formed during the acute phase response in apoA-I+/+ and apoA-I−/− mice. J. Lipid Res. 40: 1090–1103. [PubMed] [Google Scholar]
- 31.Hajri T., Elliott-Bryant R., Sipe J. D., Liang J. S., Hayes K. C., Cathcart E. S. 1998. The acute phase response in apolipoprotein A-1 knockout mice: apolipoprotein serum amyloid A and lipid distribution in plasma high density lipoproteins. Biochim. Biophys. Acta. 1394: 209–218. [DOI] [PubMed] [Google Scholar]
- 32.Webb N. R., de Beer M. C., van der Westhuyzen D. R., Kindy M. S., Banka C. L., Tsukamoto K., Rader D. L., de Beer F. C. 1997. Adenoviral vector-mediated overexpression of serum amyloid A in apoA-I-deficient mice. J. Lipid Res. 38: 1583–1590. [PubMed] [Google Scholar]
- 33.Mauldin J. P., Nagelin M. H., Wojcik A. J., Srinivasan S., Skaflen M. D., Ayers C. R., McNamara C. A., Hedrick C. C. 2008. Reduced expression of ATP-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation. 117: 2785–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Goff W., Zheng P., Brubaker G., Smith J. D. 2006. Identification of the cAMP-responsive enhancer of the murine ABCA1 gene: requirement for CREB1 and STAT3/4 elements. Arterioscler. Thromb. Vasc. Biol. 26: 527–533. [DOI] [PubMed] [Google Scholar]
- 35.Li A. C., Binder C. J., Gutierrez A., Brown K. K., Plotkin C. R., Pattison J. W., Valledor A. F., Davis R. A., Willson T. M., Witztum J. L., et al. 2004. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Invest. 114: 1564–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rigamonti E., Helin L., Lestavel S., Mutka A. L., Lepore M., Fontaine C., Bouhlel M. A., Bultel S., Fruchart J. C., Ikonen E., et al. 2005. Liver X receptor activation controls intracellular cholesterol trafficking and esterification in human macrophages. Circ. Res. 97: 682–689. [DOI] [PubMed] [Google Scholar]
- 37.Annema W., Nijstad N., Tolle M., de Boer J. F., Buijs R. V., Heeringa P., van der Giet M., Tietge U. J. 2010. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J. Lipid Res. 51: 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGillicuddy F. C., de la Llera Moya M., Hinkle C. C., Joshi M. R., Chiquoine E. H., Billheimer J. T., Rothblat G. H., Reilly M. P. 2009. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 119: 1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banka C. L., Yuan T., de Beer M. C., Kindy M., Curtiss L. K., de Beer F. C. 1995. Serum amyloid A (SAA): influence on HDL-mediated cellular cholesterol efflux. J. Lipid Res. 36: 1058–1065. [PubMed] [Google Scholar]
- 40.Tam S. P., Flexman A., Hulme J., Kisilevsky R. 2002. Promoting export of macrophage cholesterol: the physiological role of a major acute-phase protein, serum amyloid A 2.1. J. Lipid Res. 43: 1410–1420. [DOI] [PubMed] [Google Scholar]