

Abstract
Background
MicroRNAs (miRNAs) regulate gene expression by mediating gene silencing at transcriptional and post-transcriptional levels in higher plants. miRNAs and related target genes have been widely studied in model plants such as Arabidopsis and rice; however, the number of identified miRNAs in soybean (Glycine max) is limited, and global identification of the related miRNA targets has not been reported in previous research.
Results
In our study, a small RNA library and a degradome library were constructed from developing soybean seeds for deep sequencing. We identified 26 new miRNAs in soybean by bioinformatic analysis and further confirmed their expression by stem-loop RT-PCR. The miRNA star sequences of 38 known miRNAs and 8 new miRNAs were also discovered, providing additional evidence for the existence of miRNAs. Through degradome sequencing, 145 and 25 genes were identified as targets of annotated miRNAs and new miRNAs, respectively. GO analysis indicated that many of the identified miRNA targets may function in soybean seed development. Additionally, a soybean homolog of Arabidopsis SUPPRESSOR OF GENE SLIENCING 3 (AtSGS3) was detected as a target of the newly identified miRNA Soy_25, suggesting the presence of feedback control of miRNA biogenesis.
Conclusions
We have identified large numbers of miRNAs and their related target genes through deep sequencing of a small RNA library and a degradome library. Our study provides more information about the regulatory network of miRNAs in soybean and advances our understanding of miRNA functions during seed development.
Background
MicroRNAs (miRNAs) are endogenous ~21-nt noncoding RNAs derived from single-stranded RNA precursors that can form stem-loop structures [1,2]. MiRNA was first identified in Caenorhabditis elegans and subsequently found in almost all eukaryotes [3]. In higher plants, miRNAs play important roles in different developmental stages by mediating gene silencing at transcriptional and post-transcriptional levels [4-6]. Soybean is the most widely planted oil crop in the world; however, the regulation of its seed development is not well studied. The roles of miRNAs in soybean seed development remain largely unknown. Therefore, identification of new miRNAs and elucidation of their functions in seed development will help us understand the regulation of soybean lipid synthesis. Recently, the soybean genome sequence has been finished [7], which will greatly advance biological research on soybeans.
Although many soybean miRNAs were identified in previous research [8-10], the number of miRNAs known in soybean is still very small and considerably lower than that in Arabidopsis or rice. Most identified soybean miRNAs are of high abundance and conserved in many species; however, low-abundance and species-specific miRNAs may play important roles in soybean-specific processes. Generally, it is not easy to get information on these miRNAs by conventional methods. Recently, next-generation sequencing technology has been developed and widely applied to genomic studies such as gene expression pattern analysis, genome sequencing and small RNA sequencing. Because of its ultra high-throughput, many new miRNAs with low abundance could be identified using this technology.
To date, the majority of miRNA targets in soybean were predicted by bioinformatics approaches, and only a small portion were experimentally validated. A high-throughput degradome library sequencing technology has been developed for global identification of targets of miRNAs in Arabidopsis, rice and grapevine [11-18]. To detect new miRNAs participating in soybean seed development and to identify targets of soybean miRNAs globally, a small RNA library and a degradome library using RNAs from developing soybean seeds were constructed and sequenced by a Solexa analyzer. Each library generated more than 6 million short reads, and 26 new miRNAs were identified, of which 17 miRNAs belong to new families and 9 miRNAs belong to conserved families. A total of 170 genes sliced by small RNAs were detected via degradome library sequencing. Among these, 64 genes were reproduction-related genes, and the corresponding miRNAs may have a function in soybean seed development.
Results
Overview of small RNA library sequencing
The soybean small RNA library was constructed using RNAs obtained from seeds of 15-day-old after flowering and sequenced by Solexa SBS technology. We obtained more than 6 million raw reads, ranging from 18 to 30 nucleotides in length. As seen in Figure 1, the highest abundance was found for sequences with 21, 22 and 24 nucleotides (nt). After removal of low quality reads and adapter contaminants, 2,145,586 unique reads were collected and 1,495,099 (69.8%) sequences were perfectly mapped to the soybean genome using SOAP2 software (Table 1) [19]. Small RNAs were analyzed by BLAST against the known noncoding RNAs (rRNA, tRNA, snRNA, snoRNA) deposited in the Rfam and NCBI Genbank databases [20]. 25,944 distinct small RNAs belonging to these categories were removed to avoid degradation contamination. The remaining reads were used to identify the conserved and new miRNAs.
Figure 1.

Distribution of Solexa reads in the soybean small RNA library. Solexa reads with 21, 22, or 24 nucleotides were the most enriched in total small RNA sequences.
Table 1.
Different categories of small RNAs by deep sequencing
| Category | Unique reads | Total reads |
|---|---|---|
| All reads | 2,145,586 | 5,908,211 |
| Match genomea | 1,495,099 | 4,790,766 |
| Known miRNAsb | 1,695 | 677,062 |
| Rfamc | 25,944 | 450,869 |
| Unannotated | 1,467,460 | 3,662,835 |
aGenome sequences downloading from Glyma1 assembly
bKnown miRNAs deposited at miRBase database
cRfam including rRNA, tRNA, snRNA and snoRNA
Prediction and validation of new miRNAs
In total, 207 soybean miRNAs were annotated in the latest miRBase database [21,22], and most of these were identified by small RNA library sequencing. In this study, 55 annotated miRNAs were detected in a seed small RNA library. The remaining 152 miRNAs, mostly soybean specific, were not detected, possibly because of low expression levels or spatial expression pattern. Twenty-six new soybean miRNAs not previously reported were identified by bioinformatic analysis. These new miRNAs were named temporarily in the form of Soy_number, e.g., Soy_1 (Table S1 in Additional File 1). Among the 26 new miRNAs, 17 miRNAs belonged to new families that had never been found in eukaryotes (Table S1 in Additional File 1). All precursors of new miRNAs had regular stem-loop structures, and four of these, Soy_1, Soy_2, Soy_12 and Soy_20, were presented in Figure 2. These RNA structures were predicted by MFOLD software and checked manually [23].
Figure 2.

Predicted RNA hairpin structures of new miRNA precursors. Precursor structures of 4 newly identified soybean miRNAs (Soy_1, Soy_2, Soy_12, and Soy_20) were predicted by MFOLD pipeline. Mature miRNA and miRNA star sequences are highlighted in red and blue, respectively. The numbers along the structure are nucleotide sites from the 5' end of the pre-miRNA sequence.
Forty-six miRNA-star sequences (miRNA*), the complementary strands of functional mature miRNA, were also detected in this study (Table S1 in Additional File 1). These sequences are rarely found via conventional sequencing because of their quick degradation in cells. The detection of miRNA* represented further evidence for the existence of mature miRNAs. The miRNA* sequences for 38 known miRNAs and 8 new miRNAs were discovered (Figure 2, 3; Table S1 in Additional File 1). Soy_13 is the star strand of Soy_25, which belongs to the family of miR2118 [24]. Gso-miR2118 has been validated in wild soybean by northern blot in previous research [24]. In our study, Soy_13 was detected 3 times more than Soy_25 by Solexa sequencing (Table S1 in Additional File 1). Therefore, Soy_13 may be also a functional miRNA in soybean, not a miRNA* of Soy_25. In Figure 2, miRNA mature sequences and miRNA* sequences in miRNA precursors are highlighted using different colors. Their locations relative to RNA loops in precursors were not invariable. Large-scale sequencing allowed us to identify many mature miRNA variants, which represent some differences in the 5' and/or 3' ends of mature miRNA sequences (Figure 3).
Figure 3.

Diversification of mature miRNA production from miRNA precursors. Detected diverse isoforms of three conserved and one new mature miRNAs from soybean are shown. MiRNA star sequences are underlined in red. "Abundance" is the detected number of reads in small RNA library sequencing.
To validate the predicted new miRNAs, stem-loop RT-PCR was performed to examine their expression in soybean seeds [25]. Primers used in stem-loop RT-PCR are listed in Table S2 in Additional File 2. All of the 26 predicted miRNAs were found to be expressed in soybean seeds (Figure 4). The gma-miR168 was amplified as a positive control (Figure 4).
Figure 4.
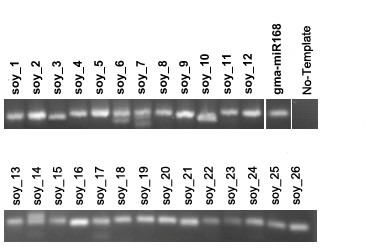
Stem-loop RT-PCR for identified new miRNAs. In total, 26 new miRNAs were confirmed by stem-loop RT-PCR with 40-cycle-amplification. The sizes of PCR products were around ~60 bp. Gma-miR168: the positive control; No Template: no RNA was added as a template in the RT reaction.
Soybean seed degradome library construction and sequencing
To identify the target genes of miRNAs in the soybean transcriptome, the widely adopted technology of degradome library sequencing was applied in this study [11-16]. MiRNAs mediate gene silencing by two mechanisms: mRNA cleavage and translation repression. In higher plants, miRNAs slice mRNAs to regulate gene expression in most cases [1,2,11]. MiRNA-directed cleavage leaves a free 5' phosphate at the 3' fraction of the sliced genes. Through poly(A) RNA purification, we constructed a 5' uncapped mRNA library. The transcriptome-wide degradome information can be collected through high-throughput sequencing. We constructed a soybean seed degradome library and obtained more than 15 million raw reads with 99% of sequences having 20 or 21 nt by Solexa sequencing. After quality filtration and adapter removal, we obtained 1,662,975 unique reads, of which 1,062,557 (64%) were perfectly matched to the soybean genome (Table 2). However, only 663,641 (40%) reads could be mapped to a single position in the soybean genome. Interestingly, 308,578 (18%) reads had two hits in the genome. We further used the published Williams 82 cDNA database as the template to map clean reads. In total, 1,044,162 unique reads were mapped to the soybean cDNAs, indicating the high quality of the present degradome library (Table 2). The reads that mapped to soybean cDNAs were subjected to further analysis.
Table 2.
Summary of degradome reads mapping statistics
| Raw reads | Unique reads | Genome mapped reads | Reads with single hit to genome | cDNA mapped readsa |
|---|---|---|---|---|
| 15168792 | 1662975 | 1062557 | 663,641 | 1044162 |
acDNA sequences downloaded from Glyma1 assembly
Identification and classification of targets for annotated miRNAs
Compared to other mRNA degradation mechanisms, miRNA mediated mRNA cleavage possesses special features. The sliced region of the mRNA should be complementary to the miRNA sequence, and the cleavage site is usually between the 10th and 11th nucleotides from the 5' end of the miRNA. These features were used to identify targets of miRNAs. We first extracted 15 nt upstream and downstream of 5' soybean cDNAs sequences mapped by degradome reads to generate 30 nt target signatures as "t-signature" [12]. These signatures were collected to find miRNA targets using CleaveLand pipeline [18]. According to the abundance of miRNA-complemented signatures relative to other signatures mapped to mRNAs, the identified targets could be sorted into 4 classes. The targets with only miRNA-directed cleavages were classified as Class I. In Class II, the cleavage signature abundance was mostly enriched among all signatures. The abundance of cleavage signatures was higher than the median in Class III targets. The rest, with a low abundance of cleavage signatures, were grouped as Class IV. Because the abundance of miRNA-directed cleavage targets in Class I and Class II was much higher than other signatures, the targets in these two classes could have low false discovery rates and be more accurate. All identified miRNA targets were classified according to these criteria.
To date, 207 soybean annotated miRNAs have been deposited in the miRBase database. Few miRNA targets have been validated by experimental methods [8-10]. In our study, 126 targets of 19 evolutionarily conserved miRNA families were identified (Table 3). Only 9 soybean-specific miRNA families were found to silence 19 genes (Table 3; the miRNAs designated by a). It should be noted that many targets of a single conserved miRNA are in pairs with very similar sequences, and the gma-miR156, gma-miR160, gma-miR164, gma-miR166, gma-miR172 and gma-miR396 had at least 10 targets, with the gma-miR396 having more than 20 targets (Table 3). On the other hand, the soybean-specific miRNAs appear to have only a limited number of targets.
Table 3.
Identified targets of known miRNAs in soybean
| miRNA | Target gene | Target annotation | Class | Abundance(TP10M) | cleavage site(nt) | target site location |
|---|---|---|---|---|---|---|
| gma-miR156 | Glyma04g37390# | SBP domain protein | I | 39 | 938 | 5'UTR |
| Glyma06g17700# | SBP domain protein | I | 39 | 1185 | CDS | |
| Glyma05g00200* | SBP domain protein | I | 42 | 1202 | CDS | |
| Glyma04g32070* | SBP domain protein | II | 42 | 130 | 3'UTR | |
| Glyma17g08840* | SBP domain protein | I | 42 | 1011 | CDS | |
| Glyma05g38180# | SBP domain protein | I | 33 | 1356 | CDS | |
| Glyma08g01450# | SBP domain protein | I | 33 | 1154 | CDS | |
| Glyma18g00890 | SBP domain protein | II | 75 | 249 | CDS | |
| Glyma12g27330 | SBP domain protein | II | 17 | 621 | 5'UTR | |
| Glyma11g36980 | SBP domain protein | II | 47 | 1223 | CDS | |
| Glyma02g30670 | SBP domain protein | I | 109 | 688/689 | CDS | |
| Glyma18g36960 | SBP domain protein | I | 14 | 723 | CDS | |
| Glyma02g13370 | SBP domain protein | I | 173 | 1219/1220 | CDS | |
| gma-miR159 | Glyma15g35860# | MYB family transcription factor | II | 27 | 937 | CDS |
| Glyma13g25720# | MYB family transcription factor | II | 27 | 838 | CDS | |
| Glyma20g11040 | MYB family transcription factor | I | 17 | 918 | CDS | |
| gma-miR160 | Glyma11g20490# | Auxin response factor | II | 134 | 1510 | CDS |
| Glyma10g35480# | Auxin response factor | I | 134 | 740 | CDS | |
| Glyma12g08110# | Auxin response factor | II | 134 | 1501 | CDS | |
| Glyma13g20370* | Auxin response factor | I | 177 | 1670 | CDS | |
| Glyma10g06080* | Auxin response factor | I | 177 | 1355 | CDS | |
| Glyma13g02410 | Auxin response factor | I | 74 | 1280 | CDS | |
| Glyma14g33730 | Auxin response factor | I | 29 | 1184 | CDS | |
| Glyma19g36570 | Auxin response factor | II | 807 | 652 | CDS | |
| Glyma04g43350 | Auxin response factor | II | 43 | 1337 | 5'UTR | |
| Glyma13g40030 | Auxin response factor | II | 67 | 1277 | CDS | |
| Glyma20g32040 | Auxin response factor | I | 19 | 1313 | CDS | |
| Glyma12g29720 | Auxin response factor | I | 25 | 1626 | CDS | |
| gma-miR162 | Glyma12g35400* | embryo-related protein | IV | 13 | 995 | CDS |
| Glyma13g35110* | embryo-related protein | IV | 13 | 963 | CDS | |
| gma-miR164 | Glyma17g10970# | NAC family transcription factor | I | 750 | 795 | CDS |
| Glyma05g00930# | NAC family transcription factor | II | 750 | 751 | CDS | |
| Glyma06g21020# | NAC family transcription factor | I | 750 | 741 | CDS | |
| Glyma04g33270# | NAC family transcription factor | I | 750 | 634 | CDS | |
| Glyma13g34950* | NAC family transcription factor | I | 153 | 747 | CDS | |
| Glyma12g35530* | NAC family transcription factor | II | 153 | 712 | CDS | |
| Glyma15g40510# | NAC family transcription factor | II | 34 | 730 | CDS | |
| Glyma08g18470# | NAC family transcription factor | II | 34 | 731 | CDS | |
| Glyma12g26190 | NAC family transcription factor | I | 87 | 778 | CDS | |
| miRNA | Target gene | Target annotation | Class | Abundance(TP10M) | cleavage site(nt) | target site location |
| gma-miR164 | Glyma06g35660 | NAC family transcription factor | I | 24 | 811 | CDS |
| gma-miR166 | Glyma15g13640#b | HD-ZIP transcription factor | II | 273 | 568/570 | CDS |
| Glyma08g21610#b | HD-ZIP transcription factor | II | 235 | 898 | CDS | |
| Glyma04g09000# | HD-ZIP transcription factor | II | 273 | 93-95 | CDS | |
| Glyma07g01950# | HD-ZIP transcription factor | II | 273 | 618/620 | CDS | |
| Glyma08g21620# | HD-ZIP transcription factor | II | 273 | 789/791 | CDS | |
| Glyma07g01940# | HD-ZIP transcription factor | II | 273 | 919/921 | CDS | |
| Glyma06g09100#b | HD-ZIP transcription factor | II | 273 | 567/569 | CDS | |
| Glyma05g30000* | HD-ZIP transcription factor | II | 59 | 1041 | CDS | |
| Glyma08g13110* | HD-ZIP transcription factor | II | 59 | 571 | CDS | |
| Glyma09g02750* | HD-ZIP transcription factor | II | 59 | 568 | CDS | |
| Glyma12g08080# | HD-ZIP transcription factor | II | 160 | 1239 | CDS | |
| Glyma11g20520# | HD-ZIP transcription factor | II | 160 | 605/607 | CDS | |
| gma-miR167 | Glyma15g09750* | Auxin response factor | II | 159 | 2444 | CDS |
| Glyma13g29320* | Auxin response factor | II | 159 | 3359 | CDS | |
| Glyma05g27580# | Auxin response factor | II | 86 | 2288 | CDS | |
| Glyma08g10550# | Auxin response factor | II | 86 | 2477 | CDS | |
| Glyma18g05330 | Auxin response factor | II | 54 | 2880 | CDS | |
| Glyma15g00770 | zinc finger family protein | I | 112 | 1815 | 5'UTR | |
| Glyma02g40650 | Auxin response factor | II | 76 | 2924 | CDS | |
| gma-miR168 | Glyma16g34300# | AGO protein | II | 74 | 534 | CDS |
| Glyma09g29720# | AGO protein | II | 74 | 409 | CDS | |
| gma-miR169 | Glyma08g45030* | NUCLEAR FACTORY | II | 31 | 1294 | 5'UTR |
| Glyma18g07890* | NUCLEAR FACTORY | I | 31 | 957 | CDS | |
| Glyma17g05920# | NUCLEAR FACTORY | I | 64 | 1262 | 5'UTR | |
| Glyma13g16770# | NUCLEAR FACTORY | II | 64 | 1022 | CDS | |
| Glyma09g07960* | NUCLEAR FACTORY | II | 33 | 931 | 5'UTR | |
| Glyma15g18970* | NUCLEAR FACTORY | II | 33 | 981 | 5'UTR | |
| Glyma19g38800 | NUCLEAR FACTORY | I | 23 | 1385 | 5'UTR | |
| gma-miR171 | Glyma08g08590# | polyubiquitin protein | IV | 13 | 195 | CDS |
| Glyma05g25610# | polyubiquitin protein | IV | 13 | 187 | CDS | |
| Glyma09g04950 | TCP family transcription factor | IV | 19 | 39 | 3'UTR | |
| gma-miR172 | Glyma19g35560* | heat shock cognate protein | IV | 47 | 282 | CDS |
| Glyma03g32850* | heat shock cognate protein | IV | 47 | 480 | CDS | |
| Glyma15g04930# | AP2 transcription factor | II | 425 | 1279 | CDS | |
| Glyma13g40470# | AP2 transcription factor | II | 348 | 1798 | CDS | |
| Glyma11g15650# | AP2 transcription factor | II | 425 | 1811 | 5'UTR | |
| Glyma12g07800# | AP2 transcription factor | II | 425 | 1763 | CDS | |
| Glyma01g39520* | AP2 transcription factor | II | 44 | 1709 | CDS | |
| Glyma11g05720* | AP2 transcription factor | II | 44 | 1777 | CDS | |
| Glyma19g36200# | AP2 transcription factor | II | 111 | 1447 | CDS | |
| Glyma03g33470# | AP2 transcription factor | II | 111 | 1243 | CDS | |
| miRNA | Target gene | Target annotation | Class | Abundance(TP10M) | cleavage site(nt) | target site location |
| gma-miR172 | Glyma17g18640 | AP2 transcription factor | III | 26 | 1973 | CDS |
| Glyma02g09600 | AP2 transcription factor | II | 78 | 1469 | CDS | |
| Glyma05g27370# | TCP family transcription factor | II | 109 | 922 | 5'UTR | |
| gma-miR319 | Glyma13g29160# | TCP family transcription factor | II | 112 | 2078 | CDS |
| Glyma08g10350# | TCP family transcription factor | II | 109 | 1172 | CDS | |
| Glyma15g09910# | TCP family transcription factor | II | 112 | 959 | CDS | |
| Glyma13g34690* | TCP family transcription factor | II | 195 | 656 | CDS | |
| Glyma12g35720* | TCP family transcription factor | II | 195 | 1223 | CDS | |
| Glyma14g06680# | Plasma membrane intrinsic protein | III | 49 | 935 | CDS | |
| Glyma02g42220# | Plasma membrane intrinsic protein | III | 49 | 1029 | 5'UTR | |
| Glyma13g36840* | TCP family transcription factor | II | 73 | 1220 | CDS | |
| Glyma12g33640* | TCP family transcription factor | II | 73 | 740 | CDS | |
| gma-miR390 | Glyma15g14670 | expressed protein | IV | 14 | 569 | CDS |
| gma-miR393 | Glyma03g36770# | Auxin signaling F-BOX protein | II | 65 | 1750 | CDS |
| Glyma19g39420# | Auxin signaling F-BOX protein | II | 65 | 1751 | CDS | |
| Glyma16g05500* | Auxin signaling F-BOX protein | II | 46 | 2279 | CDS | |
| Glyma19g27280* | Auxin signaling F-BOX protein | II | 46 | 2207 | CDS | |
| Glyma10g02630# | Auxin signaling F-BOX protein | IV | 14 | 2166 | CDS | |
| Glyma02g17170# | Auxin signaling F-BOX protein | IV | 14 | 1741 | CDS | |
| gma-miR394 | Glyma01g06230* | NADP+ | IV | 24 | 42 | CDS |
| Glyma06g01850* | NADP+ | IV | 24 | 588 | CDS | |
| gma-miR396 | Glyma03g02500# | Growth regulating factor | I | 57 | 550 | CDS |
| Glyma01g34650# | Growth regulating factor | I | 57 | 128 | CDS | |
| Glyma09g34560* | Growth regulating factor | I | 361 | 323 | CDS | |
| Glyma01g35140* | Growth regulating factor | II | 361 | 290 | CDS | |
| Glyma07g04290# | Growth regulating factor | II | 117 | 473 | CDS | |
| Glyma16g00970# | Growth regulating factor | I | 117 | 353 | CDS | |
| Glyma13g16920* | Growth regulating factor | I | 77 | 742 | CDS | |
| Glyma17g05800* | Growth regulating factor | I | 77 | 422 | CDS | |
| Glyma09g07990* | Growth regulating factor | II | 77 | 380 | CDS | |
| Glyma11g11820# | Growth regulating factor | I | 279 | 386 | CDS | |
| Glyma11g01060# | Growth regulating factor | II | 279 | 349 | CDS | |
| Glyma12g01730# | Growth regulating factor | II | 279 | 504 | CDS | |
| Glyma01g44470# | Growth regulating factor | I | 279 | 428 | CDS | |
| Glyma17g35090* | Growth regulating factor | II | 1007 | 913 | CDS | |
| Glyma17g35100* | Growth regulating factor | II | 1007 | 724 | CDS | |
| Glyma14g10090* | Growth regulating factor | II | 1007 | 704 | CDS | |
| Glyma04g40880 | Growth regulating factor | I | 46 | 233 | CDS | |
| Glyma06g13960 | Growth regulating factor | II | 46 | 831 | CDS | |
| Glyma13g22840 | Growth regulating factor | IV | 13 | 282 | 3'UTR | |
| Glyma14g10100 | Growth regulating factor | II | 373 | 711 | CDS | |
| Glyma15g19460 | Growth regulating factor | II | 69 | 347 | CDS | |
| miRNA | Target gene | Target annotation | Class | Abundance(TP10M) | cleavage site(nt) | target site location |
| gma-miR398 | Glyma15g13870 | MtN19-like protein | I | 15 | 172 | CDS |
| Glyma14g39910 | Serine-type endopeptidase | II | 87 | 1370 | CDS | |
| Glyma19g42890 | Copper/zinc superoxide dismutase | III | 30 | 174 | CDS | |
| gma-miR1509a | Glyma05g24110 | elongation factor | IV | 15 | 436 | CDS |
| gma-miR1511a | Glyma10g05580* | 60S ribosomal protein | II | 28 | 1220 | CDS |
| Glyma13g19930* | 60S ribosomal protein | III | 28 | 1318 | CDS | |
| gma-miR1514a | Glyma11g35820# | NSF attachment protein | IV | 13 | 651 | CDS |
| Glyma18g02590# | NSF attachment protein | IV | 13 | 615 | CDS | |
| Glyma07g05370 | NAC family transcription factor | II | 19 | 832 | CDS | |
| Glyma16g01940 | NAC family transcription factor | II | 25 | 844 | CDS | |
| Glyma16g01930 | NAC family transcription factor | I | 47 | 742 | CDS | |
| gma-miR1515a | Glyma12g00830 | Autophagy protein | III | 17 | 889 | CDS |
| gma-miR1516a | Glyma04g42690 | Disulfide isomerase | III | 33 | 1016 | CDS |
| gma-miR1522a | Glyma03g36390 | FAD linked oxidase family protein | III | 45 | 1826 | 5'UTR |
| gma-miR1523a | Glyma20g27950 | polyubiquitinated protein | IV | 114 | 864 | CDS |
| gma-miR1530a | Glyma10g32330# | Auxin inducible transcription factor | III | 24 | 79 | 3'UTR |
| Glyma20g35280# | Auxin inducible transcription factor | III | 24 | 445 | CDS | |
| Glyma09g41100 | expressed protein | II | 20 | 1324 | 5'UTR | |
| Glyma02g28890 | transketolase | III | 104 | 67 | CDS | |
| gma-miR1536a | Glyma19g06340# | ribulose-1,5-bisphosphate carboxylase | III | 108 | 795 | 5'UTR |
| Glyma19g06370# | ribulose-1,5-bisphosphate carboxylase | III | 108 | 668 | 5'UTR | |
| Glyma13g07610 | ribulose-1,5-bisphosphate carboxylase | III | 115 | 661 | 5'UTR | |
CDS: coding sequence; UTR: untranslated region; TP10M: transcripts per 10 million; Cleavage site: nucleotide number from 5' end of cDNA; Adjacent target genes with same # or * were matched by identical reads; alegume or soybean specific miRNAs; bMiRNA targets validated by RLM-5' RACE.
Among the 145 identified targets of known miRNAs, 114 targets (85%) belong to Class I and Class II, whereas 14 and 17 were classified into Classes III and IV, respectively (Table 3). Class I targets contained reads only from miRNA-directed cleavage, representing perfect data with no other contamination. A series of targets for known miRNAs, including gma-miR156, gma-miR159, gma-miR160, gma-miR164, gma-miR167, gma-miR169, gma-miR396, gma-miR398 and gma-miR1514, belong to this class (Tables 3, 4). More targets of soybean-specific miRNAs belong to Class III and Class IV when compared to those targets of conserved miRNAs.
Table 4.
Identified targets of new miRNAs in soybean
| miRNA | Target gene | Target annotation | Class | Abundance(TP10M) | cleavage site(nt) | Target site location |
|---|---|---|---|---|---|---|
| Soy_2 | Glyma17g02170 | F-box protein | II | 15 | 67 | CDS |
| Soy_3 | Glyma07g39750# | PPR-containing protein | II | 19 | 1633 | CDS |
| Glyma17g01050# | PPR-containing protein | III | 19 | 1659 | CDS | |
| Soy_4 | Glyma04g03110 | oxidoreductase | IV | 13 | 447 | CDS |
| Soy_5 | Glyma12g30680 | 60S ribosomal protein | III | 17 | 643 | 5'UTR |
| Soy_7 | Glyma16g25990 | G-protein | II | 15 | 1780 | CDS |
| Glyma19g37520 | copper ion binding protein | IV | 15 | 684 | CDS | |
| Soy_8 | Glyma19g28990# | tubulin | III | 17 | 920 | CDS |
| Glyma16g04420# | polyubiquitin protein | III | 17 | 931 | CDS | |
| Soy_9 | Glyma11g37920 | HD-ZIP transcription factor | IV | 19 | 629 | CDS |
| Soy_10 | Glyma19g22900 | methyltransferase | IV | 17 | 936 | 5'UTR |
| Soy_11 | Glyma05g26750# | endomembrane protein | II | 27 | 1407 | CDS |
| Glyma08g09740# | endomembrane protein | II | 27 | 1416 | CDS | |
| Glyma17g14370 | ribosomal protein | IV | 19 | 257 | CDS | |
| Soy_16 | Glyma09g30740# | PPR-containing protein | I | 14 | 616 | CDS |
| Glyma09g30680# | PPR-containing protein | IV | 14 | 460 | CDS | |
| Soy_17 | Glyma02g14400 | expressed protein | III | 15 | 955 | 5'UTR |
| Soy_19 | Glyma19g35560# | Heat shock cognate protein | IV | 47 | 282 | CDS |
| Glyma03g32850# | Heat shock cognate protein | IV | 47 | 480 | CDS | |
| Soy_21 | Glyma15g04010* | Transcription factor IIA | IV | 14 | 694 | CDS |
| Glyma13g41390* | Transcription factor IIA | IV | 14 | 1348 | 5'UTR | |
| Glyma19g03770 | transferase protein | IV | 14 | 746 | CDS | |
| Glyma03g41900 | bHLH family transcription factor | II | 55 | 1184 | CDS | |
| Soy_22 | Glyma19g41650 | peptide chain release factor | IV | 15 | 1258 | 5'UTR |
| Soy_25 | Glyma05g33260 | suppressor of gene silencing | II | 30 | 555 | CDS |
CDS: coding sequence; UTR: untranslated region; TP10M: transcripts per 10 million; Cleavage site: nucleotide number from 5' end of cDNA; Adjacent target genes with same # or * were matched by identical reads.
Validation of multiple genes matched by identical reads as targets of corresponding miRNAs
Because many soybean genes have multiple copies, some targets were matched by the same reads, as shown in Table 3. RLM-5' RACE experiments were applied to examine whether the targets mapped by the same reads were sliced by the same miRNA. For gma-miR166, 7 targets were matched by identical reads (Table 3). Among these, 4 HD-ZIP transcription factor genes were checked by 5' RACE (Figure 5). Three genes, Glyma13640, Glyma6g09100 and Glyma08g21610, were found to be cleaved by gma-miRNA166 after sequencing 6, 10 and 4 clones, respectively (Figure 5). One gene (Glyma07g01950) could not be confirmed to be cleaved by gma-miR166. Therefore, most of the genes with the identical signature could be regulated by the corresponding miRNA. By degradome sequencing, two cleavage sites were detected in 3 genes: Glyma13640, Glyma6g09100 and Glyma07g01950. However, only one cleavage site could be further validated by 5' RACE in Glyma13640 and Glyma6g09100 (Figure 5). The second cleavage site in these genes was not confirmed by 5' RACE, probably because of low frequencies.
Figure 5.

Validation of gma-miR166 targets matched by identical reads. The numbers of signatures along the sequences of targets were plotted. Red arrows indicate signatures produced by miRNA-directed cleavage. Black arrows above mRNA of targets indicate detected cleavage sites. Red numbers above the black arrows indicate cleavage probabilities (cleaved target vs total sequenced clones) through 5' RACE confirmation. Black numbers on the right or left side of each black arrow indicate detection abundance of reads. (a) Target cleavage signature, cleavage site in HD-ZIP transcription factor gene Glyma15g13640, and confirmation by RLM-5'RACE. (b) Target cleavage features in HD-ZIP transcription factor gene Glyma6g09100 and confirmation by RLM-5'RACE. (c) Cleavage features in HD-ZIP transcription factor gene Glyma08g21610 and confirmation by RLM-5'RACE. For (a), (b) and (c), only one of the two identified cleavage sites was further confirmed by RLM-5'RACE. (d) Gma-miR166 target HD-ZIP transcription factor gene (Glyma07g01950) from degradome sequencing could not be further confirmed by 5' RACE.
Most miRNAs, especially conserved ones, could target several genes. The gma-miR396 had 21 target genes, and most of these could be grouped into Class I and Class II (Table 3). Every target cDNA had three regions: 5' UTR, CDS and 3' UTR. In animals, miRNA primarily binds to the 3' UTR of a gene to suppress translation. However, in plants, miRNA mainly silences gene expression through mRNA cleavage. In soybean, the cleavage site of the miRNA was usually located in the CDS of target genes (Table 3). Because genes with full-length cDNA represent only 5% of all predicted genes in the soybean database [7], the genes sliced by miRNA in the UTR region may not be detected because of incomplete information on gene sequences. However, miRNAs mainly cleave CDS of rice genes with relatively integrated gene sequences [13].
Putative functions of annotated miRNA targets
Previous studies have found that miRNAs function in plants mainly by cleaving mRNA of transcription factors [26]. In this study, 82% of miRNA targets were transcription factors, a large number of which were auxin response factors, growth regulating factors and NAC transcription factors (Table 3). These factors may be involved in plant growth and/or responses to environmental changes. Most of the transcription factor gene targets belonged to Class I and Class II, indicating that miRNA was the key regulator of these genes.
In most cases, targets of the same miRNA belong to the same gene family (Table 3); however, some miRNAs, such as gma-miR398, can target three types of genes, including copper/zinc superoxide dismutase, MtN19-like protein and serine-type endopeptidase (Figure 6a, b, c). In previous reports [13,27,28], sucrose-inducible miR398 was found to decrease expressions of two copper superoxide dismutase genes and a copper chaperone gene in Arabidopsis and rice. The copper superoxide dismutase gene was also found to be sliced by miR398 in soybean in our research (Figure 6a; Table 3). It seems likely that the role of miRNA398 in the regulation of copper superoxide dismutase genes is conserved among Arabidopsis, rice and soybean. Two other genes were also identified as gma-miR398 targets; one is a serine-type endopeptidase and the other is an MtN19-like protein induced by bruchin treatment [29] (Table 3; Figure 6b, c). Therefore, gma-miR398 may perform additional functions in soybean by targeting more genes.
Figure 6.

Plot of signatures matched to miRNA targets and alignment of mRNA with miRNA. (a) Cleavage features in target copper/zinc superoxide dismutase gene (Glyma19g42890) by gma-miR398a. (b) Cleavage features in target MtN19-like protein gene (Glyma15g13870) by gma-miR398a. (c) Cleavage features in target serine-type endopeptidase gene (Glyma14g39910) by gma-miR398a. (d) Cleavage features in target transketolase gene (Glyma02g28890) by non-conserved gma-miR1530. Other indications are as in Figure 5.
Target genes of soybean- or legume-specific miRNAs primarily belong to Class III and Class IV, and these miRNAs regulate fewer targets than conserved miRNAs do (Table 3, the miRNAs denoted by a). The target of gma-miR1530 was found to be a transketolase gene (Figure 6d), the product of which may participate in the Calvin cycle of photosynthesis. The Calvin cycle converts carbon dioxide into organic substances in plants; this process is known as carbon fixation. Therefore, the gma-miR1530 may regulate carbon assimilation in soybean. However, the gma-miR1530 was also identified from soybean root [8]. Two auxin induced transcription factors were also detected as targets of gma-miR1530, but their signature abundance was much lower (Table 3). Considering that the degradome library was constructed using developing soybean seeds, the gma-miR1530 may be responsible for the switch from carbon assimilation to energy metabolism during seed development by silencing the transketolase gene. However, it is possible that the gma-miR1530 targets may also participate in root development.
Targets of new miRNAs from soybean
In addition to identification of the targets for known miRNAs (Table 3), targets of new miRNAs were investigated in this study (Table 4). The verification of miRNA targets provides further evidence for the existence of new miRNAs in soybean. We identified target genes for 15 new miRNAs (Table 4); these targets belonged mainly to Class III and Class IV, like the targets of soybean or legume-specific miRNAs (Table 3).
Unlike conserved miRNAs, the targets of new soybean miRNAs were not enriched in transcription factors (Table 4). Many target genes, such as G-protein and endomembrane protein, are likely involved in signal transduction, implying that the corresponding new miRNAs may participate in some specific developmental processes in soybean. Pentatricopeptide repeat proteins (PPR) were detected as the targets of Soy_3 and Soy_16. PPR-containing proteins perform functions at the post-transcriptional level in mitochondria and chloroplasts and are widely distributed in higher plants but absent in prokaryotes and archaebacteria [30,31]. They regulate gene expression in plant organelles through many processes, including RNA editing, cleavage and splicing. Soy_3 and Soy_16 may regulate plant organelle development by silencing genes encoding pentatricopeptide repeat-containing proteins.
Interestingly, the soybean homolog (Glyma05g33260) of Arabidopsis SUPPRESSOR OF GENE SLIENCING 3 (AtSGS3) was detected as the target of Soy_25 (Figure 7a). SGS3 was required for defense against virus infection through posttranscriptional gene silencing (PTGS) in plants [32-34]. AtSGS3 participates not only in mRNA degradation, but also in DNA methylation. Loss of function of AtSGS3 could reduce production of trans-acting siRNA in Arabidopsis [33]. The V2 protein of tomato yellow leaf curl virus could interact with tomato SGS3 to suppress RNA silencing for virus infection [33]. In addition to SGS3, soybean ARGONAUTE (AGO) proteins (Glyma16g34300 and Glyma09g29720), another important component in miRNA- or siRNA-mediated PTGS, were cleaved by conserved gma-miR168 (Figure 7b). Regulation of the AGO gene by miR168 was validated in Arabidopsis and rice [35].
Figure 7.

SGS3 and AGO1 were sliced by soy_25 and gma-miR168, respectively. (a) The gene for SGS3 (Suppressor of Gene Silencing 3) protein (Glyma05g33260) was identified as Soy_25 target by degradome sequencing. (b) The AGO gene (Glyma16g34300) was identified as the target of gma-miR168 by degradome sequencing.
GO analysis of targets
All targets regulated by soybean annotated miRNAs and new miRNAs identified in this study were subjected to AgriGO toolkit analysis to investigate gene ontology [36]. To date, 60,319 soybean genes have been annotated in the AgriGO database, and 159 soybean miRNA targets were recognized for GO analysis (Figure 8). As seen in Figure 8, more than 80% of these genes are involved in metabolic process, and reproduction-related genes were more enriched in miRNA targets than in soybean total genes. The enrichment of the genes involved in metabolic and reproductive processes may be consistent with the fact that both the small RNA and the degradome libraries were constructed from developing soybean seeds. The accumulation of dry matter for seed germination is the main task of developing seeds, and a large number of target genes may participate in these processes. The known and new miRNAs identified in this study may regulate expression of these target genes to control seed development and energy storage in soybeans.
Figure 8.

GO analysis of targets of known and new miRNAs in this study. Blue bars indicate the enrichment of miRNA targets in GO terms. Green bars indicate the percentage of total annotated soybean genes mapping to GO terms.
Discussion
As regulators of gene expression, miRNAs are widely present in animals and plants [37-50]. There are 243 and 511 miRNAs annotated in Arabidopsis and rice, respectively, according to the miRBase database [21,22]. Soybean is an ancient polyploid (paleopolyploid) crop plant, with a more complex and larger genome than Arabidopsis and rice [7]. In total, 207 soybean miRNAs were annotated in the miRBase database. In this study, 26 new miRNAs were identified in soybean by deep sequencing and validated by experimental approaches. Functional elucidation and target analysis of the conserved and non-conserved miRNAs could yield more clues to the different regulations of gene expressions between species.
We further studied the target genes of the miRNAs by degradome library sequencing. Unlike other species studied by degradome sequencing [11-16], in the soybean genome, only 40% of Solexa reads could be mapped to a single position. Previous reports showed soybean to be an ancient polyploidy, and the genome duplicated twice, 59 and 13 million years ago [7]. Most genes have multiple copies in the genome. In addition, the tags from degradome sequencing have only 20 or 21 nt. Therefore, the proportion of single mapped reads was not as high as that in other degradome sequencing. Extending the length of sequencing tag may improve this proportion.
Because of the high throughput of deep sequencing, a large number of reads that were not miRNA-directed cleavage products were detected in Class III and Class IV. These cleavages may have been derived from cleavage by other unidentified soybean miRNAs. Conserved miRNAs silence more targets than soybean-specific miRNAs. It is possible that conserved miRNAs play a key role in universal mechanisms of regulation in different plant species;, however, soybean-specific miRNAs may function only in regulation of gene expression during legume- or soybean-specific processes. Although the conserved miRNAs mainly regulate genes encoding transcription factors, soybean-specific miRNAs regulate various types of genes, suggesting a new feature of miRNA regulation in soybeans.
As the small RNA library was prepared from soybean developing seeds, the miRNAs with detected target genes should take part in regulation of seed development. Although most of the soybean genes were not annotated clearly, some targets related to seed development were identified in this study. The soybean seed is a storage organ, containing significant amounts of lipid and protein. Energy metabolism is very active during development of seeds, especially in chloroplasts. In the early stages of soybean seed development, photosynthesis occurs in seed chloroplasts. Subsequently, lipid accumulation becomes the major function of chloroplasts in seeds. Genes encoding transketolase and carboxylase in these processes were identified as gma-miR1530 and gma-miR1536 targets, respectively. Genes encoding PPR-containing proteins, which regulate gene expression in mitochondria and chloroplasts, were also regulated by some miRNAs. These miRNAs may affect conversion between photosynthesis and lipid accumulation in seeds by regulating genes related to metabolism and chloroplast development. Moreover, comparison of miRNA abundance in seeds and other organs of soybeans should uncover those miRNAs specifically expressed in seeds. Identification of the corresponding target genes and study of their roles will elucidate possible functions of miRNAs and target genes in relevant processes of seed development.
Only a few annotated conserved miRNAs were found to have no soybean target genes; however, many non-conserved miRNAs did not appear to silence any targets. MiRNAs regulate gene expression not only by mRNA cleavage but also by translation repression. The miRNAs with no detected targets may silence genes by repressing translation. However, we could not obtain information about translation repression by miRNA through degradome sequencing. Other methods may be used to detect such a possibility, e.g., co-expression of miRNA and the predicted target in N. benthamiana leaves [13]. Some non-conserved miRNAs are hard to detect because of low abundance or spatial expression pattern. To get more integrated information on miRNA targets, degradome libraries from different tissues, organs and different developmental stages should be constructed. Additionally, some miRNAs also function in methylation of genomic DNA or histones. More attention should be paid to the mechanism of methylation via miRNA to clarify other functions of miRNA.
In higher plants, miRNAs function mainly through silencing related gene expression. Identification of miRNA targets will help us to understand the biological effects of miRNA. By deep sequencing of a degradome library, we identified a large number of target genes regulated by corresponding miRNAs (Table 3, 4). These targets contained not only conserved families of miRNA target genes, such as MYB, ARF, NAC, GRF and TCP-type transcription factor gene families, but also non-conserved target genes, such as G-protein, SGS3 and F-box protein. The conserved targets may participate in various aspects of plant development and stress responses as in other plants and may help us to understand evolutionary relationships between soybean and other plants. Global identification of non-conserved targets provides useful information to explore the new functions of miRNAs in soybean. The regulation of SGS3 by miRNA was not found in previous studies. Further study of the relationship between SGS3 and the new miRNA Soy_25 should reveal the function of this pair in regulation of miRNA biogenesis and/or seed development in soybean and other plants.
Conclusions
In our study, a small RNA library and a degradome library were constructed from developing soybean seeds for deep sequencing. We identified 26 new miRNAs in soybean by bioinformatic analysis and experimental tests. The miRNA star sequences of 38 known miRNAs and 8 new miRNAs were also discovered, providing additional evidence for the existence of miRNAs. Degradome sequencing as a high-throughput approach for miRNA target detection was applied to identify miRNA targets in soybean. In total, 145 and 25 genes were identified as targets of annotated miRNAs and new miRNAs, respectively. Construction of degradome libraries from different developmental stages of seeds should reveal more targets of soybean miRNAs. Overall, global identification of soybean miRNA targets in this study provides more information about the regulatory network of miRNAs in soybean, and it will advance our understanding of miRNA functions during seed development.
Methods
Plant material and RNA isolation
Soybean (Glycine max) seeds of cultivar Heinong44 were directly planted in the Experimental Station of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, in Beijing in May. Seeds from soybeans 15 days after flowering (DAF) were collected and quickly frozen in liquid nitrogen. Total RNA was isolated from seeds using TRIzol reagent (Invitrogen) according to the manufacturer's instructions.
Small RNA library and degradome library construction
After total RNA isolation, low molecular weight RNAs were isolated as described previously, with some modification [51]. By polyacrylamide gel electrophoresis, the small RNAs (~17-27 nt) were purified from 100 μg of total RNA and ligated to a 5' RNA adapter and a 3' RNA adapter. A reverse transcription reaction followed by low cycle PCR was performed to obtain sufficient product for SBS sequencing. PCR products were collected by gel purification and sequenced by Solexa technology.
The soybean seed degradome library was constructed as previously described [18,19]. In brief, poly(A) RNA was extracted from 200 μg of total RNA using the Oligotex kit (Qiagen). A 5' RNA adapter containing a MmeI recognition site was ligated to the poly(A) RNA possessing a 5'-phosphate, by T4 RNA ligase (Ambion), and the ligated products were repurified using the Oligotex kit. Five PCR cycles were then performed to amplify the reverse transcription products. The PCR products were digested with MmeI and ligated to a 3' double DNA adapter. The ligation products were amplified by 20 PCR cycles and gel-purified for SBS sequencing.
The small RNA library and degradome library sequencing data were available under NCBI-GEO accession no. GSE25260.
Bioinformatic analysis of sequencing data
Small RNA reads and degradome reads were both generated from an Illumina Genome Analyzer II. The row data were preprocessed by the Fastx-toolkit pipeline to remove low quality reads and clip adapter sequences. As for the small RNA library, small RNAs ranging from 18-25 nt were collected and mapped to the soybean genome using SOAP2 [19]. The unique RNA sequences that perfectly matched the genome were subjected to subsequent analysis. RNA reads showing sequences identical to known miRNAs from the miRBase database [21,22] were picked up as the miRNA dataset of soybean. Sequences matching noncoding rRNA, tRNA, snRNA and snoRNA in the Rfam database were removed. Reads overlapping with exons of protein-coding genes were excluded to avoid mRNA contamination. The remaining sequences were considered for prediction to find new miRNAs.
As for the degradome library, only 20-21 nt sequences with high quality were collected for subsequent analysis. The raw sequences were first normalized to "reads per 10 million" (RP10M). The distinct reads that perfectly matched soybean cDNA sequences remained. The 15 nt of sequence upstream and downstream of the 5' end of matched reads were extracted to constitute 30-nt sequence tags for searching corresponding miRNA. The CleaveLand pipeline [18] was used to align the 30 nt sequence to soybean known miRNAs from miRBase and our newly identified miRNAs. All alignments with scores up to 7 and no mismatches at the cleavage site (between the 10th and 11th nucleotides) were considered candidate targets.
Prediction of new miRNAs
As miRNA precursors have a characteristic hairpin structure, 150 nt of the sequence flanking the genomic sequences of small RNAs was extracted. The MIREAP pipeline was then used to analyze their structural features to identify new miRNA candidates (https://sourceforge.net/projects/mireap/). The resulting structures, with minimal matched nucleotide pairs of miRNA and miRNA* exceeding 16 nt and with maximal size differences of miRNA and miRNA* up to 4 nt, were retained as new miRNA candidates. The filtered pre-miRNA sequences were folded again using MFOLD and checked manually [23].
Stem-loop RT-PCR
Reverse transcription reactions were performed using total RNA from soybean seeds as previously described [25]. All primers involved in stem-loop RT-PCR are listed in Table S2 in Additional File 2. The reactions contained 25 ng of RNA samples, 50 nM stem-loop RT primer (Invitrogen), 1 × RT buffer, 0.25 mM of each dNTP (Takara), 5 U/μl SuperScript II reverse transcriptase (Invitrogen) and 0.25 U/μl RNase Inhibitor (Invitrogen). The 10 μl reactions were incubated in a Biometra TProfessional Thermocycler in a 96-well plate for 30 min at 16°C, 30 min at 42°C and 5 min at 85°C and then held at 4°C.
PCR was performed using diluted cDNA products. The reactions were incubated in a Biometra TProfessional Thermocycler for 5 min at 95°C, followed by 40 cycles of 15 sec at 94°C, 30 sec at 60°C and 30 sec at 72°C. All reactions were run in duplicate. The PCR products were detected by gel electrophoresis.
RLM-5' RACE
Total RNA (200 μg) from soybean seeds was used to purify mRNA using the Oligotex kit (Qiagen). 5' RNA adaptor (5'-CGACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA-3') was ligated to the purified mRNA by T4 RNA ligase (Ambion), followed by a reverse transcription reaction. The reverse transcription product was amplified using 5' RNA adaptor primer (5'-GCACGAGGACACTGACATGGACTGA-3') and gene specific primers for 30 cycles of PCR. Twenty-five cycles of PCR were further performed with the above PCR product as templates, using a nested gene specific primer (5'-GGACACTGACATGGACTGAAGGAGTA-3') and an adapter primer. The final PCR product was detected by gel electrophoresis and extracted for sequencing.
Abbreviations
miRNA: microRNA; pre-miRNA: miRNA precursor; miRNA*: miRNA star; poly(A) RNA: Polyadenylated RNA; RT: Reverse-transcription.
Authors' contributions
QXS performed the bioinformatics analysis, analyzed the Solexa data, conducted the experiments and drafted the initial manuscript. YFL prepared the materials and RNAs. XYH was involved in Solexa sequencing. WKZ and BM contributed to the experimental design and analysis. SYC and JSZ conceived the study, obtained the funding, analyzed the data and finished the final manuscript. All authors read and approved the final manuscript.
Supplementary Material
New miRNAs identified in soybean developing seeds. Mature sequences, star sequences and precursor sequences of miRNAs. The numbers of miRNAs detected by small RNA library sequencing are also included.
Stem-loop RT-PCR Primers. All primers used in stem-loop RT-PCR.
Contributor Information
Qing-Xin Song, Email: qxsong@genetics.ac.cn.
Yun-Feng Liu, Email: yfliu@genetics.ac.cn.
Xing-Yu Hu, Email: xyhu@genetics.ac.cn.
Wan-Ke Zhang, Email: wkzhang@genetics.ac.cn.
Biao Ma, Email: mabiao@genetics.ac.cn.
Shou-Yi Chen, Email: sychen@genetics.ac.cn.
Jin-Song Zhang, Email: jszhang@genetics.ac.cn.
Acknowledgements
This work was supported by the National Key Basic Research Projects (2009CB118402), National Transgenic Research Projects (2008ZX08009-003, 2009ZX08009-054B, 2009ZX08009-115B), and National Natural Science Foundation of China (90717005, 30925006).
References
- Chen XM. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009;25:21–44. doi: 10.1146/annurev.cellbio.042308.113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297:2053–2056. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell. 2003;15:2730–2741. doi: 10.1105/tpc.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari R, Datt PB, Stitt M, Scheible WR. PHO2, microRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 2006;141:988–999. doi: 10.1104/pp.106.079707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Fu Y, Sunkar R, Barbazuk WB, Zhu JK, Yu O. New and nodulation-regulated microRNAs in soybean roots. BMC Genomics. 2008;9:160. doi: 10.1186/1471-2164-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YW, Li PC, Cao XF, Wang XJ, Zhang A, Li X. Identification and expression analysis of miRNAs from nitrogen-fixing soybean nodules. Biochem Biophys Res Commun. 2009;378:799–803. doi: 10.1016/j.bbrc.2008.11.140. [DOI] [PubMed] [Google Scholar]
- Joshi T, Yan Z, Libault M, Jeong DH, Park S, Green PJ, Sherrier DJ, Farmer A, May G, Meyers BC, Xu D, Stacey G. Prediction of new miRNAs and associated target genes in Glycine max. BMC Bioinformatics. 2010;11:S14. doi: 10.1186/1471-2105-11-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18:758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo SJ, Janardhanan P, Kannan V, Rymarquis LA, Nobuta Kan, German R, Paoli ED, Lu C, Schroth G, Meyers BC, Green PJ. Global identification of microRNA-target RNA pairs byparallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang WX, Sunkar R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010;62:742–759. doi: 10.1111/j.1365-313X.2010.04187.x. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhang QQ, Zhou HY, Ni FR, Wu XY, Qi YJ. Rice microRNA effector complexes and targets. Plant Cell. 2009;21:3421–3435. doi: 10.1105/tpc.109.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62:960–976. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- Zhou M, Gu LF, Li PC, Song XW, Wei LY, Chen ZY, Cao XF. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica) Front Biol. 2010;5:67–90. [Google Scholar]
- German MA, Luo SJ, Schroth G, Meyers BC, Green PJ. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nature Protocols. 2009;4:356–362. doi: 10.1038/nprot.2009.8. [DOI] [PubMed] [Google Scholar]
- Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25:130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Hu Z, Zhang H. Identification of microRNAs in wild soybean (Glycine soja) J Integr Plant Biol. 2009;51:1071–1079. doi: 10.1111/j.1744-7909.2009.00887.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- Dugas DV, Bartel B. Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Mol Biol. 2008;67:403–417. doi: 10.1007/s11103-008-9329-1. [DOI] [PubMed] [Google Scholar]
- Beauclair L, Yu A, Bouché N. microRNA-directed cleavage and translational repression of the copper chaperone for superoxide dismutase mRNA in Arabidopsis. The Plant J. 2010;62:454–462. doi: 10.1111/j.1365-313X.2010.04162.x. [DOI] [PubMed] [Google Scholar]
- Robert PD. Treatment of pea pods with Bruchin B results in up-regulation of a gene similar to MtN19. Plant Physiol Biochem. 2005;43:225–231. doi: 10.1016/j.plaphy.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Andrés C, Lurin C, Small LD. The multifarious roles of PPR proteins in plant mitochondrial gene expression. Physiol Plant. 2007;129:14–22. [Google Scholar]
- Schmitz-Linneweber C, Small I. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci. 2008;13:663–670. doi: 10.1016/j.tplants.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Mourrain P, Béclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, Jouette D, Lacombe AM, Nikic S, Picault N, Rémoué K, Sanial M, Vo TA, Vaucheret H. Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell. 2000;101:533–542. doi: 10.1016/S0092-8674(00)80863-6. [DOI] [PubMed] [Google Scholar]
- Glick E, Zrachya A, Levy Y, Mett A, Gidoni D, Belausov E, Citovsky V, Gafni Y. Interaction with host SGS3 is required for suppression of RNA silencing by tomato yellow leaf curl virus V2 protein. Proc Natl Acad Sci USA. 2008;105:157–161. doi: 10.1073/pnas.0709036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev. 2004;18:2368–2379. doi: 10.1101/gad.1231804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaucheret H, Vazquez F, Crété P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004;18:1187–1197. doi: 10.1101/gad.1201404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Zhou X, Ling Y, Zhang ZH, Su Z. AgriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38:W64–70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SP, Li D, Li QB, Zhao P, Xiang ZH, Xia QY. MicroRNAs of Bombyx mori identified by Solexa sequencing. BMC Genomics. 2010;11:148. doi: 10.1186/1471-2164-11-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20:3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes J, Chua NH. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007;49:592–606. doi: 10.1111/j.1365-313X.2006.02980.x. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Watanabe Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA. 2004;101:12753–12758. doi: 10.1073/pnas.0403115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XF, Fang YY, Feng L, Guo HS. Characterization of conserved and new microRNAs and their targets, including a TuMV-induced TIR-NBS-LRR class R gene-derived new miRNA in Brassica. FEBS Lett. 2008;582:2445–2452. doi: 10.1016/j.febslet.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Alves-Junior L, Niemeier S, Hauenschild A, Rehmsmeier M, Merkle T. Comprehensive prediction of new microRNA targets in Arabidopsis thaliana. Nucleic Acids Res. 2009;37:4010–4021. doi: 10.1093/nar/gkp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adai A, Johnson C, Mlotshwa S, Archer-Evans S, Manocha V, Vance V, Sundaresan V. Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 2005;15:78–91. doi: 10.1101/gr.2908205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezulian T, Palatnik JF, Huson D, Weigel D. Conservation and divergence of microRNA families in plants. Genome Biol. 2005;6:13. doi: 10.1186/gb-2005-6-11-p13. [DOI] [Google Scholar]
- Sunkar R, Zhou XF, Zheng Y, Zhang WX, Zhu JK. Identification of new and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8:25. doi: 10.1186/1471-2229-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/S0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Chen XM. MicroRNA metabolism in plants. Curr Top Microbiol Immunol. 2008;320:117–136. doi: 10.1007/978-3-540-75157-1_6. full_text. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–687. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- Axtell MJ, Bowman JL. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008;13:343–349. doi: 10.1016/j.tplants.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Arenas-Huertero C, Pérez B, Rabanal F, Blanco-Melo D, De la Rosa C, Estrada-Navarrete G, Sanchez F, Covarrubias AA, Reyes JL. Conserved and novel miRNAs in the legume Phaseolus vulgaris in response to stress. Plant Mol Biol. 2009;70:385–401. doi: 10.1007/s11103-009-9480-3. [DOI] [PubMed] [Google Scholar]
- Lu C, Meyers BC, Green PJ. Construction of small RNA cDNA libraries for deep sequencing. Methods. 2007;43:110–117. doi: 10.1016/j.ymeth.2007.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
New miRNAs identified in soybean developing seeds. Mature sequences, star sequences and precursor sequences of miRNAs. The numbers of miRNAs detected by small RNA library sequencing are also included.
Stem-loop RT-PCR Primers. All primers used in stem-loop RT-PCR.