

Abstract
Recently developed multifunctional cancer therapeutic nano-device production is based on poly(amidoamine) PAMAM generation 5 (G5) dendrimer as a carrier 1-5. Scale up synthesis of this nano-device is limited because of long reaction sequence (12 reaction steps) and long and not easy work up of the products after each reaction step. Combination of poly(propyle-imine) and poly(amidoamine) synthesis can improve the production of the drug carrier.
In this paper we give a general overview of the synthesis and characterization of a series of novel hybrid dendrimers which we coined as novel POMAM hybrid dendrimers, constructed from poly(propylene-imine) (PPI or POPAM) core and poly(amidoamine) PAMAM shells. The synthesis was accomplished by a divergent reiterating method involving repeating subsequent Michael addition and amidation reactions. Each generation of the newly synthesized dendrimer was characterized by using HPLC, GPC, NMR and AFM.
Keywords: PAMAM dendrimer, PPI or POPAM dendrimer, POMAM dendrimer, Hybrid dendrimer
Introduction
Dendrimers and hyperbranched polymers represent a novel class of structurally controlled macromolecules derived from a branches-upon-branches structural motif 6,7. Dendrimers are well defined, highly branched macromolecules that radiate from a simple organic molecule as a central core and are synthesized through a stepwise, repetitive reaction sequence that guarantees complete shells for each generation, leading theoretically to products that are unimolecular and monodisperse 8a,8b. The synthetic procedures developed for dendrimer preparation permit nearly complete control over the critical molecular design parameters, such as size, shape and surface/interior chemistry 1-8. Synthetic techniques that have proved effective include the poly(amidoamine), PAMAM, divergent strategy of Tomalia and co-workers 6,7, the convergent growth strategy of Fréchet and co-workers 9-11, and the self-assembly strategy of Zimmerman and co-workers 12. These methods have proved effective in generating macromolecules with a unique combination of properties 13-16.
The Astromol or poly(propylene-imine) dendrimers, (another amino type dendrimer synthesized by divergent strategy), were produced at DSM (Dutch State Mines). The synthesis involves a repetitive reaction sequence of Michael additions of acrylonitrile to primary amine end groups followed by catalytic hydrogenation of the nitrile groups. 1,4-diaminobutane was used as the core molecule. Detailed information on the synthesis was published elsewhere 17, 18.
Experimental
Materials
Methanol with 99.93 % purity, ether with 99+% purity, methyl acrylate (MA), and ethylenediamine (EDA) with 99.5+% were purchased from Aldrich and used as received. Another EDA with 99 % purity was distilled on a rotary evaporator at 2000 microns of mercury and with a bath temperature 36 °C. The purified EDA was transferred to a vessel and stored at −4 °C under a blanket of N2.
The Astromol poly(propylene-imine) dendrimers of different generations from DAB-dendr-(NH2)16 (POPAM G-2) to DAB-dendr-(NH2)64 (POPAM G-4), a gift from the DSM, or purchased from Aldrich were used.
Syntheses
All POMAM hybrid dendrimer syntheses (half and full generations) were conducted carefully under inert nitrogen atmosphere.
G-2:0.5, A
In a 50 ml three neck round bottom flask equipped with magnetic stirrer, pressure equalized dropping funnel and condenser, under dry N2 atmosphere, a solution of methyl acrylate (6.41 ml, 7.114 10-2mol) in 7.7 ml MeOH was cooled to 0 °C. Then a solution of 3 g (1.7785 10-3mol) DAB-Am-16 Polypropylenimine hexadecaamine Dendrimer (POPAM, Generation 2:0) in 10 ml MeOH (also cooled at 0 °C under dry argon) was added dropwise. This mixture was stirred under N2 at 36 °C for 48 hrs, and the excess methyl acrylate and methanol were evaporated by vacuum. To the residue, 3 ml of water was added and mixed carefully. After freezing, it was lyophilized to remove residue methanol and methyl acrylate, yielding methyl-ester functionalized POPAM-core dendrimer, POMAM 2:0.5, (7.7 g, 97.5 %).
G-2:0.5, B
POPAM dendrimer (1,4-diaminobutane core, G = 2:0, with 16 NH2 surface groups) (1.0203 g, 6.049 10-4 mol) in 3.5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (2.2495 g, 2.613 10-2 mol) in 3 ml MeOH cooled to 0 °C. The resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-2:0.5 was dried out at a vacuum of 500 microns of mercury to give 2.661 g (99.05 %) of the title compound.
G-2:1
POMAM hybrid dendrimer, G = 2:0.5 (1.0425 g, 2.347 10-4 mol) in 3 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (359.6 g, 5.9835 mol) in 100 ml MeOH cooled at 0 °C. This mixture was maintained at 0 °C for 48 hours. After this time, the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.246 g, 99.4 % yield of the title compound.
G-2:1.5
POMAM hybrid dendrimer (G = 2:1, with 32 primary NH2 surface groups) (1.2014 g, 2.25 10-4 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.674 g, 194 10-2 mol) in 2 ml MeOH cooled to 0 °C. The resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-2:1.5 was dried out at a vacuum of 500 microns of mercury to give 2.404 g (98.5 %) of the title compound.
G-2:2
POMAM hybrid dendrimer, G = 2:1.5 (2.186 g, 2.015 10-4 mol) in 18 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (1438.4 g, 23.934 mol) in 400 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 2.472 g, 97.02 % yield of the title compound.
G-2:2.5
POMAM hybrid dendrimer (G = 2:2, with 64 NH2 surface groups) (1.002 g, 7.92 10-5 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.31 g, 1.52 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 72 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-2:2.5 was dried out at a vacuum of 500 microns of mercury to give 1.804 g (96.2 %) of the title compound.
G-2:3
POMAM hybrid dendrimer, G = 2:2.5 (1.804 g, 7.62 10-5 mol) in 16 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (2157.6 g, 35.901 mol) in 600 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.6584 g, 79.8 % yield of the title compound.
G-2:3.5
A mixture of methyl acrylate (0.37 g, 4.3 10-3 mol) in 1 ml MeOH cooled to 0°C was added to a heterophase POMAM dendrimer (G = 2:3, with 128 NH2 surface groups) (0.3052 g, 1.12 10-5 mol) in 3 ml MeOH cooled to 0 °C. This resulting heterophase mixture was maintained and stirred at room temperature for 96 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34°C and G-2:3.5 was dried out at a vacuum of 500 microns of mercury to give 0.4387 g (79.5 %) of the title compound.
G-2:4
POMAM hybrid dendrimer, G = 2:3.5 (0.4387 g, 8.9 10-6 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (1078.8 g, 17.95 mol) in 300 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 120 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 0.4963 g, 98.7 % yield of the title compound
G-3:0.5, A
In a 50 ml three neck round bottom flask equipped with magnetic stirrer, pressure equalized dropping funnel and condenser, under dry N2 atmosphere, a solution of methyl acrylate (5.41 ml, 6.008 10-2 mol) in 6.5 ml MeOH was cooled to 0 °C. Then a solution of 3 g (7.511 10-4mol) DAB-Am-32 Polypropylenimine Dendrimer (Generation 3:0) in 10 ml MeOH (also cooled to 0 °C under dry N2) was added dropwise. This mixture was stirred under nitrogen at 34 °C for 48 hrs, and the excess methyl acrylate and methanol were evaporated by vacuum. To the residue, 3 ml of water was added and mixed carefully. After freezing it was lyophilized to remove residue methanol and methyl acrylate; yielding methyl-ester functionalized POPAM-core dendrimer, POMAM 3:0.5, (7.02 g, 98.3 %)
G-3:0.5, B
POPAM dendrimer (1,4-diaminobutane core, G = 3:0, with 32 NH2 surface groups) (1.1912 g, 3.39 10-4 mol) in 3.5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (2.2182 g, 2.577 10-2 mol) in 3 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-3:0.5 was dried out at a vacuum of 500 microns of mercury to give 3.0858 g (∼100 %) of the title compound.
G-3:1
POMAM hybrid dendrimer, G = 3:0.5 (1.0377 g, 1.15 10-4 mol) in 13 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (719.2 g, 11.967 mol) in 200 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 48 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.233 g, ∼100 % yield of the title compound.
G-3:1.5
POMAM hybrid dendrimer (G = 3:1, with 64 NH2 surface groups) (1.2728 g, 1.176 10-2 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.679 g, 1.95 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-3:1.5 was dried out at a vacuum of 500 microns of mercury to give 2.451 g (95.4 %) of the title compound.
G-3:2
POMAM hybrid dendrimer, G = 3:1.5 (2.323 g, 1.064 10-4 mol) in 18 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (2157.6 g, 35.901 mol) in 600 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 2.659 g, 98.3 % yield of the title compound.
G-3:2.5
POMAM hybrid dendrimer (G = 3:2, with 128 NH2 surface groups) (1.242 g, 4.88 10-5 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.615 g, 1.876 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 72 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-3:2.5 was dried out at a vacuum of 500 microns of mercury to give 2.185 g (94.2 %) of the title compound.
G-3:3
POMAM hybrid dendrimer, G = 3:2.5 (2.185 g, 4.56 10-5 mol) in 18 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (5753.6 g, 95.736 mol) in 1600 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.9742 g, 78.5 % yield of the title compound.
G-3:3.5
A mixture of methyl acrylate (0.37 g, 4.3 10-3 mol) in 1 ml MeOH cooled to 0 °C was added to a heterophase POMAM dendrimer (G = 3:3, with 256 NH2 surface groups) (0.3022 g, 5.5 10-6 mol) in 3 ml MeOH cooled to 0 °C. This resulting heterophase mixture was maintained and stirred at room temperature for 96 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34°C and G-3:3.5 was dried out at a vacuum of 500 microns of mercury to give 0.5088 g (93.2 %) of the title compound.
G-3:4
POMAM hybrid dendrimer, G = 3:3.5 (0.5088 g, 5.2 10-6 mol) in 15 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (2157.6 g, 35.9 mol) in 600 ml MeOH cooled to 0°C. This mixture was maintained at 0 °C for 120 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 0.5736 g, 98.4 % yield of the title compound.
G-4:0.5, A
In a 50 ml three neck round bottom flask equipped with magnetic stirrer, pressure equalized dropping funnel and condenser, under dry N2 atmosphere, a solution of methyl acrylate (5.02 ml, 5.575 10-2mol) in 6.1 ml MeOH was cooled to 0 °C. Then a solution of 3 g (3.485 10-4mol) DAB-Am-64 Polypropylenimine Dendrimer (Generation 4:0) in 10 ml MeOH (also cooled at 0 °C under dry N2) was added dropwise. This mixture was stirred under N2 at 36 °C for 48 hrs, and the excess methyl acrylate and methanol were evaporated by vacuum. To the residue, 3 ml of water was added and mixed carefully. After freezing it was lyophilized to remove excess methanol and methyl acrylate, yielding methyl-ester functionalized POPAM-core dendrimer, POMAM 4:0.5, (6.83 g, 99.8 %)
G-4:0.5, B
POPAM dendrimer (1,4-diaminobutane core, G = 4:0, with 64 NH2 surface groups) (1.0287 g, 1.435 10-4 mol) in 3.5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.7775 g, 2.065 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-4:0.5 was dried out at a vacuum of 500 microns of mercury to give 2.5112 g (96.2 %) of the title compound.
G-4:1
POMAM hybrid dendrimer, G = 4:0.5 (1.0238 g, 5.63 10-5 mol) in 13 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (1438.4 g, 23.934 mol) in 400 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 48 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34°C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.21 g, ∼100 % yield of the title compound.
G-4:1.5
POMAM hybrid dendrimer (G = 4:1, with 128 NH2 surface groups) (1.1732 g, 5.39 10-5 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.503 g, 1.75 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 48 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-4:1.5 was dried out at a vacuum of 500 microns of mercury to give 2.15 g (91.4 %) of the title compound.
G-4:2
POMAM hybrid dendrimer, G = 4:1.5 (1.865 g, 4.26 10-5 mol) in 18 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (3596 g, 59.835 mol) in 1000 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 2.116 g, 97.4 % yield of the title compound.
G-4:2.5
POMAM hybrid dendrimer (G = 4:2, with 256 NH2 surface groups) (1.015 g, 1.99 10-5 mol) in 5 ml MeOH cooled to 0 °C was added to a mixture of methyl acrylate (1.32 g, 1.53 10-2 mol) in 2 ml MeOH cooled to 0 °C. This resulting mixture was maintained and stirred at room temperature for 72 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-4:2.5 were dried out at a vacuum of 500 microns of mercury to give 1.427 g (75.4 %) of the title compound.
G-4:3
POMAM hybrid dendrimer, G = 4:2.5 (1.427 g, 1.48 10-5 mol) in 15 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (7192.0 g, 119.67 mol) in 2000 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 72 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 1.64 g, 99.8 % yield of the title compound.
G-4:3.5
A mixture of methyl acrylate (0.38 g, 4.4 10-3 mol) in 1 ml MeOH cooled to 0 °C was added to a heterophase POMAM dendrimer (G = 4:3, with 512 NH2 surface groups) (0.3107 g, 2.8 10-6 mol) in 3 ml MeOH cooled to 0 °C. This resulting heterophase mixture was maintained and stirred at room temperature for 96 hours. The MeOH and excess of methyl acrylate as volatiles were evaporated on a rotary evaporator at 34 °C and G-4:3.5 was dried out at a vacuum of 500 microns of mercury to give 0.3922 g (69.9 %) of the title compound.
G-4:4
POMAM hybrid dendrimer, G = 4:3.5 (0.3922 g, 2.0 10-6 mol) in 3 ml MeOH cooled to 0 °C was added to a mixture of ethylenediamine (2876.8 g, 47.9 mol) in 800 ml MeOH cooled to 0 °C. This mixture was maintained at 0 °C for 120 hours. After this reaction time the mixture was warmed to room temperature. The volatiles were removed from the mixture on a rotary evaporator at 34 °C with a vacuum at 2000 − 500 microns of mercury. The crude product was dissolved in MeOH and by addition of ether it was precipitated out. This purification process was repeated three times. The precipitate was dried very carefully to give 0.4411 g, 98.2 % yield of the title compound.
Characterization
Nuclear Magnetic Resonance
For 1H NMR and 13C NMR measurements, a Bruker AVANCE DRX 500 instrument was used. 30-40 mg/ml D2O solutions were prepared for NMR investigation.
Size Exclusion Cromatography
The SEC eluograms were obtained with a Alliance Waters 2690 Separation Module combined with triple detectors: Waters 2487 Dual λ Absorbance Detector, Wyatt DAWN® DSP Laser Photometer, and Optilab DSP Interferometric Refractometer at 30 °C. It was equipped with a TosoHaas TSK-GEL® Guard PWH 06762 (7.5×7.5 cm, 12 μm) (DS 1140), G 2000 PW 05761 (30×7.5cm, 10 μm) (DS 1014), G 3000 PW 05762 (10 μm) (DS 1016), and G 4000 PW 05763 (17 μm) (DS 1017) columns. The 0.1 N citric acid solution, containing 0.025 w% sodium azide (pH was adjusted to 2.72 by sodium hydroxide), was used as the mobile phase and for making macromolecules' sample solutions for SEC analysis. A nominal flow rate setting of 1.0 ml/min and injection volume of 50 μl of 1 10-4 g/ml sample solutions were used.
HPLC
For HPLC measurement Beckman System Gold® instrument was used equipped with solvent module (126) and UV detector (166). 0.1 M of trifluoroacetic acid eluent with 1 ml/min flow rate and C-18 reverse phase column was used at room temperature.
AFM Measurements
Samples on mica were examined using a TopoMetrix 2000 Discoverer instrument under ambient conditions. We found it is difficult to obtain stable images using the contact mode of imaging because the scanning tip appeared to move the samples about. This problem was minimized by using the tapping mode of imaging. Si probes having a spring constant of ca. 30 N/m were used at a resonance frequency of ca. 200 to 300 kHz. A 7 μm scanner (x, y and z directions) calibrated by TopoMetrix was used.
Sample preparation for AFM measurements
The materials were dissolved in the Milli-Q water (18 Mohm/cm) 1 10-4 to form w % solutions, then stored in a refrigerator until needed. Ultrathin films of the hybrid dendrimers were prepared by spin-coating the solutions (1 10-5 w %) on freshly cleaved mica, then air-dried at room temperature.
Results and Discussion
The preparation of POMAM hybrid dendrimers involves a typical divergent synthesis via a two-step growth sequence that consists of two reiterating reactions:
The Michael addition of amino groups to the double bond of methyl acrylate (MA) (this reaction is step growth process).
The amidation of the resulting terminal methyl ester, -COOCH3, with ethylenediamine (EDA) (this reaction is chain growth process) 6.
When poly(propylene-imine) (POPAM) dendrimers are used as the initiator cores, these syntheses may be represented by Figure 1A and B, and reaction scheme 1.
Figure 1.

A: Synthesis of half generations of POMAM hybrid dendrimers (step growth process), B: synthesis of full generations of POMAM hybrid dendrimers (chain growth process).
Scheme 1.
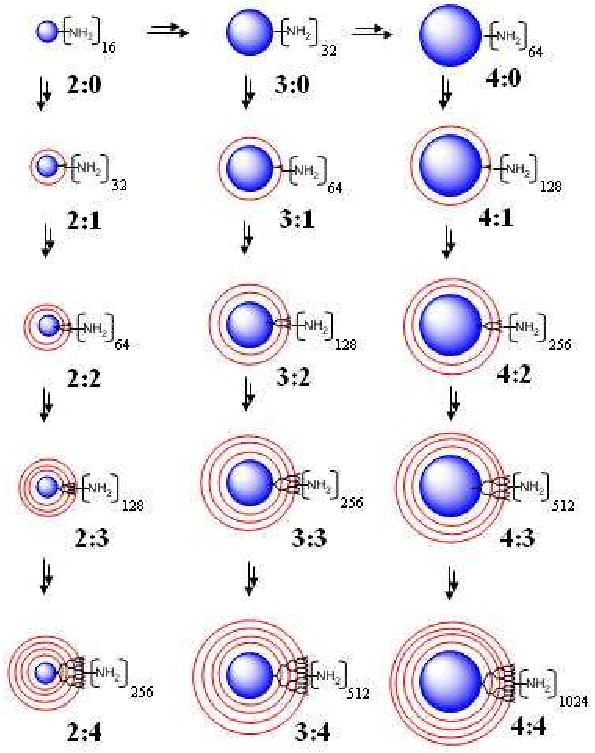
Synthetic strategy for synthesis of variety of POMAM hybrid dendrimers (first number refers generation number of the POPAM core second number indicates number of the PAMAM shells).
In the first step of this process, POPAM dendrimers generations 2, 3, and 4 with 16, 32 and 64 primary amines on the surface (or full generation of the POMAM dendrimers) are allowed to react under inert nitrogen atmosphere with excess amount of MA at room temperature for 24-72 hours (depending on the number of primary amines on the surface). The resulting compounds are referred to as half generation of POMAM hybrid dendrimers. The branching point at the tertiary amine is formed in this high yield step (step growth process). The next step; also high yield, (chain growth process) involves reacting multi-esters with excess EDA produce the PAMAM shells around the POMAM half generation dendrimers so that the POMAM hybrid dendrimers are formed with a doubled amount of primary amines on the surface. These amidation reactions are performed under inert nitrogen atmosphere in methanol and require 24-120 hours reaction times (depending on the number of primary amines on the surface) at ∼2°C for completion.
The 13C NMR spectroscopy provides a sensitive measurement of the magnetic environment for every carbon atom in the compound. The high symmetry of the POPAM, PAMAM and POMAM dendrimer structure places all of the terminal groups in an almost equivalent magnetic environment, giving very simple spectra for molecules of such high molecular weights. Any deviations from this symmetry (as a result of not complete Michal addition, ester hydrolysis or retro Michel addition), perhaps caused by errors in synthesis or degradation of the material, are indicated by additional signals in the 13C NMR spectrum, corresponding to carbon atom in the vicinity of the structural defect.
Scheme 1 shows the strategy followed in the synthetic work (reaction conditions are given in the experimental section).
Figure 2 provides a representative example of a 13C NMR spectrum of POMAM 2:1 dendrimer. This spectrum essentially is the superposition of the spectra of POPAM and PAMAM dendrimers. The peak at ∼175 ppm clearly indicates the PAMAM shell incorporation with the POPAM dendritic core.
Figure 2.
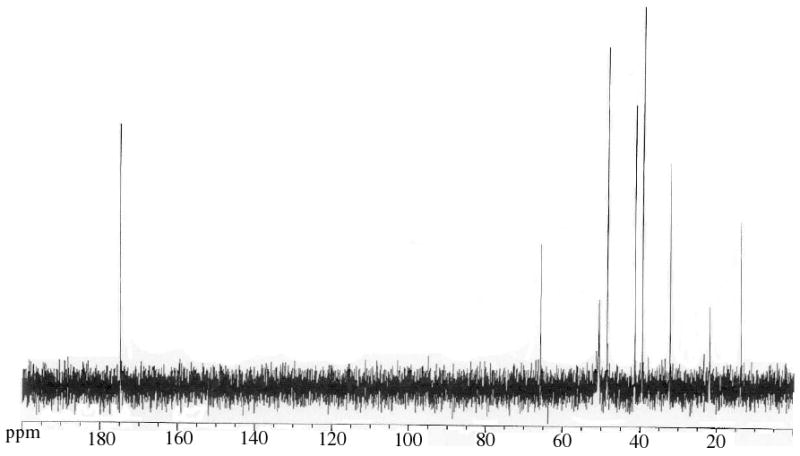
13C NMR spectrum of the POMAM 2:1 hybrid dendrimer.
Figure 3 shows theoretical (Figure 3A) and experimental (Figure 3B) molecular weight dependence on the generation number of the POPAM core and number of PAMAM shells. As the non-linear surface function indicates, molecular weights are highly influenced by both number of PAMAM shells and generation number of the POPAM cores.
Figure 3.
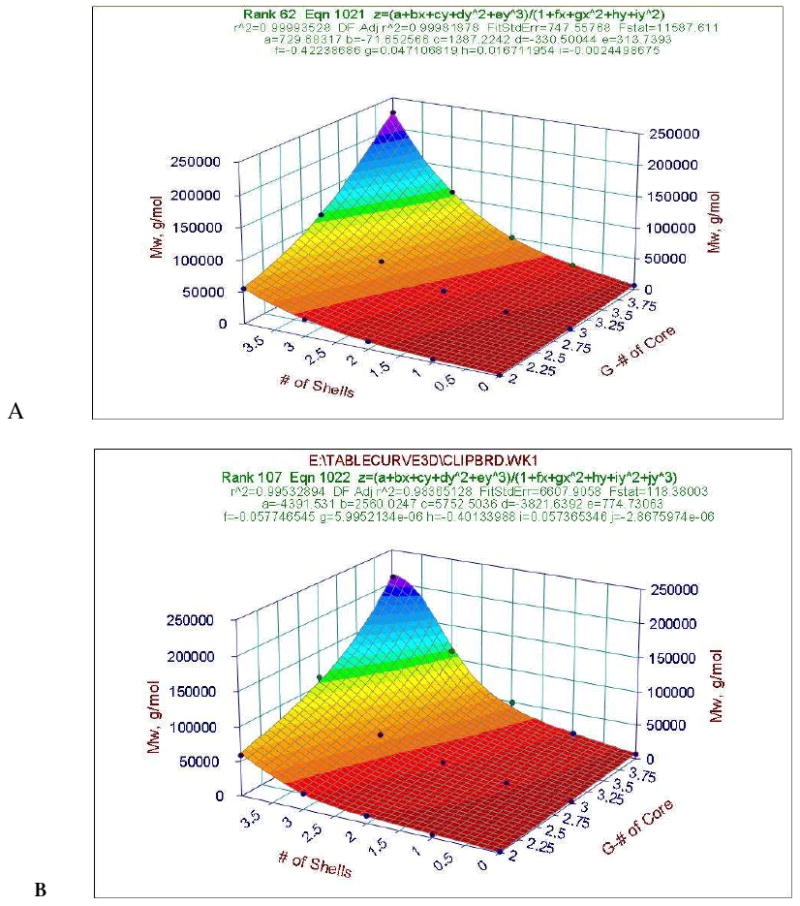
Theoretical (A) and experimental (B) molecular weight dependence on the generation number of the POPAM core and number of PAMAM shells.
Experimental molecular weights were determined by GPC. It was observed that lower generations of POMAM hybrid dendrimers (2:1, 2:2, 3:1, 3:2, 4:1, and 4:2) exhibit monodispers with very narrow distribution, while at the higher generations (2:3, 2:4, 3:3, 3:4, 4:3, and 4:4) bimodal narrow distribution was observed. The second peak with higher molecular weight was 10 % below the area of that lower molecular weight peak. Under the conditions used for GPC, aggregation may take place, which results in a higher molecular weight component. Interestingly, this aggregation behavior of the higher molecular weight hybrid dendrimer was not observed on HPLC eluogram (see later). Figure 4A, the RI eluogram of the POMAM 2:2 hybrid dendrimer (with 64 primary amino groups on the surface {theoretical value}), is presented as a representative example for comparison with the PAMAM dendrimers (G0-G7) in Figure 4B. This result clearly indicates the more compact structure of the hybrid dendrimer, as the hydrodynamic volume of the POMAM 2:2 hybrid dendrimer lies between the hydrodynamic volumes of PAMAM G3 and PAMAM G4.
Figure 4.
GPC-RI eluogram of the POMAM 2:2 hybrid dendrimer (A) and its comparation with PAMAM dendrimers (G-0 to G-7) (B)
Figure 5 shows the relation of hydrodynamic diameter of the POMAM hybrid dendrimers as a function of the generation number of the core and number of shells. The hydrodynamic diameter of the POMAM hybrid dendrimers exhibits an almost linear relationship to the generation number of the core and number of shells, reaching as high a value as ∼83 Å.
Figure 5.
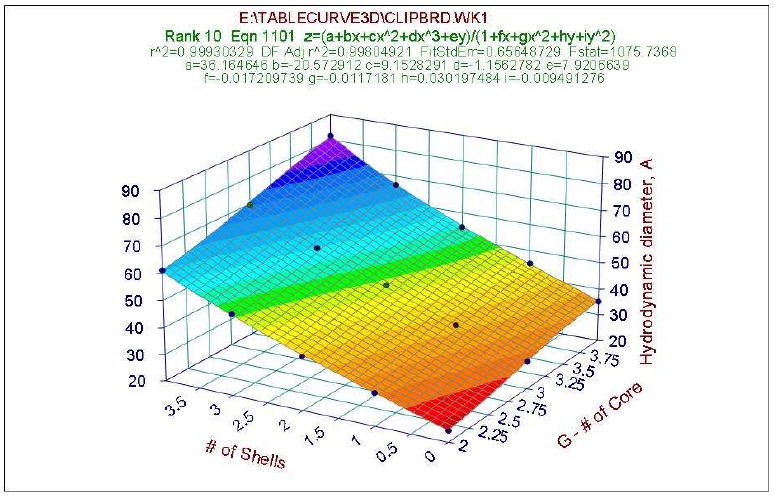
Hydrodynamic diameter as function of the generation number of the core and number of shells
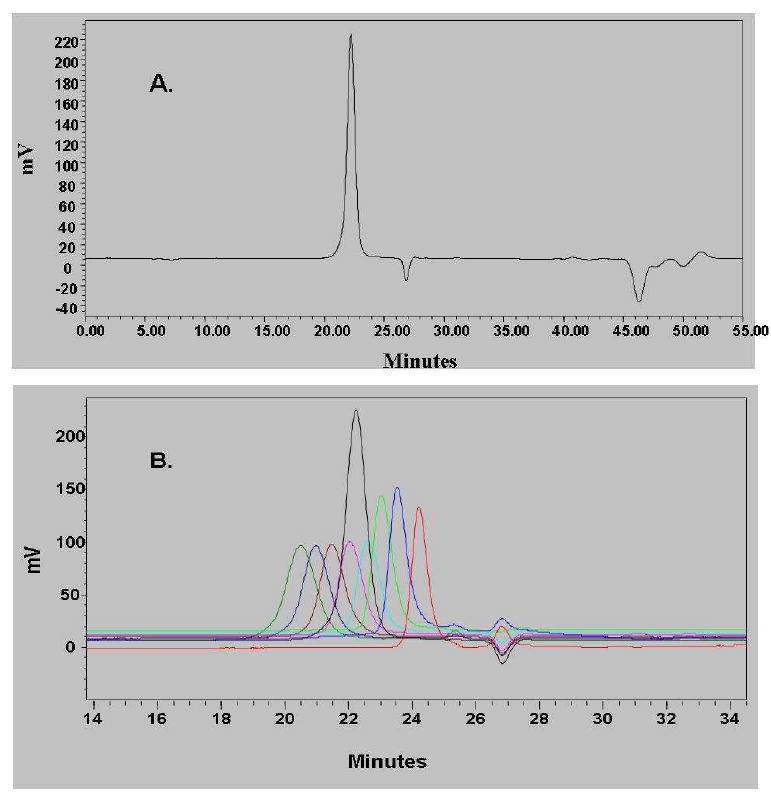
Figure 6A presents representative examples of the HPLC eluograms for POMAM hybrid dendrimers of generations 2:4, 3:4 and 4:4. It is clear that the dendrimers are relatively pure. A very small amount of higher molecular weight material is present due to aggregation (it is negligible). Interestingly, this aggregation can be observed at lower generations in the HPLC eluograms, but not in the GPC eluogram. This result suggests that aggregation behavior is highly influenced by the environment of the molecules. Figure 6B shows how POMAM dendrimers asymptotically approach liquid chromato-graphycal properties of the PAMAM dendrimers.
Figure 6.
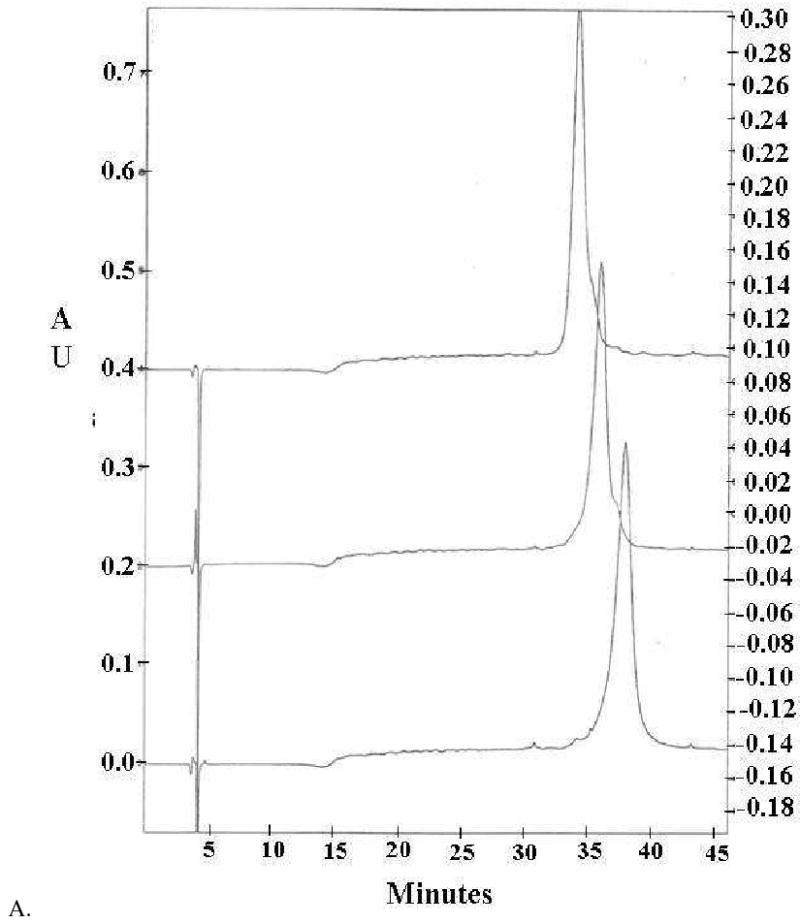
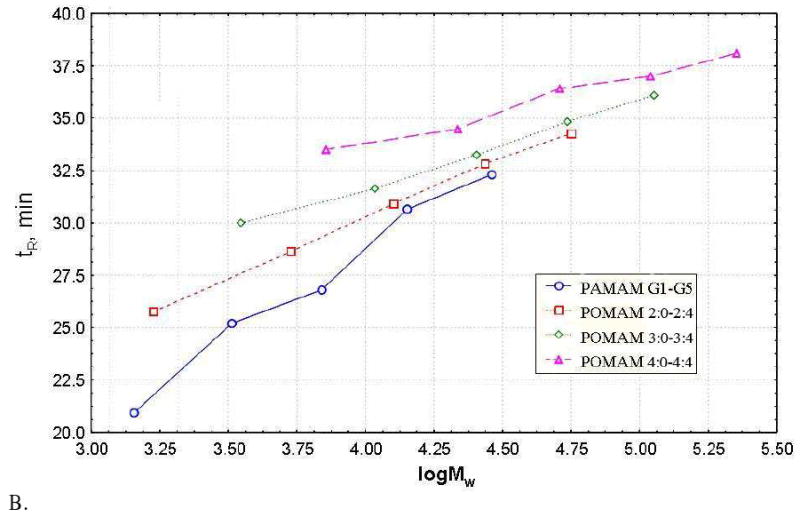
HPLC eluograms of the POMAM hybrid dendrimers (generations 2:4, 3:4 and 4:4) (A). Retention time vs. logarithm of molecular weight of PAMAM and POMAM dendrimers (B).
The first challenge in achieving high-resolution AFM images of individual POMAM hybrid dendrimer molecules was obtaining films in which the individual molecules were separated from one another. Dendrimer molecules possess a high concentration of surface functional groups, which often causes them to aggregate. High-resolution images of individual PAMAM or POPAM and the hybrid POMAM dendrimers of generations lower than G5 are difficult to obtain because of this problem 20. However, the larger dendrimer molecules, (particularly the hybrid dendrimer molecules) can be spread on a mica surface to show individual molecules; provided the spreading is performed from ultra-dilute solutions. Spin-coating techniques also help in preparing uniform films. Figure 7 shows the tapping mode AFM image of a representative hybrid dendrimer (POMAM 4:2).
Figure 7.

Tapping mode AFM image of hybrid dendrimer (POMAM 4:2) molecules. (Sample preparation: one drop of a 1 10-4 w % solution was spread on a freshly cleaved mica surface by spin coating and then dried at room temperature).
The image shows many globular particles randomly deposited on the mica surface. The particles appear to the substantially uniform in size, i.e., they are essentially mono-dispersed. Also, the images apparently are of single molecules (although a few obvious clusters or aggregates can be identified).
It is well known that ionic strength, surface charge density, and concentration (molecular environment) are the crucial parameters controlling the degree of structural organization of the solution 21. Addition of monovalent salt (KCl or NaCl) screens the long-range electrostatic interactions and leads to a gas-like structure i.e., disaggregated arrangement. Full generation of dendrimers have primary amino groups in high density on the surface. In this case, the surface charge densities of these molecules can be manipulated by varying the pH of the solution, and the dendrimers can be viewed as nanoscopic polyelectrolyte particles.
It is important to point out that POMAM dendrimer molecules involve two sorts of amine groups. The primary amine end groups (-NH2) are on the periphery of POMAM dendrimers, and their number is equal to 2(n+m+2), with n the generation of POPAM dendrimer core, m the number of PAMAM dendrimer shells. The tertiary amine groups (>N-) are situated at the branching points in the molecule, and their number is equal to 2(n+m+2) - 2. These two types of amine groups, if isolated, are characterized by different pK values 19, which are the negative logarithms of the acidic dissociation constant for the protonated primary (pKpr) and tertiary (pKt) amine groups correspondingly. The basicity of the primary amine group is higher compared with that of the tertiary one, i.e., (pKpr) > pKt. The experimental titration and experimental mass spectroscopy information (data not shown) is beyond the breadth of this paper and will published soon.
Conclusion
The primary objective of this study was the synthesis of novel, well-defined POMAM hybrid dendrimer with POPAM core and PAMAM shells. The goal was achieved with the use of POPAM G-2, G-3, and G-4 well characterized dendrimers with 16, 32, and 64 primary amines on the surface. The reiterating synthetic studies using these multifunctional cores revealed that monodisperse POMAM hybrid dendrimers can be prepared using the MA addition and EDA amidation two-step process, under controlled conditions. The 13C NMR, molecular weights, and hydrodynamic diameters determined were as expected close to the theoretical values. AFM imaging has shown relatively uniform size of the particles. It is known, as previously experimentally demonstrated, that poly(amidoamine) PAMAM dendrimers are efficient gene delivery reagents for in vitro and in vivo applications. These PAMAM dendrimers are not toxic in biomedical applications.
Preliminary experiments with POMAM hybrid dendrimers have shown similar transfer activity and are not toxic either in comparison with POPAM dendrimers.
Acknowledgments
This material is based on the work supported by the National Cancer Institute (Project Contract # NOI-CO-97111). We acknowledge the cooperation of the NMR Center, Department of Pharmacology, University of Michigan, and the help of Scott Woehler, Jing Li, Andrea Wetmore for recording the high-resolution proton and carbon NMR, GPC, AFM and HPLC.
References
- 1.Majoros IJ, Thomas TP, Mehta C, Baker JR., Jr PAMAM Dendrimer-Based Multi-Functional Engineered Nanodevice for Cancer Therapy. Journal of Medicinal Chemistry. 2005;48(19):5892–5899. doi: 10.1021/jm0401863. [DOI] [PubMed] [Google Scholar]
- 2.Thomas TP, Majoros IJ, Kotlyar A, Kukowska-Latallo JF, Bielinska A, Myc A, Baker JR., Jr Targeting and Inhibition of Cell Growth by an Engineered Dendritic Nanodevice. Journal of Medicinal Chemistry. 2005;48(11):3729–3735. doi: 10.1021/jm040187v. [DOI] [PubMed] [Google Scholar]
- 3.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR., Jr Nanoparticle Targeting of Anticancer Drug Improves Therapeutic Response in Animal Model of Human Epithelial Cancer. Cancer Research. 2005;65(12):5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 4.Majoros IJ, Myc A, Thomas TP, Mehta C, Baker JR., Jr PAMAM Dendrimer-Based Multi-Functional Engineered Nano-Device for Cancer Therapy. II: Synthesis, Characterization, and Functionality. Biomacromolecules. 2006;7(2):572–9. doi: 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]
- 5.Majoros István J, Baker James R., Jr, editors. Dendrimer Based Nanomedicine. World Scientific Publishing Co.; 2008. [Google Scholar]
- 6.Tomalia DA, Naylor AM, Goddard WA., III Starburst Dendrimers: Molecular-Level Control of Size, Shape, Surface Chemistry, Topology, and Flexibility from Atoms to Macroscopic Matter. Angew Chem Intl Edit. 1990;29:138–175. [Google Scholar]
- 7.Naylor AM, Goddard WA, III, Keiffer GE, Tomalia DA. Starburst dendrimers. 5. Molecular shape control. J Am Chem Soc. 1989;111:2339–2341. [Google Scholar]
- 8a.Tomalia DA, Hedstrand DM, Ferritto MS. Comb-burst dendrimer topology: new macromolecular architecture derived from dendritic grafting. Macromolecule. 1999;24:1435–1438. [Google Scholar]
- 8b.Dvornic PR, Tomalia TA. Dendritic Polymers, Divergent Synthesis: Starburst Poly(amidoamine) Dendrimers. In: Salamone JC, editor. The Polymeric Materials Encyclopedia: Synthesis, Properties and Applications. CRC Press; Boca Raton: 1996. [Google Scholar]
- 9.Hawker CJ, Wooley KL, Fréchet JMJ. Unimolecular micelles and globular amphiphiles: dendritic macromolecules as novel recyclable solubilization agents. J Chem Soc Perkins Trans. 1993;12:1287–1297. [Google Scholar]
- 10.Fréchet JMJ. Functional Polymers and Dendrimers: Reactivity, Molecular Architecture, and Interfacial Energy. Science. 1994;263:1710–1715. doi: 10.1126/science.8134834. [DOI] [PubMed] [Google Scholar]
- 11.Fréchet JMJ, Henmi M, Gitsov I, Aochima S, Leduc MR, Grubbs RB. Self-Condensing Vinyl Polymerization: An Approach to Dendritic Materials. Science. 1995;269(5227):1080–1083. doi: 10.1126/science.269.5227.1080. [DOI] [PubMed] [Google Scholar]
- 12.Zimmerman SC, Zeng FW, Richert DEC, Kolotuchin SV. Self-Assembling Dendrimers. Science. 1996;271:1095–1098. doi: 10.1126/science.271.5252.1095. [DOI] [PubMed] [Google Scholar]
- 13.Bell TW. Molecular Trees: A New Branch of Chemistry. Science. 1996;271:1077–1078. doi: 10.1126/science.271.5252.1077. [DOI] [PubMed] [Google Scholar]
- 14.van Hest JCM, Delnoye DAP, Baars MWPL, van Genderen MHP, Meijer EW. Polystyrene-Dendrimer Amphiphilic Block Copolymers with a Generation-Dependent Aggregation. Science. 1995;268:1592–1595. doi: 10.1126/science.268.5217.1592. [DOI] [PubMed] [Google Scholar]
- 15.Jansen FGAJ, Meijer EW, de Brabander-van den Berg EMM. The Dendritic Box: Shape-Selective Liberation of Encapsulated Guests. J Am Chem Soc. 1995;117:4417–4418. [Google Scholar]
- 16.Jansen FGAJ, de Brabander-van den Berg EMM, Meijer EW. Encapsulation of Guest Molecules into a Dendritic Box. Science. 1994;266:1226–1229. doi: 10.1126/science.266.5188.1226. [DOI] [PubMed] [Google Scholar]
- 17.De Brabander EMM, Meijer EW. Poly(propylene imine) Dendrimers: Large-Scale Synthesis by Hetereogeneously Catalyzed Hydrogenations. Angew Chem Int Ed. 1993;32(9):1308–1311. [Google Scholar]
- 18.De Brabander EMM, Brackman J, Mure-Mak M, de Man H, Hogeweg M, Keulen J, Scherrenberg R, Coussens B, Mengerink Y, van der Wal S. Polypropylenimine dendrimers: improved synthesis and characterization. Macromol Symp. 1996;102:9. [Google Scholar]
- 19.Van Genderen MMP, Baars MWPL, Elissen-Roman C, de Brabander-van den Berg EMM, Meijer EW. Natural abundance 15N-NMR spectroscopic investigations of poly(propyleneimine) dendrimers. Polym Mater Sci Eng. 1995;73:336. [Google Scholar]
- 20.Jackson CL, Chanzy HD, Booy FP, Brake BJ, Tomalia DA, Bauer BJ, Amis EJ. Visualization of Dendrimer Molecules by Transmission Electron Microscopy (TEM): Staining Methods and Cryo-TEM of Vitrified Solutions. Macromolecules. 1998;31:6259–6265. [Google Scholar]
- 21.Nisato G, Ivkov R, Amis EJ. Structure of Charged Dendrimer Solutions As Seen by Small-Angle Neutron Scattering. Macromolecules. 1999;32:5895–5900. [Google Scholar]