

Abstract
In contrast to tropane-based compounds such as benztropine and cocaine, non-tropane-based photoaffinity ligands for the dopamine transporter (DAT) are relatively unexplored. Towards addressing this knowledge gap, ligands were synthesized in which the piperidine nitrogen of 3- and 4-iodomethylphenidate was substituted with a benzyl group bearing a photoreactive azide. Analog (±)-3a demonstrated modest DAT affinity and a radioiodinated version was shown to bind covalently to rat striatal DAT and hDAT expressed in cultured cells. Co-incubation of (±)-3a with nonradioactive D-(+)-methylphenidate or (−)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane (β-CFT, WIN-35,428, a cocaine analog) blocked DAT labeling. Compound (±)-3a represents the first successful example of a DAT photoaffinity ligand based on the methylphenidate scaffold. Such ligands are expected to assist in mapping non-tropane ligand-binding pockets within plasma membrane monoamine transporters.
Keywords: methylphenidate, Ritalin, Concerta, photoaffinity labeling, dopamine transporter, cocaine, attention-deficit hyperactivity disorder
1. Introduction
Threo-methylphenidate ((±)-1a, Scheme 1) is a well-known stimulant acting through dopaminergic and adrenergic pathways. Both immediate-release (Ritalin) and long-acting (Concerta) preparations of methylphenidate remain the mainstay of treatment for adult attention-deficit hyperactivity disorder (ADHD), showing more than 75% efficacy in controlling the symptoms of the disease.1 In addition to being prescribed for narcolepsy and a number of off-label uses including lethargy, depression, and obesity, methylphenidate analogs continue to attract significant attention towards the development of cocaine abuse therapeutics.2
Scheme 1.

Rational design of (±)-threo-N-(azido-benzyl)-iodomethylphenidates (3) via molecular hybridization of halogenated methylphenidates (1b, 1f) with N-benzylmethylphenidates (2).
In contrast to amphetamines, which cause a direct release of norepinephrine and dopamine into the synapse, methylphenidate acts as a mild central nervous system stimulant by inhibiting the dopamine and norepinephrine transporter proteins (DAT and NET, respectively), thus blocking the reuptake of dopamine and norepinephrine into the presynaptic neuron.3 However, in sharp contrast to the beneficial therapeutic effects associated with methylphenidate, behavioral and pharmacological studies have also implicated the DAT as the primary target associated with the reward/reinforcing properties of cocaine and amphetamines as abused psychostimulants.4, 5 As a result, the long-term goal of our research is to understand how the DAT discriminates abused versus therapeutic compounds at the molecular level.6
The lack of clinically available medications to battle psychostimulant abuse can be linked, in part, to limited information on the 3-D structure and function of the DAT. Indeed, the search for therapeutics has resulted in a host of structurally disparate ligands capable of selectively binding to the DAT and inhibiting dopamine uptake;7 however, details regarding discrete ligand-binding pockets and the transport inhibition mechanism remain poorly understood. Furthermore, inhibitor structure-activity relationships (SAR) and site-directed mutagenesis studies imply that structurally divergent DAT inhibitors bind to different domains/binding sites within the DAT, or with differential conformational preferences, both of which could affect their behavioral profile in cocaine abuse animal models.8–10 Radiolabeled (3H, 125I) molecular probes serving as photoaffinity (-N3) and affinity (-NCS) ligands represent important tools towards determining DAT conformational states and mapping of inhibitor-/substrate-binding sites.
The chemical development of DAT affinity and photoaffinity ligands has focused on cocaine and benztropine analogs as tropane-based ligands11–19 or their conformationally flexible piperidine20 and piperazine21–26 analogs. In contrast and due to having received significantly less attention,27 the work herein focused on developing DAT photoaffinity ligands based on therapeutically relevant non-tropane compounds.28 These ligands expand the battery of complementary chemical probes that may be useful for mapping DAT inhibitor-binding pockets at the molecular level. With the crystal structure of the bacterial homolog LeuT serving as a template for homology modeling of DAT 3-D structure, such high resolution ligand-binding mapping is finally in sight.29 Herein we report our initial studies with respect to rational design, synthesis, pharmacological evaluation, and photoaffinity labeling of methylphenidate-based DAT irreversible ligands.
2. Results and Discussion
2.1. Probe Design and Synthesis
With respect to photoaffinity probe design based on DAT inhibitors, the overwhelming number of known compounds in this group contain an aromatic 4-azido-3-iodo-substituted ring motif.12–17, 19–21, 24, 25, 28 However, this motif may inherently lead to low efficiency (e.g., <1%) of incorporation of the aryl azide into the protein due to its juxtaposition with the sterically bulky iodine.30 Towards overcoming this potential problem, we decided to focus on the design of compounds in which the photoreactive azide group and 125I radiotracer tag are on different parts of the methylphenidate scaffold.
We chose 4-iodomethylphenidate ((±)-1b, Scheme 1) as a lead compound given that this analog displays ~6-fold higher DAT affinity versus methylphenidate (DAT IC50 for inhibition of [3H]-WIN-35,428 (a cocaine analog) binding, (±)-1a = 83.0 ± 7.9 nM, (±)-1b = 14.0 ± 0.1 nM).31 We envisioned the 4-position of methylphenidate’s aromatic ring as a logical place to anchor a bulky iodine as a future radiotracer tag within rationally designed photoaffinity probes. Additionally, we considered N-benzylmethylphenidate analogs ((±)-2) as lead compounds since they display either improved ((±)-2a = 52.9 ± 2.3 nM, (±)-2b = 41.2 ± 3.4 nM, (±)-2c = 76.3 ± 2.7 nM, (±)-2d = 31.2 ± 5.7 nM, (±)-2e = 79.1 ± 1.4 nM) or retained ((±)-2f = 106 ± 24 nM, (±)-2g = 113 ± 3.0 nM) DAT affinity versus methylphenidate.32 Furthermore, improved DAT affinity was observed for methylphenidate analogs bearing halogens at the 3-position of the aromatic ring ((±)-1c = 40.5 ± 4.5 nM, (±)-1d = 5.10 ± 1.6 nM, (±)-1e = 4.18 ± 0.17 nM).31 This prompted us to investigate this position as another logical place to anchor a bulky iodine as a future radiotracer tag (i.e., 3-iodomethylphenidate ((±)-1f)). Given these established structure-activity relationships for methylphenidate analogs, a series of photoaffinity ligands ((±)-3) was designed via molecular hybridization of iodomethylphenidates ((±)-1b, (±)-1f) with N-benzylmethylphenidates ((±)-2). These target probes feature systematic placement of a photoreactive azide on the aromatic ring of the N-benzyl group (Scheme 1).
The preparation of 4-iodomethylphenidate ((±)-1b) has been previously described;33 however, this synthesis affords a poor yield (6 steps, 9% overall yield) and originates from methylphenidate itself, a relatively expensive starting material. Towards synthesizing a variety of probes based on either (±)-1b or (±)-1f, we first demanded short syntheses of these iodomethylphenidates involving cheap, readily available starting materials. The application of methodology originally developed by Axten et al.,34 and later modified by Gutman et al.,35 met our needs (Scheme 2). Ketoamide 4a was prepared from its corresponding ethyl ester derivative36 by treatment with piperidine in ethanol,34 whereas ketoamide 4b was accessed via regioselective meta-iodination of 1-(phenylglyoxylyl)piperidine35 using N-iodosuccinimide. Tosylhydrazones 5a and 5b were subsequently generated by allowing the ketoamides to react with p-toluenesulfonyl hydrazide in acidic ethanol under reflux. Next, thermal cyclization of the tosylhydrazones in refluxing toluene was performed under basic aqueous conditions using Aliquat 336 as a phase transfer catalyst.35 Subsequent recrystallization from ether provided diastereomerically pure racemic threo-β-lactams (±)-6 as the major product in moderate yield (55–60%). In turn, these threo-β-lactams were converted to iodomethylphenidates (±)-1b and (±)-1f by ring opening with methanol under acidic conditions. These threo-iodomethylphenidates were determined to be ≥95% diastereomerically pure by 1H NMR upon comparison to the known data for enantiomerically pure threo- and erythro-methylphenidate and its para-substituted derivatives.37 Finally, target photoaffinity probes (±)-3 were accessed from the iodomethylphenidates by either N-alkylation with an azidobenzyl bromide38 (e.g., (±)-3a), or a sequence of N-alkylation with a nitrobenzyl bromide, nitro reduction, and diazotization followed by azide displacement (e.g., (±)-3e).
Scheme 2.

Synthesis of iodomethylphenidates (±)-1b and (±)-1f and representative approaches to target probes 3.
2.2. Pharmacology and Structure-Activity Relationships
With the target methylphenidate compounds in hand, ligand affinities (Ki values) were determined for inhibition of [3H]-WIN-35,428 binding to a human dopamine transporter (hDAT) stably expressed in N2A neuroblastoma cells. [3H]-Dopamine uptake inhibition potencies in the same cells under the same conditions were also determined (Table 1). Racemic threo-methylphenidate ((±)-1a) was synthesized34, 35 and pharmacologically evaluated for comparison to the novel compounds. A ~5.5-fold improvement in DAT affinity was observed upon substituting the 3-position of methylphenidate’s aromatic ring with iodine (i.e., (±)-1f), further supporting the observation that halogens are tolerated at this position31 and suggesting an anchoring position for 125I as a radiotracer tag. The affinity of 4-iodomethylphenidate ((±)-1b) was approximately two-fold higher than methylphenidate in our hands, whereas previous pharmacological results indicated an approximate 6-fold increase in DAT affinity.31 However, substituting the piperidine nitrogen of these iodomethylphenidates with a N-benzyl group bearing an aromatic azide resulted in a significant decrease in DAT binding affinity (~15-fold to 110-fold lower binding affinity than methylphenidate). Out of the series of hybrid compounds, N-(p-azido-benzyl)-4-iodomethylphenidate ((±)-3a) displayed the highest DAT affinity (Ki = 363 ± 28 nM). With respect to DAT binding affinity and the position of the azide group on the N-benzyl aromatic ring, the para position appears to be about the same (15 and 26-fold lower binding affinity than methylphenidate) as compared to the ortho position (21 and 44-fold lower binding affinity than methylphenidate). Substitution of the azide at the meta position resulted in a much greater decrease in DAT binding affinity (110 and 82-fold lower binding affinity than methylphenidate). There was no obvious advantage in terms of DAT binding affinity associated with the position of the iodine in these hybrid analogs (average Ki: 3-iodo compounds = 1280 nM; 4-iodo compounds = 1210 nM). Thus, these designed azido-N-benzyl-iodomethylphenidates behaved unlike previously reported methylphenidates in which N-benzyl substitution either increased or retained DAT affinity.32
Table 1.
Inhibition of [3H]-WIN-35,428 binding and [3H]-dopamine uptake by compounds at hDAT N2A neuroblastoma cells.
| Compound | [3H]-WIN Binding Inhibition, Ki (nM)* | [3H]-DA Uptake Inhibition, IC50 (nM)* |
|---|---|---|
| (±)-1a, threo-methylphenidate | 25 ± 1 | 156 ± 58 |
| (±)-1b, 4-I-methylphenidate | 14 ± 3a | 11 ± 2b |
| (±)-1f, 3-I-methylphenidate | 4.5 ± 1a | 14 ± 5b |
| (±)-3a, p-N3-N-Bn-4-I-methylphenidate | 363 ± 28a | 2764 ± 196b,c |
| (±)-3b, m-N3-N-Bn-4-I-methylphenidate | 2754 ± 169a | 7966 ± 348b,c |
| (±)-3c, o-N3-N-Bn-4-I-methylphenidate | 517 ± 65a | 1232 ± 70b,c |
| (±)-3d, p-N3-N-Bn-3-I-methylphenidate | 658 ± 70a | 1828 ± 261b,c |
| (±)-3e, m-N3-N-Bn-3-I-methylphenidate | 2056 ± 73a | 4627 ± 238b,c |
| (±)-3f, o-N3-N-Bn-3-I-methylphenidate | 1112 ± 163a | 2696 ± 178b,c |
Each Ki or IC50 value represents data from at least three independent experiments with each data point on the curve performed in duplicate.
P<0.05 vs. Ki of (±)-1a, threo-44 methylphenidate;
P<0.05 vs. IC50 of (±)-1a, threo-methylphenidate;
P<0.05 vs. its corresponding Ki.
With respect to inhibition of dopamine uptake (Table 1), IC50 values were typically 2- to 8-fold higher than the binding Ki values for each compound even though the two assays were conducted under identical conditions. Using the Cheng-Prusoff equation, conversion of uptake inhibition constants from IC50 to Ki did not significantly change the value, allowing for direct comparison of binding and uptake results. A 3- to 4-fold difference between inhibitor binding and dopamine uptake inhibition potencies has been previously observed with rDAT/CHO cells in this laboratory for WIN-35,428, cocaine, mazindol, and methylphenidate.10 Interestingly, 4-iodomethylphenidate ((±)-1b) displayed essentially the same value for binding and uptake inhibition (Table 1), a pattern previously seen for benztropine and the related compounds GBR-12,909 and rimcazole.10 Cocaine and benztropine have been suggested to occupy non-identical DAT sites or conformations; 8–10 the present result may imply that 4-iodomethylphenidate also interacts with the DAT in a fashion different from the other compounds in Table 1.
With respect to identifying candidate probes for photoaffinity labeling experiments, ideal compounds are those that are bioactive in the same range as the parent compound. However, compounds with as much as 1000-fold lower activity can still be useful.39 With this in mind and the observation that compound (±)-3a retained modest DAT affinity, we decided to further pursue development of this compound into a potential DAT photoaffinity probe.
Wash-resistant binding experiments on nonradioactive azido compounds frequently give false positives in the assessment of covalent attachment.17 Therefore, the synthesis of radioiodo compound [125I]-(±)-3a was attempted to determine if photoactivation produced covalent ligation to the DAT. Synthesis of [125I]-(±)-3a was achieved by converting nonradioactive iodo compound (±)-3a into a tributylstannyl derivative ((±)-7) followed by radioiodination with [125I]-NaI (1.53 mCi) in the presence of Chloramine-T (Scheme 3). This sequence ended with reversed-phase HPLC isolation to provide [125I]-(±)-3a in 61% isolated yield, high purity (98%), and high specific activity (2099 mCi/μmol). The radioligand exhibited a chromatographic profile identical to that of non-radioactive (±)-3a (Figure 1). [125I]-(±)-3a (tR = 18.4 min) was well resolved from radioactive and non-radioactive side-products by preparative HPLC (Figure 1A). The major non-radioactive material was assigned as the chloro analog of (±)-3a (tR = 13.3 min) based upon model studies conducted using an excess of Chloramine-T but no radioiodine. Purified [125I]-(±)-3a co-eluted with a fully characterized sample of non-radioactive (±)-3a under the same reversed-phase HPLC conditions (Figure 1B).
Scheme 3.

Radiosynthesis of [125I]-(±)-3a.
Figure 1.
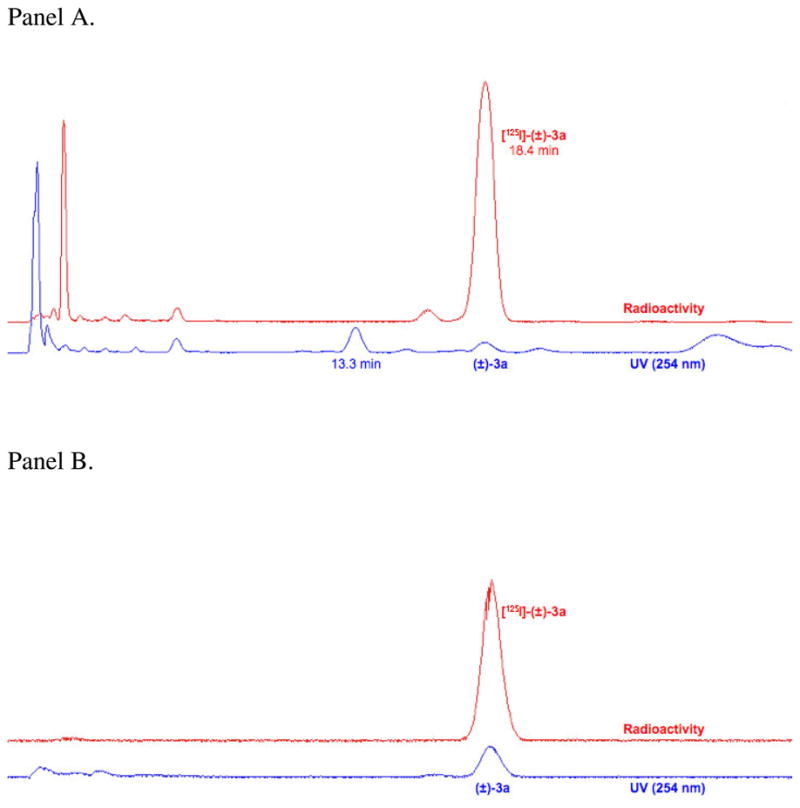
Panel A: Reversed-phase HPLC chromatogram for isolation of [125I]-(±)-3a. The radioligand exhibited an appropriate retention time based on the non-radioactive standard and was well resolved from radioactive and non-radioactive side-products. Panel B: Co-elution of purified [125I]-(±)-3a and a standard sample of (±)-3a under the same HPLC conditions used for preparative work.
2.4. DAT Photoaffinity Labeling
To determine if the DAT was able to be irreversibly labeled with [125I]-(±)-3a, we used procedures previously developed for the analysis of other DAT photoaffinity labels.13, 14, 17, 20, 28, 30, 40–43 Briefly, rat striatal membranes and HEK 293 cells expressing 6Xhis-hDAT were incubated with 30 nM [125I]-(±)-3a in the absence or presence of 10 μM or 100 μM β-CFT ((−)-2-β-carbomethoxy-3-β-(4-fluorophenyl)tropane, WIN-35,428, a cocaine analog) or D-(+)-methylphenidate (D-MPH). The membranes and cells were then detergent-solubilized and the lysates were immunoprecipitated with DAT antibody and analyzed by SDS-PAGE/autoradiography. Labeled proteins of ~80 kDa were obtained from both rat striatal tissue (Figure 2) and HEK hDAT cells (not shown), demonstrating the incorporation of [125I]-(±)-3a into the DAT. Incorporation of the ligand was blocked by 75–90% by both β-CFT and D-MPH, demonstrating the appropriate pharmacological specificity of [125I]-(±)-3a attachment to the DAT. Similar to results previously reported for tropane, GBR, and benztropine DAT photoaffinity ligands,42, 43 analysis of total cell lysates showed that several proteins undergo adduction with [125I]-(±)-3a (not shown). However, these do not represent the DAT because they do not immunoprecipitate with DAT antibody as shown for the protein in Figure 2.
Figure 2.

Photoaffinity labeling of DAT with [125I]-(±)-3a. Rat striatal membranes were photoaffinity labeled with 30 nM [125I]-(±)-3a in the absence or presence of 10 μM or 100 μM β-CFT or D-MPH. Membranes were solubilized and DATs were immunoprecipitated followed by analysis by SDS-PAGE and autoradiography. The relevant portion of a representative autoradiograph is pictured followed by a histogram that quantitates relative band intensities. Mean ± S.E. of three independent experiments is shown; ***, p < 0.001 vs. control.
3. Conclusions
We have designed, synthesized, and pharmacologically evaluated novel photoaffinity ligands based on the well-known ADHD drug threo-methylphenidate. In particular, (±)-3a represents the first successful example of a DAT photoaffinity ligand based on the methylphenidate scaffold, thus representing an important contribution to the growing arsenal of probes useful for characterizing DAT function and 3-D structure. There is evidence that structurally diverse DAT inhibitors bind to non-identical DAT sites or conformations,8–10 suggesting that novel irreversible ligands based on methylphenidate may yield new monoamine transporter structure-function information. The 3-iodo analog ((±)-1f) was synthesized as a new DAT inhibitor and pharmacologically found to possess high affinity. Our results potentially warrant the exploration of an 123I-labeled derivative of this compound as a prospective SPECT radiopharmaceutical for brain dopamine transporters.33 Additionally, we found that ligand (±)-3a bound with modest affinity to the DAT and its 125I analog was shown to bind covalently to rDAT and hDAT expressed in cultured cells. Since the (R,R)-(+)-enantiomer of threo-methylphenidate has been found to be the more biologically active compound,44 future directions include resolving (±)-3a into its enantiomerically pure components towards obtaining a more specific and improved DAT photoaffinity probe. Additional DAT irreversible ligands based on methylphenidate (particularly those with improved DAT affinity), their pharmacological characterization, binding site prediction via docking within 3-D DAT homology models,29 and detailed elucidation of DAT binding domains for comparison to established tropane-based probes will be investigated in due course.
4. Experimental
Reaction conditions and yields were not optimized. All reactions were performed in flame-dried glassware under argon unless otherwise noted. All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and used without further purification. Flash column chromatography was performed according to the method of Still et al.45 using Fisher S826-25 silica gel sorbent (70-230 mesh) and eluting solvent mixtures as specified. Thin-layer chromatography (TLC) was performed using TLC silica gel 60 F254 plates obtained from EMD Chemicals, Inc. and compounds were visualized under UV light and/or I2 stain. Proportions of solvents used for TLC are by volume. 1H and 13C NMR spectra were recorded on either a Bruker 400 or 500 MHz spectrometer. Chemical shifts for 1H and 13C NMR spectra are reported as parts per million (δ ppm) relative to tetramethylsilane (0.00 ppm) as an internal standard. Coupling constants are measured in Hz. HRMS samples were analyzed at Old Dominion University (Norfolk, VA) by positive ion electrospray on a Bruker 12 Tesla APEX-Qe FTICR-MS with an Apollo II ion source. Combustion analyses of selected solid compounds were performed by Atlantic Microlab, Inc. (Norcross, GA) and agree within 0.4% of calculated values. Melting point determinations were conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Infrared spectra were recorded using a Perkin-Elmer Spectrum RZ I FT-IR spectrometer. On the basis of NMR and combustion data, all compounds were ≥95% pure. A radioisotope dose calibrator (Capintec CRC-15W) was used for radioactivity measurements and similar counting geometries were employed for each determination.
4.1. 1-(4-Iodophenylglyoxylyl)piperidine (4a)
A solution of ethyl 2-(4-iodophenyl)-2-oxoacetate36 (0.23 g, 0.75 mmol) in EtOH (3 mL) was added to a stirred mixture of piperidine (0.19 g, 2.25 mmol) in EtOH (3 mL). The reaction mixture was stirred at 70°C for 3 hours, cooled to room temperature, diluted with H2O then 1M aq. HCl, and extracted with EtOAc. The organic layer was dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 1:9) to provide 0.22 g of 4a as a colorless oil (63%). Rf = 0.23 (EtOAc:hexanes, 1:9). 1H NMR (CDCl3, 400 MHz): δ 7.82 (d, 2H, J = 8.2 Hz), 7.59 (d, 2H, J = 8.2 Hz), 3.63 (t, 2H, J = 5.6 Hz), 3.21 (t, 2H, J = 5.5 Hz), 1.63–1.59 (m, 4H), 1.50–1.45 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 191.0, 164.8, 138.3, 132.5, 130.7, 103.2, 47.0, 42.2, 26.2, 25.4, 24.3. HRMS calcd for C13H14INO2Na+ 365.9961, found 365.9961.
4.2. 1-(3-Iodophenylglyoxylyl)piperidine (4b)
N-iodosuccinimide (0.23 g, 1 mmol) was added to 96% H2SO4 (3 mL) and cooled to 0°C. The resulting suspension was stirred for 20–30 minutes until it became a homogeneous black solution. 1-(Phenylglyoxylyl)piperidine34, 35 (0.11 g, 0.5 mmol) was added to the solution and the mixture was stirred at 0°C for 20 min. The reaction mixture was poured into 10 mL of an ice-H2O mixture and treated with Na2SO3 to adjust the pH to 7. The mixture was then extracted with CH2Cl2, washed with brine, dried (MgSO4), and concentrated to obtain 0.13 g of 4b as a yellow oil (77%). Rf = 0.45 (EtOAc:hexanes, 3:7). 1H NMR (CDCl3, 400 MHz): δ 8.27 (s, 1H), 7.95 (d, 1H, J = 7.8 Hz), 7.89 (d, 1H, J = 7.8 Hz), 7.26 (t, 1H, J = 7.8 Hz), 3.69 (t, 2H, J = 4.8 Hz), 3.27 (t, 2H, J = 5.5 Hz), 1.80-1.60 (m, 4H), 1.60-1.50 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 190.2, 164.5, 143.2, 138.0, 134.9, 130.6, 128.7, 94.5, 47.0, 42.2, 26.1, 25.4, 24.2. HRMS calcd for C13H14INO2Na+ 365.9961, found 365.9960.
4.3. 1-(4-Iodophenylglyoxylyl)piperidine p-toluenesulfonylhydrazone (5a)
A solution of ketone 4a (0.93 g, 2.71 mmol) in EtOH (10 mL) was added to a stirred mixture of p-toluenesulfonyl hydrazide (0.55 g, 2.93 mmol) in ethanol (10 mL) containing H2SO4 (4 mg, 0.04 mmol). The reaction was refluxed at 85°C overnight then cooled to room temperature. The white precipitated solid was filtered, washed with cold MeOH and cold hexanes, then dried under reduced pressure to give 1.01 g of 5a (74%). Rf = 0.45 (EtOAc:hexanes, 4:6). 1H NMR (CDCl3, 400 MHz): δ 8.40 (s, 1H), 7.83 (d, 2H, J = 8.3 Hz), 7.71 (d, 2H, J = 8.7 Hz), 7.31 (d, 2H, J = 8.6 Hz), 7.26 (d, 2H, J = 8.1 Hz), 3.70-3.66 (m, 2H), 3.17 (t, 2H, J = 5.6 Hz), 2.38 (s, 3H), 1.66-1.64 (m, 4H), 1.50-1.40 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 161.5, 149.5, 144.2, 137.9, 135.0, 131.7, 129.6, 127.9, 127.7, 97.2, 47.3, 42.3, 26.3, 25.5, 24.1, 21.6. HRMS calcd for C20H22IN3O3SNa+ 534.0319, found 534.0318. Anal. Calcd for: C20H22IN3O3S: C, 46.97; H 4.34; N, 8.22; I, 24.82; S, 6.27. Found: C, 46.92; H 4.31; N, 8.23; I, 24.63; S, 6.42. MP: 137–138°C.
4.4. 1-(3-Iodophenylglyoxylyl)piperidine p-toluenesulfonylhydrazone (5b)
Ketone 4b (7.3 g, 21.2 mmol) was added to a stirred mixture of p-toluenesulfonyl hydrazide (4.26 g, 22.89 mmol) in EtOH (194 mL) containing H2SO4 (0.03 g, 0.3 mmol). The mixture was refluxed at 85°C overnight then cooled to room temperature. The precipitated solid was filtered, washed with cold MeOH and cold hexanes, then dried under reduced pressure to give 7.8 g of 5b (72%). Rf = 0.1 (EtOAc:hexanes, 3:7). 1H NMR (CDCl3, 400 MHz): δ 8.93 (s, 1H), 7.94 (s, 1H), 7.78 (d, 2H, J = 8.3 Hz), 7.69 (d, 1H, J = 7.9 Hz), 7.51 (d, 1H, J = 7.9 Hz), 7.19 (d, 2H, J = 8.1 Hz), 7.09 (t, 1H, J = 7.9 Hz), 3.7-3.6 (m, 2H), 3.2-3.1 (m, 2H), 2.34 (s, 3H), 1.76-1.52 (m, 4H), 1.48-1.38 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 161.3, 148.4, 144.1, 139.2, 135.0, 134.7, 134.2, 130.3, 129.4, 127.9, 127.9, 125.5, 94.5, 47.2, 42.3, 26.2, 25.4, 24.0, 21.5. HRMS calcd for C20H22IN3O3SNa+ 534.0319, found 534.0314. MP: 158–159°C.
4.5. (±)-7-(4-Iodophenyl)-1-azabicyclo[4.2.0]octan-8-one ((±)-6a)
Hydrazone 5a (1 g, 2 mmol) was added to a stirred mixture of 50% aq. NaOH (0.17 mL, 2.08 mmol) and Aliquat 336 (8 mg, 0.02 mmol) in toluene (20 mL). The reaction was refluxed at 130°C overnight then cooled to room temperature, diluted with H2O, and extracted with toluene. The organic layer was dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 2:8) to afford (±)-6a as a yellow solid. Recrystallization from Et2O provided 0.39 g of (±)-6a (60%). 1H NMR (CDCl3, 500 MHz): δ 7.64 (d, 2H, J = 8.3 Hz), 7.03 (d, 2H, J = 8.4 Hz), 3.92 (d, 1H, J = 4.3 Hz), 3.91-3.88 (m, 1H), 3.34-3.30 (m, 1H), 2.82-2.76 (m, 1H), 2.18-2.14 (m, 1H), 1.94-1.91 (m, 1H), 1.71-1.67 (m, 1H), 1.45-1.38 (m, 3H). 13C NMR (CDCl3, 100 MHz): δ 165.5, 137.7, 135.3, 129.3, 92.6, 62.8, 56.5, 39.0, 30.4, 24.4, 22.1. HRMS calcd for C13H14INONa+ 350.0012, found 350.0009. Anal. Calcd for: C13H14INO: C, 47.73; H 4.31; N, 4.28; I, 38.79. Found: C, 47.84; H 4.29; N, 4.23; I, 38.57. MP: 127–128°C.
4.6. (±)-7-(3-Iodophenyl)-1-azabicyclo[4.2.0]octan-8-one ((±)-6b)
50% aq. NaOH (1.3 mL, 15.96 mmol) was added to a stirred mixture of hydrazone 5b (7.8 g, 15.2 mmol) and Aliquat 336 (61 mg, 0.16 mmol) in toluene (140 mL). The mixture was refluxed at 130°C overnight then cooled to room temperature, diluted with H2O, and extracted with toluene. The organic layer was dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 2:8) to afford (±)-6b as a yellow solid. Further recrystallization from Et2O provided 2.7 g of (±)-6b (55%). Rf = 0.13 (EtOAc:hexanes, 2:8). 1H NMR (CDCl3, 400 MHz): δ 7.63-7.57 (m, 2H), 7.25 (d, 1H, J = 7.7 Hz), 7.05 (t, 1H, J = 7.8 Hz), 4.05-3.80 (m, 2H), 3.40-3.30 (m, 1H), 2.85-2.70 (m, 1H), 2.20-2.10 (m, 1H), 2.05-1.90 (m, 1H), 1.72-1.65 (m, 1H), 1.45-1.35 (m, 3H). 13C NMR (CDCl3, 100 MHz): δ 165.4, 137.9, 136.3, 136.1, 130.4, 126.6, 94.6, 62.6, 56.5, 39.0, 30.4, 24.3, 22.1. HRMS calcd for C13H14INONa+ 350.0012, found 350.0013. MP: 126–127°C.
4.7. (±)-threo-4-Iodomethylphenidate Hydrochloride ((±)-1b).33
(±)-threo-β-Lactam 6a (90 mg, 0.28 mmol) was refluxed in 1.25M HCl in MeOH (20 mL) at 85°C for 7 hours then concentrated under reduced pressure to afford 107 mg of (±)-1b as a yellow solid (100%). This threo-iodomethylphenidate was determined to be ≥95% diastereomerically pure by 1H NMR upon comparison to the known data for enantiomerically pure threo- and erythro-methylphenidate and its para-substituted derivatives.37 1H NMR (CD3OD, 400 MHz): δ 7.76 (d, 2H, J = 7.9 Hz), 7.11 (d, 2H, J = 8.0 Hz), 3.95 (d, 1H, J = 9.6 Hz), 3.83 (t, 1H, J = 9.9 Hz), 3.73 (s, 3H), 3.45 (d, 1H, J = 12.4 Hz), 3.10 (t, 1H, J = 11.8 Hz), 1.89-1.71 (m, 3H), 1.53-1.40 (m, 3H). 13C NMR (CD3OD, 100 MHz): δ 172.8, 139.6, 134.9, 131.7, 95.0, 58.9, 54.7, 53.6, 46.7, 27.6, 23.3, 22.8. HRMS calcd for C14H18INO2Na+ 382.0274, found 382.0275. MP: 188–189°C.
4.8. (±)-threo-3-Iodomethylphenidate Hydrochloride ((±)-1f)
(±)-threo-β-Lactam 6b (0.19 g, 0.58 mmol) was refluxed in 1.25M HCl in MeOH (41 mL) for 7 hours then concentrated under reduced pressure to afford 229 mg of (±)-1f (100%). This threo-iodomethylphenidate was determined to be ≥95% diastereomerically pure by 1H NMR upon comparison to the known data for enantiomerically pure threo-and erythro-methylphenidate and its para-substituted derivatives.37 1H NMR (CD3OD, 400 MHz): δ 7.74 (d, 1H, J = 9.6 Hz), 7.70 (t, 1H, J = 1.7 Hz), 7.31 (d, 1H, J = 8.4 Hz), 7.18 (t, 1H, J = 7.8 Hz), 3.89-3.81 (m, 2H), 3.74 (s, 3H), 3.43 (d, 1H, J = 12.8 Hz), 3.10 (t, 1H, J = 12.9 Hz), 1.91-1.79 (m, 2H), 1.74-1.63 (m, 1H), 1.57-1.46 (m, 2H), 1.41-1.32 (m, 1H). 13C NMR (CD3OD, 100 MHz): δ 172.8, 139.0, 138.6, 137.4, 132.2, 128.9, 95.6, 58.9, 54.7, 53.6, 46.7, 27.8, 23.3, 22.7. HRMS calcd for C14H18INO2Na+ 38 2.0274, found 382.0271. MP: 191–192°C.
4.9. (±)-threo-N-(p-Azido-benzyl)-4-iodomethylphenidate ((±)-3a)
(±)-threo-4-iodomethylphenidate hydrochloride ((±)-1b) (0.11 g, 0.27 mmol) was added to a suspension of K2CO3 (0.15 g, 1.08 mmol) in DMF (6 mL). The resulting suspension was stirred at room temperature for 10 minutes, then p-N3-N-BnBr38 (60 mg, 0.29 mmol) was added and the mixture was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added and the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 5:95) to give 90 mg of (±)-3a as a yellow solid (67%). Rf = 0.26 (EtOAc:hexanes, 5:95). 1H NMR (CDCl3, 400 MHz): δ 7.64 (d, 2H, J = 8.3 Hz), 7.27 (d, 2H, J = 8.3 Hz), 7.14 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.4 Hz), 4.11 (d, 1H, J = 11.5 Hz), 3.89 (d, 1H, J = 13.6 Hz), 3.74 (d, 1H, J = 13.6 Hz), 3.65 (s, 3H), 3.45-3.41 (m, 1H), 2.97-2.91 (m, 1H), 2.55-2.50 (m, 1H), 1.58-1.47 (m, 4H), 1.33-1.25 (m, 1H), 1.06-1.02 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.5, 138.4, 137.7, 137.1, 136.6, 130.7, 129.9, 118.7, 93.1, 62.3, 55.9, 52.6, 51.9, 44.8, 21.1, 20.7, 19.5. HRMS calcd for C21H23IN4O2H+ 491.0938, found 491.0930. Anal. Calcd for: C21H23IN4O2: C, 51.44; H 4.73; N, 11.43; I, 25.88. Found: C, 51.72; H 4.67; N, 11.48; I, 25.62. MP: 108–110°C. IR: azide, 2109 cm−1.
4.10. (±)-threo-N-(m-Azido-benzyl)-4-iodomethylphenidate ((±)-3b)
(±)-threo-4-iodomethylphenidate hydrochloride ((±)-1b) (0.15 g, 0.38 mmol) was added to a suspension of K2CO3 (0.13 g, 0.9 mmol) in DMF (5 mL). The mixture was stirred at room temperature for 10 minutes, then m-NO2-N-BnBr (82 mg, 0.38 mmol) was added. The reaction was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added then the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3:hexanes, 1:1) to give 0.13 g of (±)-threo-N-(m-nitro-benzyl)-4-iodomethylphenidate as a yellow oil (69%). Rf = 0.21 (CHCl3:hexanes, 1:1). 1H NMR (CDCl3, 400 MHz): δ 8.18 (s, 1H), 8.10 (d, 1H, J = 8.1 Hz), 7.65-7.60 (m, 3H), 7.47 (t, 1H, J = 7.9 Hz), 7.16 (d, 2H, J = 8.4 Hz), 4.13 (d, 1H, J = 11.6 Hz), 4.05 (d, 1H, J = 14.3 Hz), 3.86 (d, 1H, J = 14.3 Hz), 3.72 (s, 3H), 3.48-3.44 (m, 1H), 3.03-2.96 (m, 1H), 2.54-2.51(m, 1H), 1.60-1.50 (m, 4H), 1.37-1.34 (m, 1H), 1.10-1.06 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.6, 148.4, 142.7, 137.8, 136.4, 134.5, 130.7, 128.9, 123.3, 122.0, 93.2, 62.7, 56.0, 52.4, 52.1, 44.9, 21.2, 20.5, 19.5. HRMS calcd for C21H23IN2O4Na+ 517.0594, found 517.0594. (±)-threo-N-(m-nitro-benzyl)-4-iodomethylphenidate (0.1 g, 0.2 mmol) was added to MeOH (5 mL) at 0°C followed by concentrated HCl (1 mL) and SnCl2 (0.15 g, 0.78 mmol). The mixture was allowed to stir at room temperature overnight then quenched with H2O (5 mL). The pH was then brought to 11 with 1M aq. NaOH and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3: MeOH, 9:1) to give 90 mg of (±)-threo-N-(m-amino-benzyl)-4-iodomethylphenidate as a colorless oil (81%). Rf = 0.29 (CHCl3:MeOH, 9:1). 1H NMR (CDCl3, 400 MHz): δ 7.63 (d, 2H, J = 8.4 Hz), 7.15 (d, 2H, J = 8.4 Hz), 7.09 (t, 1H, J = 7.7 Hz), 6.68-6.65 (m, 2H), 6.56 (d, 1H, J = 7.8 Hz), 4.10 (d, 1H, J = 11.4 Hz), 3.82 (d, 1H, J = 13.5 Hz), 3.69 (d, 1H, J = 13.4 Hz), 3.66 (s, 3H), 3.48-3.40 (m, 1H), 2.97-2.91 (m, 1H), 2.57-2.52 (m, 1H), 1.54-1.42 (m, 4H), 1.32-1.29 (m, 1H), 1.05-1.02 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.6, 146.3, 141.4, 137.7, 136.8, 130.8, 128.9, 118.9, 115.4, 113.7, 93.0, 62.5, 56.4, 52.7, 51.9, 45.1, 21.3, 20.9, 19.6. HRMS calcd for C21H25IN2O2Na+ 487.0852, found 487.0859. A solution of (±)-threo-N-(m-amino-benzyl)-4-iodomethylphenidate (62 mg, 0.13 mmol) in concentrated HCl (0.5 mL) and H2O (5 mL) at 0°C was treated with NaNO2 (10 mg, 0.15 mmol). The mixture was stirred in the dark for 10 min at 0°C then carefully treated with NaN3 (18 mg, 0.27 mmol). The reaction was allowed to stir in the dark for 2 h at 0°C then diluted with H2O and CHCl3. The organic layer was separated, washed with brine, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3) to give 65 mg of (±)-3b as a yellow oil (85%). Rf = 0.30 (CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.63 (d, 2H, J = 8.4 Hz), 7.29-7.25 (m, 1H), 7.14 (d, 2H, J = 8.4 Hz), 7.05-7.01 (m, 2H), 6.89 (d, 1H, J = 7.9 Hz), 4.11 (d, 1H, J = 11.5 Hz), 3.91 (d, 1H, J = 13.8 Hz), 3.76 (d, 1H, J = 13.8 Hz), 3.68 (s, 3H), 3.45-3.41 (m, 1H), 2.99-2.93 (m, 1H), 2.55-2.50 (m, 1H), 1.64-1.48 (m, 4H), 1.33-1.25 (m, 1H), 1.06-1.03 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.3, 142.4, 139.9, 137.7, 136.6, 130.7, 129.3, 125.1, 119.0, 117.6, 93.1, 62.5, 56.3, 52.5, 52.0, 44.9, 21.1, 20.6, 19.5. HRMS calcd for C21H23IN4O2H+ 491.0938, found 491.0940. IR: azide, 2109 cm−1.
4.11. (±)-threo-N-(o-Azido-benzyl)-4-iodomethylphenidate ((±)-3c)
(±)-threo-4-iodomethylphenidate hydrochloride ((±)-1b) (80 mg, 0.2 mmol) was added to a suspension of K2CO3 (0.11 g, 0.81 mmol) in DMF (5 mL). The mixture was stirred at room temperature for 10 minutes then o-N3-N-BnBr38 (42 mg, 0.2 mmol) was added. The reaction was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added and the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (hexanes) to give 48 mg of (±)-3c as a colorless oil (49%). Rf = 0.28 (hexanes). 1H NMR (CDCl3, 400 MHz): δ 7.63 (d, 2H, 8.4 Hz), 7.38 (d, 1H, J = 7.6 Hz), 7.29-7.25 (m, 1H), 7.14-7.09 (m, 4H), 4.09 (d, 1H, J = 11.5 Hz), 3.92 (d, 1H, J = 14.4 Hz), 3.68 (d, 1H, J = 14.4 Hz), 3.59 (s, 3H), 3.43-3.38 (m, 1H), 2.99-2.93 (m, 1H), 2.58-2.52 (m, 1H), 1.62-1.46 (m, 4H), 1.38-1.33 (m, 1H), 1.06-1.03 (m, 1H). 13C NMR (CDCl3, 100 MHz): 173.4, 138.0, 137.7, 136.6, 131.5, 130.7, 130.3, 127.9, 124.6, 117.8, 173.4, 138.0, 137.7, 136.6, 131.5, 130.7, 130.3, 127.9, 124.6, 117.8, 93.0, 62.4, 52.6, 51.9, 50.7, 45.5, 21.3, 20.7, 19.9. HRMS calcd for C21H23IN4O2H+ 491.0938, found 491.0930. IR: azide, 2116 cm−1.
4.12. (±)-threo-N-(p-Azido-benzyl)-3-iodomethylphenidate ((±)-3d)
(±)-threo-3-iodomethylphenidate hydrochloride ((±)-1f) (0.14 g, 0.35 mmol) was added to a suspension of K2CO3 (0.19 g, 1.43 mmol) in DMF (7 mL). The mixture was stirred at room temperature for 10 minutes then p-N3-N-BnBr 38 (80 mg, 0.39 mmol) was added. The reaction was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added and the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 5:95) to give 0.11 g of (±)-3d as a yellow gum (63%). Rf = 0.23 (EtOAc:hexanes, 5:95). 1H NMR (CDCl3, 400 MHz): δ 7.74 (s, 1H), 7.60 (d, 1H, J = 7.9 Hz), 7.36 (d, 1H, J = 7.8 Hz), 7.26 (d, 2H, J = 8.6 Hz), 7.05 (t, 1H, J = 7.8 Hz), 6.97 (d, 2H, J = 8.4 Hz), 4.08 (d, 1H, J = 11.5 Hz), 3.89 (d, 1H, J = 13.6 Hz), 3.74 (d, 1H, J = 13.6 Hz), 3.66 (s, 3H), 3.46-3.42 (m, 1H), 2.97-2.90 (m, 1H), 2.53-2.49 (m, 1H), 1.58-1.48 (m, 5H), 1.07-1.03 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.4, 139.2, 138.4, 137.0, 136.6, 130.3, 129.9, 128.0, 118.7, 94.5, 62.5, 55.9, 52.5, 51.9, 44.6, 21.1, 20.6, 19.4. HRMS calcd for C21H23IN4O2H+ 491.0938, found 491.0945. IR: azide, 2111 cm−1.
4.13. (±)-threo-N-(m-Azido-benzyl)-3-iodomethylphenidate ((±)-3e)
(±)-threo-3-iodomethylphenidate hydrochloride ((±)-1f) (0.3 g, 0.76 mmol) was added to a suspension of K2CO3 (0.25 g, 1.8 mmol) in DMF (5 mL). The mixture was stirred at room temperature for 10 minutes then m-NO2-N-BnBr (0.16 g, 0.76 mmol) was added. The reaction was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added then the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3:hexanes, 1:1) to give 0.26 g of (±)-threo-N-(m-nitro-benzyl)-3-iodomethylphenidate as a yellow oil (70%). Rf = 0.31 (CHCl3:hexanes, 1:1). 1H NMR (CDCl3, 400 MHz): δ 8.18 (s, 1H), 8.10 (d, 1H, J = 8.1 Hz), 7.76 (s, 1H), 7.60 (d, 2H, J = 7.9 Hz), 7.47 (t, 1H, J = 7.9 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.06 (t, 1H, J = 7.8 Hz), 4.10 (d, 1H, J = 11.6 Hz), 4.05 (d, 1H, J = 14.3 Hz), 3.86 (d, 1H, J = 14.3 Hz), 3.72 (s, 3H), 3.48-3.45 (m, 1H), 3.01-2.96 (m, 1H), 2.54-2.50 (m, 1H), 1.61-1.52 (m, 4H), 1.38-1.34 (m, 1H), 1.10-1.07 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.4, 148.3, 142.6, 138.9, 137.4, 136.7, 134.4, 130.4, 128.9, 127.9, 123.2, 122.0, 94.6, 62.8, 56.0, 52.4, 52.1, 44.7, 21.2, 20.5, 19.4. HRMS calcd for C21H23IN2O4H+ 495.0775, found 495.0780. (±)-threo-N-(m-nitro-benzyl)-3-iodomethylphenidate (0.26 g, 0.53 mmol) was added to MeOH (5mL) at 0°C then treated with concentrated HCl (2 mL) and SnCl2 (0.39 g, 2.05 mmol). The mixture was allowed to stir at room temperature overnight then quenched with H2O (5 mL). The pH was brought to 11 with 1M aq. NaOH and extracted with EtOAc. The organic layers were washed with brine, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3:MeOH, 95:5) to give 0.2 g of (±)-threo-N-(m-amino-benzyl)-3-iodomethylphenidate as a colorless oil (81%). Rf = 0.27 (CHCl3:MeOH, 95:5). 1H NMR (CDCl3, 400 MHz): 7.75 (s, 1H), 7.59 (d, 1H, 7.75 (s, 1H), 7.59 (d, 1H, J = 8.5 Hz), 7.37 (d, 1H, J = 7.8 Hz), 7.09-7.02 (m, 2H), 6.68-6.65 (m, 2H), 6.56 (d, 1H, J = 7.9 Hz), 4.08 (d, 1H, J = 11.4 Hz), 3.82 (d, 1H, J = 13.6 Hz), 3.69 (d, 1H, J = 13.7 Hz), 3.66 (s, 3H), 3.48-3.42 (m, 1H), 2.97-2.90 (m, 1H), 2.57-2.52 (m, 1H), 1.56-1.46 (m, 4H), 1.33-1.28 (m, 1H), 1.06-1.02 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.4, 146.3, 141.3, 139.2, 137.5, 136.4, 130.2, 128.8, 127.9, 118.7, 115.2, 113.5. 94.5, 62.5, 56.3, 52.6, 51.0, 44.8, 21.2, 20.8, 19.5. HRMS calcd for C21H25IN2O2H+ 465.1033, found 465.1026. A solution of (±)-threo-N-(m-amino-benzyl)-3-iodomethylphenidate (0.18 g, 0.39 mmol) in 2N HCl (6 mL) at 0°C was treated with NaNO2 (30 mg, 0.43 mmol). The mixture was stirred in the dark for 10 min at 0°C, carefully treated with NaN3 (50 mg, 0.78 mmol), stirred in the dark at 0°C for 2 h, then diluted with H2O and CHCl3. The organic layer was separated, washed with brine, dried (MgSO4), filtered, concentrated, and chromatographed (CHCl3) to give 0.16 g of (±)-3e as a yellow oil (95%). Rf = 0.35 (CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.75 (s, 1H), 7.59 (d, 1H, J = 8.4 Hz), 7.37 (d, 1H, J = 7.9 Hz), 7.29-7.25 (m, 1H), 7.06-7.01 (m, 3H), 6.89 (d, 1H, J = 7.9 Hz), 4.08 (d, 1H, J = 11.5 Hz), 3.93 (d, 1H, J = 13.8 Hz), 3.76 (d, 1H, J = 13.8 Hz), 3.68 (s, 3H), 3.46-3.41 (m, 1H), 2.98-2.92 (m, 1H), 2.55-2.50 (m, 1H), 1.64-1.48 (m, 4H), 1.35-1.31 (m, 1H), 1.07-1.03 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.3, 142.4, 139.9, 139.1, 137.5, 136.6, 130.3, 129.3, 127.9, 125.0, 118.9, 117.5, 94.5, 62.5, 56.2, 52.5, 52.0, 44.7, 21.1, 20.6, 19.4. HRMS calcd for C21H23IN4O2Na+ 513.0757, found 513.0763. IR: azide, 2113 cm−1.
4.14. (±)-threo-N-(o-Azido-benzyl)-3-iodomethylphenidate ((±)-3f)
(±)-threo-3-iodomethylphenidate hydrochloride ((±)-1f) (0.1 g, 0.27 mmol) was added to a suspension of K2CO3 (0.15 g, 1.08 mmol) in DMF (5 mL). The mixture was stirred at room temperature for 10 minutes then o-N3-N-BnBr38 (58 mg, 0.27 mmol) was added. The reaction was allowed to stir at room temperature in the dark for 30 h. Et2O (20 mL) was added then the mixture was decanted followed by rinsing with Et2O (2 × 20 mL). The combined organic layers were washed with H2O, dried (MgSO4), filtered, concentrated, and chromatographed (CH2Cl2:hexanes, 1:1) to give 67 mg of (±)-3f as a yellow oil (51%). Rf = 0.23 (CH2Cl2:hexanes, 7:3). 1H NMR (CDCl3, 400 MHz): δ 7.74 (t, 1H, J = 1.7 Hz), 7.59 (d, 1H, J = 7.9 Hz), 7.39 (t, 2H, J = 8.3 Hz), 7.32-7.27 (m, 1H), 7.14-7.09 (m, 2H), 7.03 (t, 1H, J = 7.8 Hz), 4.07 (d, 1H, J = 11.5 Hz), 3.93 (d, 1H, J = 14.5 Hz), 3.67 (d, 1H, J = 14.5 Hz), 3.60 (s, 3H), 3.44-3.40 (m, 1H), 2.99-2.92 (m, 1H), 2.57-2.51 (m, 1H), 1.58-1.52 (m, 4H), 1.39-1.33 (m, 1H), 1.07-1.03 (m, 1H). 13C NMR (CDCl3, 100 MHz): δ 173.3, 139.3, 137.9, 137.6, 136.5, 131.5, 130.3, 128.0, 127.9, 124.6, 117.8, 94.5, 62.5, 52.6, 51.9, 50.7, 45.4, 21.3, 20.7, 19.8. HRMS calcd for C21H23IN4O2H+ 491.0938, found 491.0932. IR: azide, 2115 cm−1.
4.15. (±)-threo-N-(p-Azido-benzyl)-4-(tri-n-butylstannyl) methylphenidate ((±)-7)
A mixture of (±)-threo-N-(p-azido-benzyl)-4-iodomethylphenidate ((±)-3a) (105 mg, 0.21 mmol), Pd(PPh3)2Br2 (10 mg, 0.02 mmol), and bis(tri-n-butyltin) (0.15 mL, 0.39 mmol) in toluene (10 mL) was heated at 105°C for 6 hours. The mixture was then cooled to room temperature, diluted with sat. aq. K2CO3 solution, and extracted with EtOAc. The organic phase was washed with brine, dried (MgSO4), filtered, concentrated, and chromatographed (EtOAc:hexanes, 1:9) to give 49 mg of (±)-7 as a colorless oil (41%). Rf = 0.35 (EtOAc:hexanes, 1:9). 1H NMR (CDCl3, 400 MHz): δ 7.39 (d, 2H, J = 7.9 Hz), 7.32 (d, 2H, J = 7.9 Hz), 7.28 (d, 2H, J = 8.4 Hz), 6.97 (d, 2H, J = 8.4 Hz), 4.11 (d, 1H, J = 11.5 Hz), 3.90 (d, 1H, J = 13.6 Hz), 3.76 (d, 1H, J = 13.6 Hz), 3.65 (s, 3H), 3.49-3.45 (m, 1H), 2.99-2.94 (m, 1H), 2.54-2.51 (m, 1H), 1.56-1.49 (m, 9H), 1.34-1.29 (m, 9H), 1.03 (t, 6H, J = 8.0 Hz), 0.88 (t, 9H, J = 7.3 Hz). 13C NMR (CDCl3, 100 MHz): δ 174.1, 141.0, 138.3, 137.3, 136.7, 136.4, 129.9, 128.2, 118.7, 62.5, 55.8, 53.1, 51.8, 44.8, 29.0, 27.4, 21.1, 20.8, 19.5, 13.7, 9.5. HRMS calcd for C33H50N4O2SnH+ 655.3029, found 655.3033. IR: azide, 2110 cm−1.
4.16. [125I]-(±)-threo-N-(p-Azido-benzyl)-4-iodomethylphenidate ([125I]-(±)-3a)
[125I]-NaI (15 μL, 1.53 mCi) was treated with a solution of tri-n-butylstannyl precursor (±)-7 (25 μL, 6.0 mM) in MeOH followed by aqueous N-chloro-4-toluenesulfonamide (Chloramine-T) trihydrate (15 μL, 5.0 mM) and a solution of MeOH (85 μL) containing 3% glacial HOAc. After 2 min at ambient temperature, the mixture was quenched with Na2S2O5 (10 μL, 50 mM) then taken up in a syringe along with a rinse (200 μL) of the vessel with the ternary HPLC mobile phase consisting of MeOH (35%), CH3CN (35%), and an aqueous solution (30%) containing Et3N (2.1% v/v) and HOAc (2.8% v/v). A Waters C-18 Nova-Pak column (radial compression module, 8 × 100 mm, 6 μm) was employed for separation at a flow rate of 3 mL/min. The HPLC system was equipped with a flow- through radioactivity detector and a UV absorbance detector (254 nm). Radioactive material (tR = 18.4 min) corresponding to [125I]-(±)-3a was well resolved from non-radioactive and radioactive side products. [125I]-(±)-3a was collected in 5 mL of HPLC mobile phase, diluted to 40 mL with distilled water, and then passed through an activated (MeOH/water) solid phase extraction cartridge (Waters Sep-Pak Light t-C-18) that was flushed with water (2.0 mL) and then with air. The cartridge retained essentially all radioactivity. Elution with MeOH (1.5 mL) gave 0.93 mCi (61%) of [125I]-(±)-3a. This material co-eluted with a standard sample of (±)-3a under the HPLC conditions described above and displayed 98% radiochemical purity. After 20 weeks, > 2 half-lives, of storage at −20 °C in the dark, about 80% radiochemical purity was observed by HPLC. A specific radioactivity of 2099 mCi/μmol was calculated for [125I]-(±)-3a using HPLC to determine the mass associated with the absorbance peak for carrier in a purified sample of known radioactivity (0.21 mCi). The UV response for non-radioactive (±)-3a was linear (r2 = 1.0) over a six-point standard curve (30–750 pmol). The major non-radioactive product observed during HPLC purification was tentatively assigned as the chloro analog of (±)-3a (tR = 13.3 min) based upon HPLC analyses of model reactions using an excess of Chloramine-T but no radioiodine.
4.17. Pharmacology
[3H]-WIN-35,428 binding inhibition assays were performed with N2A neuroblastoma cells stably transfected with human wildtype DAT cDNA. Cell monolayers were grown to confluence at 37°C, 5% CO2 in 24 well plates with Opti-MEM media (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 U/mL streptomycin (from Fisher Scientific), and 450 μg/mL g/mL G-418 (Clontech). Confluent monolayers were first washed 2 × 1 mL with 22°C “KRH buffer” (25 mM HEPES, pH 7.3, 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, and 5.6 mM glucose) supplemented with 50 mM ascorbic acid (KRH/AA). The washed and aspirated monolayers were incubated with 500 μl [3H]-WIN-35,428 (1 nM) and nonradioactive competitor (0.1 nM – 10 μM) for 15 minutes at 22°C, followed by removal by aspiration and 2 × 1 mL KRH/AA washes. Non-specific binding was determined by using 10 μM mazindol as the competitor. Monolayers were solubilized by incubation with 1 mL 1% SDS at 22°C for 1 hour with gentle shaking. Cell lysates were transferred into vials containing 5 mL ScintiSafe fluid (Fisher), mixed thoroughly by inversion and vortexing, and analyzed via scintillation counting for determination of remaining tritium radioactivity.
[3H]-Dopamine uptake inhibition assays were conducted identical to the [3H]-WIN-35,428 binding inhibition assays described above with the exceptions that 10 nM [3H]-dopamine replaced the WIN radioligand and the nonradioactive inhibitor drug was added 10 min before the 5 min [3H]-dopamine uptake interval commenced.
Ki values for nonlinear regression of [3H]-WIN-35,428 displacement curves were determined with GraphPad Prism 5.0 (GraphPad, La Jolla, CA). The algorithm converts Ki values from IC50 values using the Cheng-Prusoff equation: Ki = IC50/1 + [Ligand]/Kd.46 The regression best fit the data points using one site competitive binding curves. Irreversible binding was not otherwise addressed in these assays (it is unknown whether the time course employed is adequate for covalent linkage, nor was UV irradiation employed).
4.18. DAT Photoaffinity Labeling
Assessment of irreversible labeling of the DAT by [125I]-(±)-3a was performed using previously published procedures. 13, 14, 17, 20, 28, 30, 40–43 Rat striatal membranes were prepared as previously described and suspended at 20 mg/mL original wet weight in sucrose phosphate buffer (SP, 0.32 M sucrose, 10 mM Na2HPO4, pH 7.4). HEK-293 cells expressing 6Xhis hDAT were grown to 90% confluency in 6-well plates and washed with KRH buffer. For both preparations, [125I]-(±)-3a was added to a final concentration of 30 nM followed by incubation for 1 h at 4°C. For pharmacological displacement studies, 10 μM or 100 μM β-CFT or D-(+)-methylphenidate was included in the binding mixture. Membranes or cells were irradiated with shortwave ultraviolet light (254 nm, Fotodyne UV Lamp model 3-6000) for 45 s at a distance of 15–20 mm to photoactivate the radioligand. Membranes or cells were washed twice with 1 mL of ice-cold SP or KRH buffer and lysed with RIPA buffer (50 mM NaF, 2 mM EDTA, 125 mM Na3PO4, 1.25% Triton X-100, and 1.25% sodium deoxycholate) for 15 min at 0 °C. Lysates were centrifuged at 20,000 × g for 15 min at 4°C to remove insoluble material and the supernatants were transferred to clean tubes for immunoprecipitation. Lysates were subjected to immunoprecipitation as described previously13, 14, 17, 20, 28, 30, 40–43 using antiserum 16 generated against amino acids 42–59 of rDAT or anti-his monoclonal antibody (Sigma-Aldrich) for his-tagged hDAT.
Immunoprecipitated samples were separated on 4–20% SDS-polyacrylamide gels followed by autoradiography using Hyperfilm MP film (GE Healthcare) for 1–4 days at −80°C. Band densities were quantified using LumiAnalyst software (Roche) and expressed as a fraction of control samples that received no inhibitor.
Acknowledgments
This work was funded by a Hunkele Dreaded Disease Award (D.J.L.), the Mylan School of Pharmacy at Duquesne University (D.J.L.), and NIH grants DA27081 (D.J.L), DA16604 (C.K.S.), and DA15175 (R.A.V.). We thank NIDA Drug Supply for contribution of certain nonradioactive DAT ligand compounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nair R, Moss SB. Neuropsychiatric Disease and Treatment. 2009;5:421. doi: 10.2147/ndt.s4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For recent examples see: Kim DI, Deutsch HM, Ye X, Schweri MM. J Med Chem. 2007;50:2718. doi: 10.1021/jm061354p.Froimowitz M, Gu Y, Dakin LA, Nagafuji PM, Kelley CJ, Parrish D, Deschamps JR, Janowsky A. J Med Chem. 2007;50:219. doi: 10.1021/jm0608614.
- 3.Solanto MV. Behav Brain Res. 1998;94:127. doi: 10.1016/s0166-4328(97)00175-7. [DOI] [PubMed] [Google Scholar]
- 4.Fleckenstein AE, Volz TJ, Riddle EL, Gibb JW, Hanson GR. Annu Rev Pharmacol Toxicol. 2007;47:681. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 5.Woolverton WL, Johnson KM. Trends Pharmacol Sci. 1992;13:193. doi: 10.1016/0165-6147(92)90063-c. [DOI] [PubMed] [Google Scholar]
- 6.For significant work aimed at determining the DAT binding sites for cocaine and benztropines, plus the relationship between DAT conformational changes and cocaine-like subjective effects of uptake inhibitors see: Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. Nature Neuroscience. 2008;11:780. doi: 10.1038/nn.2146.Bisgaard H, Larsen MAB, Mazier S, Beuming T, Newman AH, Weinstein H, Shi L, Loland CJ, Gether U. Neuropharmacology. 2010 doi: 10.1016/j.neuropharm.2010.08.021.Loland CJ, Desai RI, Zou MF, Cao J, Grundt P, Gerstbrein K, Sitte HH, Newman AH, Katz JL, Gether U. Mol Pharmacol. 2008;73:813. doi: 10.1124/mol.107.039800.
- 7.a.) Runyon SP, Carroll FI. Curr Top Med Chem. 2006;6:1825. doi: 10.2174/156802606778249775. [DOI] [PubMed] [Google Scholar]; b.) Trudell ML, Izenwasser S, editors. In Dopamine Transporters: Chemistry, Biology, and Pharmacology. Wiley; Hoboken, NJ: 2008. pp. 123–304. [Google Scholar]
- 8.Reith MEA, Berfield JL, Wang L, Ferrer JV, Javitch JA. J Biol Chem. 2001;276:29012. doi: 10.1074/jbc.M011785200. [DOI] [PubMed] [Google Scholar]
- 9.Newman AH, Kulkarni SS. Med Res Rev. 2002;22:1. doi: 10.1002/med.10014. [DOI] [PubMed] [Google Scholar]
- 10.Ukairo OT, Bondi CD, Newman AH, Kulkarni SS, Kozikowski AP, Pan S, Surratt CK. J Pharmacol Exp Ther. 2005;314:575. doi: 10.1124/jpet.105.085829. [DOI] [PubMed] [Google Scholar]
- 11.For naphthyl-containing phenylisothiocyanate tropane analogs see: Murthy V, Davies HML, Hedley SJ, Childers SR. Biochem Pharmacol. 2007;74:336. doi: 10.1016/j.bcp.2007.04.019.Murthy V, Martin TJ, Kim S, Davies HML, Childers SR. J Pharmacol Exp Ther. 2008;326:587. doi: 10.1124/jpet.108.138842.
- 12.For 3-iodo-4-azidococaine and characterization of its binding site within the sigma-1 receptor see: Chen Y, Hajipour AR, Sievert MK, Arbabian M, Ruoho AE. Biochemistry. 2007;46:3532. doi: 10.1021/bi061727o.
- 13.For [125I]-3-β-(4′-azido-3′-iodo-biphenyl-4-yl)-8-methyl-8-aza-bicyclo-[3.2.1]octane-2β-carboxylic acid methyl ester (JHC-2-48) see: Newman AH, Cha JH, Cao J, Kopajtic T, Katz JL, Parnas ML, Vaughan R, Lever JR. J Med Chem. 2006;49:6621. doi: 10.1021/jm0603973.
- 14.For (-)-N-[4-(3′-[125I]iodo-4′-azidophenyl)butyl]-2β-carbomethoxy-3β-(4-chlorophenyl)tropane (MFZ-2-24) and (-)-N-[4-(3′-[125I]iodo-4′-38 isothiocyanophenyl)butyl]-2β-carbomethoxy-3β-(4-chlorophenyl)tropane (MFZ-3-37) see: Lever JR, Zou MF, Parnas ML, Duval RA, Wirtz SE, Justice JB, Vaughan RA, Newman AH. Bioconjugate Chem. 2005;16:644. doi: 10.1021/bc0497214.
- 15.For azido and isothiocyanate analogs of (S)-2β-substituted-3α-(bis[4-fluorophenyl]methoxy)tropanes and (R)-2β-substituted-3β-(3,4-dichlorophenyl)tropanes see: Zou MF, Kopajtic T, Katz JL, Newman AH. J Med Chem. 2003;46:2908. doi: 10.1021/jm0300375.
- 16.For azido and isothiocyanate analogs of 3α-[bis(4′-fluorophenyl)methoxy]tropane see: Zou MF, Kopajtic T, Katz JL, Wirtz S, Justice JB, Newman AH. J Med Chem. 2001;44:4453. doi: 10.1021/jm0101904.
- 17.For an azido N-substituted 3α-[bis(4′-fluorophenyl)methoxy]tropane see: Agoston GE, Vaughan R, Lever JR, Izenwasser S, Terry PD, Newman AH. Bioorg Med Chem Lett. 1997;7:3027.
- 18.For azido and isothiocyanate substituted 3-carbamoylecgonine methyl ester analogues see: Kline RH, Eshleman AJ, Wright J, Eldefrawi ME. J Med Chem. 1994;37:2249. doi: 10.1021/jm00040a019.
- 19.For irreversible ligands derived from (-)-cocaine or 3β-phenyltropan-2β-carboxylic acid methyl ester (WIN-35,065-2) see: Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW, Kuhar MJ. J Med Chem. 1992;35:1813. doi: 10.1021/jm00088a017.
- 20.For a 4-[2-(diphenylmethoxy)ethyl]-1-benzyl piperidine based on GBR-12909 see: Dutta AK, Fei XS, Vaughan RA, Gaffaney JD, Wang NN, Lever JR, Reith MEA. Life Sci. 2001;68:1839. doi: 10.1016/s0024-3205(01)00981-x.
- 21.For azido and isothiocyanato analogues of [3-(4-phenylalkylpiperazin-1-yl)propyl]bis(4-fluorophenyl)amines see: Cao J, Lever JR, Kopajtic T, Katz JL, Pham AT, Holmes ML, Justice JB, Newman AH. J Med Chem. 2004;47:6128. doi: 10.1021/jm049670w.
- 22.For isothiocyanate analogs of 9-[3-(cis-3,5-dimethyl-1-piperazinyl)propyl]-carbazole (Rimcazole) see: Husbands SM, Izenwasser S, Loeloff RJ, Katz JL, Bowen WD, Vilner BJ, Newman AH. J Med Chem. 1997;40:4340. doi: 10.1021/jm9705519.
- 23.For an azido-diphenylpiperazine analog, 3-azido[3H]GBR-12935, see: Berger SP, Martenson RE, Laing P, Thurkauf A, Decosta B, Rice KC, Paul SM. Mol Pharmacol. 1991;39:429.
- 24.For 1-(2-[bis-(4-fluorophenyl)-methoxyethyl)-4-(2-[4-azido-3-[125I]iodophenyl]ethyl)piperazine (FAPP) see: Sallee FR, Fogel EL, Schwartz E, Choi SM, Curran DP, Niznik HB. FEBS Lett. 1989;256:219. doi: 10.1016/0014-5793(89)81752-1.
- 25.For 125I-1-[2-(diphenylmethoxy)ethyl]-4-[2-(4-azido-3-iodophenyl)ethyl]piperazine (DEEP) see: Grigoriadis DE, Wilson AA, Lew R, Sharkey JS, Kuhar MJ. J Neurosci. 1989;9:2664. doi: 10.1523/JNEUROSCI.09-08-02664.1989.
- 26.For irreversible affinity labels based on aryl 1,4-dialkylpiperazines related to GBR-12783 see: Deutsch HM, Schweri MM, Culbertson CT, Zalkow LH. Eur J Pharmacol. 1992;220:173. doi: 10.1016/0014-2999(92)90745-p.
- 27.For Fourphit, an isothiocyanate phencyclidine derivative that inhibits the binding of [3H]-methylphenidate to rDAT see: Schweri MM, Thurkauf MV, Mattson MV, Rice KC. J Pharmacol Exp Ther. 1992;261:936.
- 28.For a photoaffinity ligand based on pyrovalerone, a structural variant of bupropion (Wellbutrin, Zyban), see: Lapinsky DJ, Aggarwal S, Huang Y, Surratt CK, Lever JR, Foster JD, Vaughan RA. Bioorg Med Chem. 2009;17:3770. doi: 10.1016/j.bmc.2009.04.057.
- 29.Indarte M, Madura JD, Surratt CK. Proteins. 2008;70:1033. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 30.Vaughan RA, Parnas ML, Gaffaney JD, Lowe MJ, Wirtz S, Pham A, Reed B, Dutta SM, Murray KK, Justice JB. J Neurosci Methods. 2005;143:33. doi: 10.1016/j.jneumeth.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 31.Deutsch HM, Shi Q, Gruszecka-Kowalik E, Schweri MM. J Med Chem. 1996;39:1201. doi: 10.1021/jm950697c. [DOI] [PubMed] [Google Scholar]
- 32.Misra M, Shi Q, Ye X, Gruszecka-Kowalik E, Bo W, Liu Z, Schweri MM, Deutsch HM, Venanzi CA. Bioorg Med Chem. 2010;18:7221. doi: 10.1016/j.bmc.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 33.Pan D, Gatley SJ, Chen R, Ding Y-S. J Labelled Compounds and Radiopharm. 1996;38:523. [Google Scholar]
- 34.Axten JM, Krim L, Kung HF, Winkler JD. J Org Chem. 1998;63:9628. [Google Scholar]
- 35.Gutman A, Zaltsman I, Shalimov A, Sotrihin M, Nisnevich G, Yudovich L, Fedotev I. US2004180928. Patent. (A1)
- 36.Kiesewetter DO. Tetrahedron: Asymmetry. 1993;4:2183. [Google Scholar]
- 37.Thai DL, Sapko MT, Reiter CT, Bierer DE, Perel JM. J Med Chem. 1998;41:591. doi: 10.1021/jm970620j. [DOI] [PubMed] [Google Scholar]
- 38.Mornet R, Leonard NJ, Theiler JB, Doree M. J Chem Soc Perkin Trans I. 1984;5:879. [Google Scholar]
- 39.Dorman G, Prestwich GD. Trends Biotechnol. 2000;18:64. doi: 10.1016/s0167-7799(99)01402-x. [DOI] [PubMed] [Google Scholar]
- 40.Parnas ML, Gaffaney JD, Zou MF, Lever JR, Newman AH, Vaughan RA. Mol Pharmacol. 2008;73:1141. doi: 10.1124/mol.107.043679. [DOI] [PubMed] [Google Scholar]
- 41.Vaughan RA, Sakrikar DS, Parnas ML, Adkins S, Foster JD, Duval RA, Lever JR, Kulkarni SS, Newman AH. J Biol Chem. 2007;282:8915. doi: 10.1074/jbc.M610633200. [DOI] [PubMed] [Google Scholar]
- 42.Vaughan RA, Agoston GE, Lever JR, Newman AH. J Neurosci. 1999;19:630. doi: 10.1523/JNEUROSCI.19-02-00630.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vaughan RA, Kuhar MJ. J Biol Chem. 1996;271:21672. doi: 10.1074/jbc.271.35.21672. [DOI] [PubMed] [Google Scholar]
- 44.Shafi’ee A, Hite G. J Med Chem. 1969;12:266. doi: 10.1021/jm00302a015. [DOI] [PubMed] [Google Scholar]
- 45.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
- 46.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]