

Abstract
Covalent modifications of intracellular proteins, such as phosphorylation, are generally thought to occur as secondary or tertiary responses to neurotransmitters, following the intermediation of membrane receptors and second messengers such as cyclic AMP. By contrast, the gasotransmitter nitric oxide directly S-nitrosylates cysteine residues in diverse intracellular proteins. Recently, hydrogen sulfide has been acknowledged as a gaso-transmitter, which analogously sulfhydrates cysteine residues in proteins. Cysteine residues are also modified by palmitoylation in response to neurotransmitter signaling, possibly in reciprocity with S-nitrosylation. Neurotransmission also elicits sumoylation and acetylation of lysine residues within diverse proteins. This review addresses how these recently appreciated protein modifications impact our thinking about ways in which neurotransmission regulates intracellular protein disposition.
Introduction
Neurotransmitter influences on cellular function have classically been ascribed to second and tertiary messengers such as cyclic AMP and protein phosphorylation. The advent of gasotransmitters, particularly nitric oxide (NO), but including carbon monoxide (CO) and hydrogen sulfide (H2S), has markedly altered conceptualizations about the path from the synapse to intracellular protein modification. Gases do not bind to cell surface receptors, hence do not require the intermediation of conventional membrane receptors and second messenger machinery such as G-proteins and adenylyl cyclase. Instead, the gases directly interact with targets, such as guanylyl cyclase [1]. Probably more prevalent is the S-nitrosylation (hereafter designated ‘nitrosylation’) by NO of cysteine residues in a wide range of target proteins. Specificity of signaling derives from NO synthase (NOS) binding to its targets directly or via scaffolding proteins such as CAPON (carboxy-terminal PDZ ligand of nNOS) [2]. H2S also modifies cysteines in target proteins, forming persulfide bonds, a process designated sulfhydration [3] (Glossary). In addition to nitrosylation and sulfhydration, cysteines in a wide range of proteins can be modified by fatty acids. Most of these alterations, such as prenylation, farnesylation and geranylation, are semi-permanent, serving to anchor proteins to membranes. By contrast, palmitoylation is dynamic and turns over with a half-life as short as 1–2 h [4].
Lysine residues in a variety of intracellular proteins are also modified in response to neurotransmitter signaling systems. Acetylation of nuclear histones has long been known to regulate transcription [5]. More recently, many non-nuclear, non-histone proteins have been shown to be acetylated, with this process being regulated by neuro-transmission [6]. Sumoylation involves the attachment of the 11-kDa protein SUMO (small ubiquitin-like modifier) to lysines in target proteins in a fashion analogous to ubiquitination. Recent studies implicate sumoylation in neural events, including neurological disorders such as Huntington’s disease (HD) [7].
This review will focus upon recent advances in neural signaling via protein modification. Because of space constraints and many excellent previous reviews on phosphorylation and ubiquitination, we will not deal with these modifications here, but focus instead on alterations of cysteine and lysine residues by nitrosylation, sulfhydration, palmityolation, sumoylation and acetylation.
S-nitrosylation
Stamler and associates [8] pioneered the concept of nitrosylation as a signaling system. The biotin-switch technique, which can monitor basal levels of nitrosylation, has permitted demonstration that many brain proteins are physiologically nitrosylated, because nitrosylation is lost in neuronal NOS (nNOS)-deleted mice [9]. We will focus on a limited number of nitrosylation targets that illustrate specific themes (Table 1).
Table 1.
Neural roles of nitrosylation
| Protein targets | Alteration in function | Physiological/pathophysiological relevance | Refs |
|---|---|---|---|
| COX2 | Increase in activity, binds nNOS | Decrease in NMDA-mediated neurotoxicity | [34,35] |
| Dexras | Increase in activity | Enhanced iron uptake and NMDA-mediated neurotoxicity | [23] |
| Drp1 | Increase in GTPase activity and mitochondrial fission | Increase in cell death in Alzheimer’s disease | [33] |
| GAPDH | Decrease in activity, nuclear translocation | Increase in cell death in MPTP parkinsonism | [25,27] |
| GOSPEL | Increase in binding to GAPDH, prevent nuclear translocation of GAPDH | Decrease in cell death in NMDA-mediated neurotoxicity | [26] |
| NMDAR | Decrease in activity | Decrease in NO production | [10] |
| NSF | Increase in activity and decrease in platelet granules | Increase in AMPAR surface expression | [17] |
| Parkin | Decrease in activity | Increase in cell death in Parkinson’s disease and Alzheimer’s disease | [28] |
| Stargazin | Increase in interaction with AMPAR | Increase in surface expression of AMPAR | [19] |
| Serine racemase | Decrease in activity | Increase in stroke | [20] |
COX2, cyclo-oxygenase-2; Drp1, dynamin related protein-1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GOSPEL, GAPDH’s competitor of Siah protein enhances life; NMDAR, N-methyl-D-aspartate receptor; NSF, N-ethylmaleimide sensitive factor; AMPAR, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
The NR2A subunit of the NMDA receptor (NMDAR), an ionotropic glutamate receptor, was one of the first characterized nitrosylation targets. Nitrosylation of cysteine residue 399 within NR2A inhibits NMDAR activity, which affords a negative feedback system for glutamate neuro-transmission [10,11]. NMDARs are linked via the scaffolding postsynaptic density protein 95 (PSD95) to nNOS. The PDZ ligand of NMDAR binds with a PDZ domain of PSD95 which, in turn, associates with the PDZ domain of nNOS [12]. This linkage affords a pathway whereby calcium ions, entering cells through NMDARs, bind to calmodulin, which is tightly associated with nNOS. Calmodulin activates nNOS, which leads to the generation of NO, which stimulates cyclic GMP formation. NO also feeds back to nitrosylate and inhibits NMDARs [13].
NMDARs function in close concert with another subtype of the ionotropic glutamate receptor family, AMPA receptors (AMPAR). NMDARs and AMPARs are physically linked by transmembrane AMPA receptor regulatory proteins (TARPS) and PSD95 [14]. Synaptic plasticity, evident in long-term potentiation (LTP) and long-term depression (LTD), is regulated by the cycling of AMPARs into and out of the plasma membrane [15,16], a process which is modulated by several proteins. One of these, N-ethylmaleimide sensitive factor (NSF), enhances surface expression of AMPARs [16]. Its binding to the GluR2 subunit of AMPARs is disrupted by ATP degradation, consistent with the known ATPase function of NSF [16]. NSF is basally nitrosylated, with its nitrosylation abolished in nNOS-deleted mice [17]. NMDAR activation stimulates NO formation, with associated nitrosylation of NSF augmenting the binding of NSF to the GluR2 subunit, resulting in an enhancement of the surface expression of AMPARs [17].
Stargazin is an auxiliary subunit of AMPARs that is crucially involved in their surface expression [18]. As occurs with NSF, NMDAR transmission leads to nitrosylation of stargazin, increasing its binding to AMPARs (in this instance the GluR1 subunit), whose surface expression is thereby enhanced [19]. Thus, nitrosylation impacts crosstalk between ionotropic glutamate transmission at multiple levels.
Serine racemase (SR) generates D-serine, a coagonist with glutamate at NMDARs. Postsynaptic stimulation of NO formation feeds back to presynaptic cells to nitrosylate SR and decrease D-serine availability to postsynaptic NMDARs [20]. SR knockout mice are less susceptible to NMDAR-mediated neurotoxicity and stroke [20].
NMDAR transmission is also influenced by nitrosylation of a small G-protein, Dexras1, a member of the Ras subfamily identified as a protein induced by the glucocorticoid dexamethasone [21]. Dexras1 binds to neuronal nNOS via the adaptor protein CAPON, eliciting nitrosylation and activation of Dexras1 [22]. Nitrosylation of Dex-ras1 physiologically stimulates iron uptake by DMT1 (Divalent Metal Transporter 1), which is linked to Dexras1 via a scaffolding protein PAP7 (PBR associated protein 7). Because selective iron chelation prevents NMDA neurotoxicity in cortical cultures, the NMDA-NO-Dexras1-PAP7-DMT1-iron uptake signaling cascade also appears to influence NMDA neurotoxicity [23].
In addition to its role in the physiological transmission of information, NO in excess is pathophysiological. NO is generated in the periphery largely in response to activation of inducible NOS (iNOS), whereas in the brain such stress involves NMDARs activating nNOS. The large amount of NO generated by iNOS in response to cell stressors facilitates the inflammatory response initiated by macrophages and other cells and can also lead to cell death [24]. Overactivation of nNOS by NMDAR activation during cerebral ischemia triggers NO formation which is cytotoxic [24]. Cell death can be mediated by a novel signaling cascade involving nitrosylation. The generated NO nitrosylates the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) at cysteine-150, abolishing catalytic activity and conferring upon GAPDH the ability to bind to the E-3 ligase Siah1 [25]. The nuclear localization signal of Siah1 enables it to ferry GAPDH to the nucleus where events involving another protein modification, acetylation (to be discussed below), lead to cell death. GOSPEL, a 52-kDa cytosolic protein, physiologically binds GAPDH in competition with Siah, retaining GAPDH in the cytosol and preventing its nuclear translocation (Figure 1). GOSPEL is neuroprotective, because its overexpression prevents NMDA-glutamate excitotoxicity, whereas its depletion enhances death in primary neuron cultures [26]. S-nitrosylation of GOSPEL at cysteine 47 enhances GAPDH–GOSPEL binding and the neuroprotective actions of GOSPEL [26].
Figure 1.
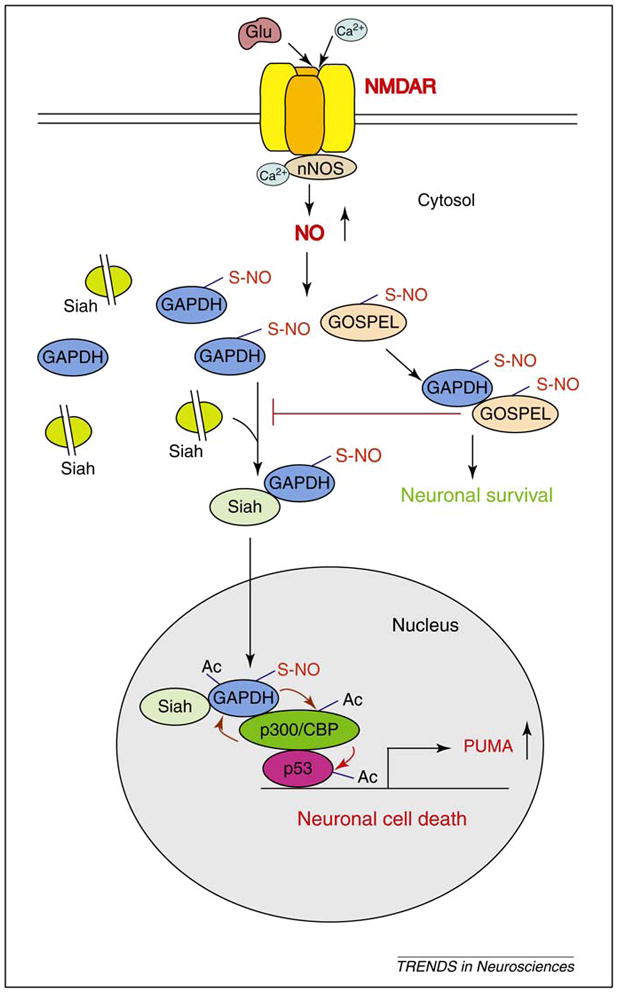
An apoptotic cell death cascade involving S-nitrosylation (S-NO) of GAPDH (glyceraldehyde-3-phosphate dehydrogenase), its nuclear translocation and acetylation of nuclear targets. Diverse apoptotic stimuli, including the pathological overactivation of NMDARs, can lead to the activation of nNOS. The subsequent generation of NO results in the S-NO of GAPDH, which abolishes its catalytic activity and confers on it the ability to bind to Siah1 (seven in absentia homolog 1), an E3-ubiquitin-ligase with a nuclear localization signal (NLS). Siah1, a protein with a rapid turnover rate, is degraded by the ubiquitin proteasome system (represented by Siah with dashed line in between two parts) so that under basal conditions tissue levels are extremely low. GAPDH stabilizes the rapidly turning over Siah1 (oval shaped without dashed line), augmenting its nuclear levels. The GAPDH-Siah1 protein complex, in turn, translocates to the nucleus, where GAPDH is acetylated at lysine residue 160 by the acetyltransferase p300/CREB binding protein (CBP) through direct protein interaction, which, in turn, stimulates the acetylation and catalytic activity of p300/CBP. Consequently, downstream targets of p300/CBP, such as p53, are activated and cause neuronal cell death through the transcriptional activation of apoptotic-inducing genes such as PUMA (p53 upregulated modulator of apoptosis). PUMA is a pro-apoptotic member of the Bcl-2 protein family which mediates apoptosis associated with p53 activity. GOSPEL (GAPDH’s competitor Of Siah Protein Enhances Life), physiologically binds GAPDH, in competition with Siah, retaining GAPDH in the cytosol and preventing its nuclear translocation. S-nitrosylation of GOSPEL enhances GAPDH–GOSPEL binding and the neuroprotective actions of GOSPEL.
The GAPDH-NO signaling system might participate in the pathophysiology of neurological diseases. GAPDH nitrosylation in the brain is augmented in the mouse MPTP model of Parkinson’s disease (PD) [25]. MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is a neurotoxin that causes symptoms of PD by destroying dopamine neurons in the substantia nigra. Deprenyl, a monoamine oxidase inhibitor used to treat PD, is neuroprotective independently of monamine oxidase inhibition. Its neuroprotective actions reflect its potent inhibition of GAPDH nitrosylation, which is demonstrable in the brains of MPTP-treated mice [27].
Nitrosylation could also impact PD via parkin, an ubiquitin E3 ligase whose mutation elicits an autosomal-recessive form of PD [28]. The E3 ligase activity of parkin, which is neuroprotective, stems from the ring finger domain of the protein, which contains several conserved cysteines. In MPTP parkinsonism and in brains of patients with PD, parkin is nitrosylated, which impairs its E3 ligase activity and leads to neuroprotective effects [29].
Several enzymes exert denitrosylase activity, one of which is superoxide dismutase 1 (SOD1) [30]. Mutations of SOD1 are responsible for some familial cases of amyotrophic lateral sclerosis (ALS) via a toxic gain-of-function. Also, thioredoxin/thioredoxin reductases can elicit both basal and stimulus-coupled protein denitrosylation, a process regulated dynamically by a thioredoxin-interacting protein (Txnip), which inhibits thioredoxin activity [31].
Protein-disulfide isomerase (PDI) prevents protein mis-folding by preserving appropriate alignment of crucial disulfide bonds. Nitrosylation of PDI inhibits this process, and increased levels of nitrosylated PDI accumulate in brains of patients with PD and Alzheimer’s disease (AD) [32]. NO might also influence mitochondrial fission with neuropathological consequences. Nitrosylation of Drp-1 enhances its dimerization and GTPase activity, which facilitates mitochondrial fission [33]. Increased nitrosylation of Drp-1 has been associated with neuronal injury and has been observed in AD patients [33].
NO generated by iNOS plays a prominent role in inflammation. It also regulates prostaglandin formation by cyclo-oxygenase-2 (COX2) via nitrosylation [34]. Thus, iNOS binds selectively to COX2 with the generated NO nitrosylating COX2 and activating prostaglandin formation [34]. NMDA neurotoxicity appears to involve an analogous process, because NMDA stimulation augments nNOS-COX2 binding and prostaglandin formation. Selective disruption of nNOS-COX2 binding prevents NMDA neurotoxicity [35].
Nuclear factor-kappa B (NF-κB) is a dimeric transcription factor composed of two members, p50 and RelA/p65 [40]. It is essential for neuronal survival and its activation could protect neurons against oxidative-stresses or ischemia-induced neurodegeneration [36–38], although its activation can contribute to inflammatory reactions and apoptotic cell death after brain injury and stroke [37,39]. Nitrosylation of the p65 subunit inhibits NF-κB-dependent gene transcription, and nuclear levels of nitrosylated p65 correlate with decreased DNA binding of the p50–p65 heterodimer [40].
Sulfhydration
Because of its high chemical reactivity, H2S has long been studied for its toxic actions [41–43]. Until recently, there was no definitive evidence for its physiological formation in mammalian tissues. H2S can be generated from cysteine by the actions of cystathionine γ-lyase (cystathionase; CSE) [42–45] or cystathionine β-synthase (CBS) [46]. In mice with targeted deletion of CSE, H2S formation is abolished in numerous peripheral tissues but not the brain [47]. In brain, CBS is highly expressed in the hippocampus and the cerebellum and appears primarily responsible for H2S formation [48].
Here, we will focus on the biosynthesis of H2S, its signaling mechanisms and on a limited number of neural H2S targets. Influences of H2S upon neuronal activity in the brain have been explored extensively by Kimura and colleagues [49]. This group noted that physiological concentrations of H2S enhance LTP, and that this effect is abolished by NMDA antagonists [48]. In addition to its actions upon neurons, H2S also appears to influence astrocytes [50].
H2S might also serve as a neuroprotectant. Glutamate neurotoxicity in brain cultures involves, at least in part, inhibition of cystine uptake [51]. The cystine/glutamate anti-porter couples influx of cystine, an H2S precursor, with efflux of glutamate. H2S can inhibit monoamine oxidase, leading to norepinephrine elevations in the hippocampus, striatum and brainstem but not in the cortex and cerebellum [52]. H2S ameliorates hippocampal damage caused by febrile seizures [53] and it has been demonstrated to be neuroprotective in a 6-hydroxydopamine model of PD [54]. H2S appears to signal predominantly by sulfhydrating cysteines in its target proteins, analogous to S-nitrosylation by NO [3,55]. For instance, GAPDH, whose catalytic activity is abolished by nitrosylation at C150, is sulfhydrated at the same position, with sulfhydration resulting in a 700% enhancement of catalytic activity [3,55]. Actin is also sulfhydrated physiologically. H2S donors augment actin polymerization more than 30% [3,55], an effect reversed by dithiothreitol (DTT), which abolishes sulfhydration. Sulfhydration also leads to rearrangement of the actin cytoskeleton [55]. These actions could regulate the influence of actin on synaptic dynamics.
Palmitoylation
Palmitoylation is one of four principal types of lipid modifications of proteins. The other three lipid modifications are isoprenylation [56] and myristoylation [57] of cytosolic proteins and modifications of membrane proteins by glycophosphatidyl inositol (GPI). However, because palmitoylation is quantitatively much more prominent than these other modifications, most authors prefer to use the term ‘palmitoylation’ to refer to all four of these lipid modifications, rather than the more accurate term of S-acylation.
Here, we will address only palmitoylation reactions of particular relevance to synaptic transmission (Table 2). Palmitoylation impacts several proteins of the synaptic vesicle fusion apparatus. Vesicular exocytosis involves interactions of a family of SNARE (soluble N-ethylmaleimide-sensitive fusion protein-attachment protein receptor) proteins present on the membranes of vesicles and on the plasma membrane. The most prominent vesicular SNARE is termed synaptobrevin-2 (VAMP2) [58], whereas SNAP23/25 (Synaptosomal-associated protein 23/25) and syntaxin 1 [58] are membrane-associated SNAREs. All of these exocytotic proteins appear to be palmitoylated [58–62]. Whereas syntaxin 1 and VAMP2 have membrane-spanning domains, SNAP25 is a soluble protein that requires palmitoylation on a cluster of cysteine residues to facilitate stable membrane attachment [59,60].
Table 2.
Neural features of palmitoylation
| Protein targets | Protein family | Palmitoylating enzyme | Physiological/pathophysiological relevance | Refs |
|---|---|---|---|---|
| β2-adrenergic receptor | G-protein coupled receptor | ND | Regulates phosphorylation and internalization of β-arrestin | [66] |
| CDC42 | Small GTPase | ND | Neurite outgrowth and dendritic spine formation | [60,62] |
| GABAAR (γ2 subunit) | Neurotransmitter | DHHC 3,7 | Inhibition of synaptic transmission | [76] |
| AMPAR (GluR1, GluR2 subunits) | Ion channel | DHHC 3 | Regulates AMPAR surface expression | [69] |
| Gαq, Gαs | G-protein signaling | DHHC 7 | Regulates G-protein assembly and activity | [64,65] |
| Huntingtin | Polyglutamine | DHHC 17 | Huntington’s disease | [83] |
| NMDAR | Neurotransmitter receptor | ND | Enhances tyrosine phosphorylation and increases surface expression | [68] |
| PSD95 | Scaffolding protein | DHHC 2, 15, 3, 7 | Synaptic targeting and postsynaptic protein assembly | [71] |
| SNAP25 | Vesicle trafficking | DHHC 17 | Neurotransmitter release, Huntington’s disease | [59] |
| Synaptobrevin | Vesicle trafficking | ND | Multimerization of synaptobrevin | [60] |
GABAAR, gamma-aminobutyric acid receptor; DHHC, aspartate-histidine-histidine-cysteine; NMDAR, N-methyl-D-aspartate receptor; PSD95, postsynaptic density protein 95; SNAP25, synaptosomal-associated protein 25; CDC42, cell division control protein 42; AMPAR, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor; ND, not determined.
Voltage-dependent potassium ion channels and G-proteins have been demonstrated to be palmitoylated [63]. Activation of beta-adrenergic receptors markedly accelerates the depalmitoylation of Gαs, which translocates the G-protein from the membrane to a cytosolic localization. This increases its interactions with RGS proteins (Regulators of G-protein Signaling), whose GTPase-stimulating activity further diminishes G-protein signaling [64,65]. Adrenergic agonists elicit depalmitoylation of beta-adrenergic receptors, which promotes their phosphorylation and internalization via β-arrestin [66]. The influence of palmitoylation on G-protein coupled receptors can vary, with depalmitoylation of alpha-adrenergic receptors preventing the reduction in number of receptors associated with desensitization [67].
Ionotropic glutamate receptors are also regulated by palmitoylation. The NMDAR subunits NR2A and NR2B have two separate clusters of palmitoylation sites. Palmitoylation in the first cluster has been shown to enhance tyrosine phosphorylation by Src kinases, leading to increased surface expression [68,69]. Palmitoylation of the second cluster, evidently by distinct enzymes, elicits receptor accumulation in the Golgi apparatus, with reduced NMDAR surface expression [68,69].
AMPARs are also palmitoylated in two domains: one in the second transmembrane domain at cysteine-610 and another in the juxtatransmembrane region of the C-terminal cytoplasmic tail at cysteine-836 of GluR2 [69]. Juxta-membrane palmitoylation does not directly affect AMPAR trafficking to the cell surface but does decrease interaction with the protein band 4.1N [69]. Depalmitoylation of the second site of GluR1 stabilizes surface expression through interactions with protein 4.1N [70].
Palmitoylation is catalyzed by a family of palmitoyl acyl transferases (PATs), most if not all of which display a consensus sequence aspartate-histidine-histidine-cysteine (DHHC) [71]. One of these, DHHC3, is a potential PAT for palmitoylating the transmembrane domain site of AMPAR, which could negatively regulate AMPAR trafficking from the Golgi complex to the plasma membrane [69]. Palmitoylation of PSD95 is particularly dynamic and regulates the surface expression of AMPARs [71]. PSD95 is palmitoylated at cysteines-3 and -5 at the N-terminus of the protein. Their mutation, which prevents palmitoylation, markedly diminishes surface expression of AMPAR [72]. Turnover of palmitate on PSD95 during synaptic activity is rapid with a half-life of under an hour [72], consistent with a role in regulating synaptic plasticity. By contrast, palmitate associated with synaptotagmin-2 and SNAP25 turns over very slowly and presumably functions primarily as a structural signal [73]. DHHC3 and DHHC2 are both required for postsynaptic accumulation of PSD95, but only DHHC2 mediates palmitoylation of PSD95 in response to reduced synaptic activity [74]. Suppression of neuronal activity rapidly mobilizes dendritic DHHC2 close to the postsynaptic membranes, thereby inducing robust palmitoylation and enhancing synaptic accumulation of PSD95. The increase in synaptic AMPARs elicited by blockade of synaptic activity requires DHHC2-mediated PSD95 palmitoylation [74].
DHHC3 has also been implicated in GABAA receptor (GABAAR)-mediated inhibitory synaptic transmission. Palmitoylation of the γ2 subunit of GABAARs occurs on cysteine residues in the large intracellular loop and is required for efficient postsynaptic clustering of GABAAR at inhibitory synapses [75,76]. Gephyrin, a scaffolding protein for glycine receptors and GABAARs, is expected to be palmitoylated, as indicated by synaptic palmitoyl proteome analysis [71,77]. Similarly to PSD95 at excitatory synapses [71], palmitate cycling on gephyrin might modulate the efficacy of GABAAR-mediated synaptic transmission in an activity-dependent manner.
Palmitoylation might also impact synaptic transmission through direct effects on dendritic spine morphology. Glutamatergic stimulation induces rapid depalmitoylation of Cell Division Control Protein-42 Homolog (CDC42), a member of the Rho family implicated in dendritic spine disposition. Depalmityolation of CDC42 leads to its dislocation from dendritic spines in cultured hippocampal neurons, possibly contributing to rapid spine collapse [60,84].
DHHCs have also been implicated in neurological and psychiatric disorders. DHHC17 palmitoylates huntingtin and regulates its trafficking and other functions [83]. Moreover, palmitoylation of huntingtin is diminished in a mouse model of HD [83]. DHHCs have also been associated with schizophrenia. Microdeletions of the 22q11.2 locus, a common chromosomal abnormality, are associated with a high incidence of emotional problems, with approximately 30% involving schizophrenia or related disorders [78,79]. This area of the genome possesses 27 genes, one of which is DHHC8, a genetic variant of which is associated with schizophrenia [80]. Thus, psychiatric disturbances associated with 22q11.2 microdeletion might reflect a loss of DHHC8 and associated palmitoylation [81]. PSD95 appears to be a physiological substrate of DHHC8, because deletion of the enzyme markedly reduces endogenous palmitoylation of PSD95 [81]. The enzymology of depalmitoylation is as yet unclear, although candidate depalmitoylating enzymes have been reported [82].
Sumoylation
Early studies focused on sumoylation of nuclear proteins, particularly those in nuclear promyelocytic leukemia protein (PML) bodies, whereas it is now appreciated that proteins in all intracellular compartments can be sumoylated [85]. Poly-ubiquitination in almost all instances targets proteins for degradation, whereas sumoylation has diverse functions including protecting proteins from ubiquitination, regulating protein–protein interactions, and transporting proteins between cytosol and the nucleus [86–88]. The sumoylation enzymatic pathway is analogous to ubiquitination, with SUMO attachment to proteins involving a series of enzymatic steps (Figure 2) [89–91].
Figure 2.

The SUMO modification cycle. SUMO precursor proteins contain a C-terminal extension which is cleaved by SUMO-specific carboxyl-terminal hydrolase activity of a sentrin-specific protease (SENP) enzyme to produce a carboxyl-terminal diglycine motif in a process called ‘SUMO maturation’. The mature SUMO is activated by an E1 SUMO activating enzyme (SAE1/2) which utilizes ATP and links the C-terminal glycine of SUMO by a thioester bond to a cysteine in the E1 activating enzyme. During the conjugation step, the SUMO moiety is transferred from the E1 activating enzyme to Ubc9, the SUMO E2 enzyme, which then attaches the SUMO to a lysine residue in the target protein that is typically, but not always, found within the consensus sequence ΨKxE (Ψ represents hydrophobic amino acids). SUMO E3 proteins stimulate protein sumoylation by associating with both UBC9 and substrates to increase the efficiency of the modification reaction by a process called target recognition and ligation. The SUMO E3 proteins identified to date include members of the family of Protein Inhibitor of Activated STAT (PIAS) family of proteins (PIAS1, PIAS3, PIASx and PIASy), the polycomb protein 2 (Pc2), and Ran-binding protein 2 (RANBP2). SUMO groups are removed from proteins by SENPs, of which there are six human forms. Rhes, a small G-protein, is selectively localized to the corpus striatum and binds directly to both E1 and Ubc9, enhancing cross-sumoylation as well as thioester transfer from E1 to Ubc9.
Although regulation of nuclear events by sumoylation might have bearing on neural function, the best characterized neuronal targets are non-nuclear. Neural sumoylation targets are largely membrane proteins including transporters, neurotransmitter receptors, regulators of neuronal development, and proteins involved in the pathophysiology of neurodegenerative diseases [92]. In the interest of brevity, we will focus only on a few examples, which highlight particular themes (Table 3).
Table 3.
Impact of sumoylation on neuronal activity
| Protein targets | Physiological/pathophysiological relevance | Refs |
|---|---|---|
| α-Synuclein | Pathophysiology of Parkinson’s disease | [105] |
| Ataxin1 | Pathophysiology of SCA type 1 | [104] |
| DJ-1 | Pathophysiology of Parkinson’s disease | [109] |
| Kainate receptor (GluR6 subunit) | Regulates kainate receptor endocytosis and synaptic activity | [98] |
| mHtt | Solubilization of huntingtin and increase in toxicity | [101,102] |
| MEF2 | Regulates cerebellar granule cell differentiation | [94] |
| Parkin | Pathophysiology of Parkinson’s disease | [106] |
| Potassium channel (K2P1) | Microglial cell proliferation, production and release of NO following brain injury | [96] |
mHtt, mutant Huntington; MEF2, myocyte enhancer factor-2; SCA type 1, spinocerebellar ataxia type 1.
In cerebellar neuronal preparations, myocyte enhancer factor-2 (MEF2) is required for dendritic development, specifically of dendritic claws, the branched tips of granule cell dendrites where glutamate inputs synapse [93]. Sumoylation of MEF2 enhances synapse formation, whereas replacement of the sumo group by an acetyl moiety reduces synaptic density [94].
At least two neuronal potassium channels are regulated by sumoylation. K2P1 channels are background plasma membrane leak channels whose sumoylation under basal conditions abolishes basal channel function [95], although there is some controversy about the importance of sumoylation for K2P1 [96]. The voltage-gated Kv1.5 potassium channel participates in microglial generation of NO following brain injury [97]. Selective mutations blocking its sumoylation markedly alter channel function [98].
Sumoylation influences a variety of glutamate receptors, including metabotropic receptors and the ionotropic kainate receptors. Sumoylation of the GluR6 subunit of kainate receptors is triggered by synaptic activation with glutamate or kainate [99]. Sumoylation leads to rapid internalization of GluR6-containing receptors, resulting in decreased synaptic activity. This is a physiological process, because the SENP1 desumoylating enzyme, which removes SUMO from proteins, increases spontaneous miniature excitatory post-synaptic currents [100]. Internalization of GluR6 is also enhanced by ubiquitination. However, GluR6 ubiquitination and sumoylation involve distinct lysine residues, although they are only three amino acids apart [99].
Sumoylation plays a role in neurodegenerative disorders, particularly those involving polyglutamine repeats, such as in HD and spinocerebellar ataxia (SCA). HD is caused by genetically dominant mutations in the gene encoding the protein huntingtin (Htt). HD is brain-specific, with profound abnormal movements related to selective, gross degeneration of the corpus striatum and lesser damage to the cerebral cortex eliciting dementia [100]. Sumoylation reduces pathology in Drosophila HD models, where it is more prominent than ubiquitination [101]. The striatal selectivity of HD pathophysiology could be explained by interactions of mHtt with the small G-protein Rhes (Ras Homologue Enriched in Striatum) [102]. Rhes binds mHtt with much greater avidity than wild-type Htt. Moreover, Rhes functions as an E3 ligase to stimulate sumoylation of mHtt [103]. Such sumoylation augments the neurotoxicity of mHtt by decreasing its aggregation [102]. Rhes is a major determinant of protein sumoylation in the striatum, because sumoylation of multiple proteins is markedly and selectively diminished in the striatum of Rhes-deleted mice [102]. Earlier studies identifying aggregation of mHtt had assumed that such aggregation was associated with neurotoxicity, whereas subsequent studies have established that the disaggregated soluble form of mHtt is most probably the pathogenic species [104]. Synaptic NMDAR activity induces mHtt inclusions via a T complex-1 (TCP-1) ring complex (TRiC)-dependent mechanism, rendering neurons more resistant to mHtt-mediated cell death. By contrast, stimulation of extrasynaptic NMDARs increases the vulnerability of mHtt-containing neurons to cell death by impairing the neuroprotective CREB-PGC-1alpha cascade and increasing levels of Rhes [104].
SCA is a dominantly inherited progressive condition with atrophy of the Purkinje cell layer of the cerebellum associated with expanded polyglutamines in ataxin1. Ataxin1 is sumoylated at several lysines, with sumoylation diminished by the expansion of polyglutamines [105]. SCA type 3 patients and mouse models of SCA all display substantial levels of SUMO1 in affected tissues [106].
PD might also be impacted by sumoylation. Alpha-synuclein is mutated in certain familial forms of the disease. It is known to be sumoylated, although the role of sumoylation in alpha-synuclein function has not been clarified [107]. Parkin is an E3 ubiquitin ligase associated with autosomal-recessive juvenile parkinsonium [107]. SUMO interacts non-covalently with parkin and increases its ubiquitin-E3-ligase activity [108]. Parkin regulates the turnover of the SUMO E3 ligase RanBP2 by catalyzing its ubiquitination and proteasomal degradation [109]. DJ-1 is a multifunctional protein involved in the transcriptional regulation of genes [110]. Loss-of-function in DJ-1, either by deletion or point mutation, leads to early onset of PD [111]. DJ-1 is modified by SUMO1 and also interacts with members of the PIAS family [111], which act as SUMO E3 ligases.
Acetylation and deacetylation
Protein acetylation in discrete brain areas can affect behavior. For instance, chronic social defeat stress in mice leads to a selective increase of acetylated histone H3 in the nucleus accumbens (Nac) [112]. The increased histone H3 acetylation is associated with decreased levels of histone deacetylase2 (HDAC2), and similar effects are detected in the nucleus accumbens of post-mortem depressed patients [112]. Infusion of HDAC inhibitors into the Nac elicits anti-depressant effects in the social defeat model [112]. Many other beneficial therapeutic effects have been suggested for HDAC inhibitors [113,114].
Brain-derived neurotrophic factor (BDNF) influences multiple genetic programs that are important for synaptic plasticity [115]. Recently, it was shown that nitrosylation of HDAC2 induces its release from chromatin, which increases acetylation of histones surrounding neurotrophin-dependent gene promoters and promotes transcription [116].
There has been increasing appreciation that many non-nuclear proteins are acetylated. A broad screen identified 3500 acetylation sites in 1700 acetylated proteins with the resultant ‘acetylome’ approaching the size of the ‘phosphoproteome’ [117]. Most of the acetylated proteins were found to be non-nuclear. Cell death, which is influenced by histone acetylation, is also regulated by acetylation of the non-nuclear protein GAPDH. As described above, a NO-GAPDH-Siah1 signaling cascade leads to nuclear translocation of GAPDH and cell death. What nuclear events elicit the associated apoptosis? In the nucleus, the acetylating enzymes p300/CBP acetylate GAPDH, causing it to activate auto-acetylation of p300/CBP, which, in turn, acetylate and activate p53 (Figure 1) [118]. Thus, it appears that the ‘executioner’ of the NO/GAPDH pathway is acetylated and activated p53.
Acetylation directly regulates actions of neurotoxic proteins. Acetylation of mHtt at lysine-444 facilitates its trafficking into autophagosomes, leading to clearance of mHtt by autophagy, thereby reversing the toxic effects of mHtt in rat primary striatal and cortical neurons, as well as in a Caenorhabditis elegans model of HD [119]. mHtt that is resistant to acetylation at this position accumulates to high levels and leads to neurodegeneration [118]. In SCA, polyglutamine proteins inhibit transcription, either by directly inhibiting histone acetylase co-activator activity, or as a consequence of histone ‘masking’ by binding to histones or blocking the sites that are available for acetylation [120].
Concluding remarks
It has long been known that neurotransmitters signal through a variety of secondary and tertiary messengers to impact a myriad of cellular functions including nuclear ones. The advances described here show that transmitters themselves, particularly gasotransmitters such as NO and H2S, directly modify target proteins. The extent of such modifications is substantial, with sulfhydration impacting up to 25% of certain proteins and with a major portion of all proteins examined being sulfhydrated [55]. Thus, S-nitrosylation and sulfhydration could be as prevalent and as functionally important as phosphorylation.
Increasingly appreciated are interactions between the various post-translational modifications which could have bearing on neurotransmission (Box 1). For example, cysteines are modified by palmitoylation as well as nitrosylation and sulfhydration, and there is early evidence that nitrosylation and palmitoylation are reciprocal processes. Modifications of lysines, particularly acetylation and sumoylation, might also display reciprocity, although there is thus far only modest evidence. The prevalence of sumoylation and acetylation of non-nuclear proteins is striking, as is the occurrence of these modifications in disease-related proteins, as exemplified by the role of mHtt sumoylation in the neurotoxicity of HD. As these modifications are further characterized, clinical relevance is probable, both for understanding pathophysiology and for the development of new therapeutic agents.
Box 1. Outstanding questions.
Excessive activation of NMDARs has been implicated in neuronal damage in many neurological disorders, ranging from acute hypoxic-ischemic brain injury to chronic neurodegenerative diseases that include Parkinson’s disease, Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and HIV-associated dementia. Might post-translational modifications of NMDARs, particularly nitrosylation, play a role in these diseases?
Nitrosylation and sulfhydration generally involve the same cysteines in proteins and seem to be functionally counterbalanced. Are the two modifications dynamically and reciprocally regulated in the brief time span of synaptic events?
Post-translational modifications occur on individual cysteine, serine, and lysine residues within proteins. Because multiple modifications can occur on each of these amino acids, how do the combinational interactions between the different modifications regulate synaptic signaling?
A striatal specific protein, Rhes, sumoylates mutant Huntington (mHtt) and enhances its cytoxicity by disaggregating and solubilizing mHtt. Might drugs that prevent Rhes-mHtt binding retard the progression of Huntington’s disease?
Acknowledgments
This work was supported by United States Public Health Services (USPHS) grants DA-000266 and MH-18501 and Research Scientist Award DA-00074 to S.H.S.
Glossary
- Acetylation
histone acetylation involves the attachment of acetyl groups to lysine residues in the N-terminal tails of histone proteins. It is believed that the acetylation of lysine residues decreases the affinity of histones for DNA, thereby making DNA more accessible for transcription. The opposite reaction (deacetylation) removes acetyl groups from lysine residues in the N-terminal tails of histone proteins. A large number of non-histone proteins are also acetylated to control diverse cell processes
- Biotin Switch assay
demonstration of physiological nitrosylation of numerous proteins under basal conditions by endogenously generated NO was rendered feasible by development of the biotin switch assay. In this procedure free thiols are blocked by the sulfhydryl-reactive compound, methyl methane thiolsulfonate; the nitrosylated thiols are then exposed by treatment with ascorbate, labeled with biotin, coupled to streptavidin, and nitrosylated proteins are separated by gel electrophoresis
- Palmitoylation
the most prominent modification of cysteines in proteins is palmitoylation, Protein palmitoylation is an enzymatic reaction, which is mediated by palmitoyltransferases (PAT). Palmitate, a 16-carbon fatty acid, forms an ester linkage with free thiols of cysteines. Other fatty acids, such as oleate, arachidonate, myristate and stearate can also attach in a similar manner to cysteine residues in proteins, however, these forms of modifications are beyond the scope of the current review
- S-nitrosylation
nitric oxide (NO) is produced enzymatically in most or all cell types and tissues. Addition of an NO group to the thiol side chain of cysteine residues within proteins and peptides is termed S-nitrosylation. Nitrosylation provides an NO ‘cap’ to the reactive sulfhydryl of cysteines in proteins, thus limiting chemical reactivity of this side chain
- Sulfhydration
sulfhydration (SHY) is a physiological process wherein hydrogen sulfide (H2S) attaches an additional sulfur to the thiol (−SH) groups of cysteine residues within proteins, yielding a hydropersulfide group (−SSH). The process is mediated by H2S enzymatically generated from L-cysteine by cystathione γ-lyase (CSE) or cystathionine β-synthase (CBS). This is not to be confused with S-thiolation or S-thionylation, in which a protein thiol forms a mixed disulfide with a small-molecular weight thiol such as glutathione or cysteine. S-thiolation blocks the protein thiol rendering it non-reactive, whereas S-sulfhydration yields an −SSH moiety which has enhanced chemical reactivity
- Sumoylation
SUMO (small ubiquitin-related modifier), also known as ‘centrin’, is a 101 amino acid protein. In a similar fashion to ubiquitin, SUMO can be covalently attached to the Σ-amino group of lysine side chains on proteins via an enzymatic reaction known as sumoylation. Four mammalian SUMO isoforms have been identified (SUMO 1–4), although their differential functions are not altogether clear yet
References
- 1.Garthwaite J. New insight into the functioning of nitric oxide-receptive guanylyl cyclase: physiological and pharmacological implications. Mol Cell Biochem. 2010;334:221–232. doi: 10.1007/s11010-009-0318-8. [DOI] [PubMed] [Google Scholar]
- 2.Jaffrey SR, et al. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–124. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 3.Mustafa AK. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prescott GR, et al. Palmitoylation of the synaptic vesicle fusion machinery. J Neurochem. 2009;110:1135–1149. doi: 10.1111/j.1471-4159.2009.06205.x. [DOI] [PubMed] [Google Scholar]
- 5.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spange S, et al. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 7.Anderson DB, et al. Protein SUMOylation in neuropathological conditions. Drug News Perspect. 2009;22:255–265. doi: 10.1358/dnp.2009.22.5.1378636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hess DT, et al. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 9.Jaffrey SR, et al. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 10.Choi YB, et al. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 11.Brenman JE, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 12.Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Lipton SA, et al. Cysteine regulation of protein function – as exemplified by NMDA-receptor modulation. Trends Neurosci. 2002;25:474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 14.Derkach VA, et al. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- 15.Kullmann DM, et al. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron. 1996;17:461–474. doi: 10.1016/s0896-6273(00)80178-6. [DOI] [PubMed] [Google Scholar]
- 16.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 17.Huang Y, et al. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, et al. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 19.Selvakumar B, et al. S-nitrosylation of stargazin regulates surface expression of AMPA-glutamate neurotransmitter receptors. Proc Natl Acad Sci U S A. 2009;106:16440–16445. doi: 10.1073/pnas.0908949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustafa AK, et al. Nitric oxide S-nitrosylates serine racemase, mediating feedback inhibition of D-serine formation. Proc Natl Acad Sci U S A. 2007;104:2950–2955. doi: 10.1073/pnas.0611620104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kemppainen RJ, Behrend EN. Dexamethasone rapidly induces a novel ras superfamily member-related gene in AtT-20 cells. J Biol Chem. 1998;273:3129–3131. doi: 10.1074/jbc.273.6.3129. [DOI] [PubMed] [Google Scholar]
- 22.Fang M, et al. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 23.Cheah JH, et al. NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron. 2006;51:431–440. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang PL, et al. Nitric oxide and cerebral ischemic preconditioning. Cell Calcium. 2004;36:323–329. doi: 10.1016/j.ceca.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Hara MR, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 26.Sen N, et al. GOSPEL: a neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron. 2009;63:81–91. doi: 10.1016/j.neuron.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hara MR, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–3889. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung KK, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 29.Yao D, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci U S A. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jourd’heuil D, et al. Effect of superoxide dismutase on the stability of S-nitrosothiols. Arch Biochem Biophys. 1999;361:323–330. doi: 10.1006/abbi.1998.1010. [DOI] [PubMed] [Google Scholar]
- 31.Benhar M, et al. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science. 2008;320:1050–1054. doi: 10.1126/science.1158265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uehara T, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 33.Cho DH, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SF, et al. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 35.Tian J, et al. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci U S A. 2008;105:10537–10540. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Yu Z, et al. Lack of the p50 subunit of nuclear factor-kappaB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci. 1999;19:8856–8865. doi: 10.1523/JNEUROSCI.19-20-08856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meffert MK, et al. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- 39.Guerrini L, et al. Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci U S A. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall HE, et al. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci U S A. 2004;101:8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura H, et al. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal. 2005;7:795–803. doi: 10.1089/ars.2005.7.795. [DOI] [PubMed] [Google Scholar]
- 42.Pereira SG, Oakley F. Nuclear factor-kappaB1: regulation and function. Int J Biochem Cell Biol. 2008;40:1425–1430. doi: 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 44.Wang R. Hydrogen sulfide: a new EDRF. Kidney Int. 2009;76:700–704. doi: 10.1038/ki.2009.221. [DOI] [PubMed] [Google Scholar]
- 45.Łowicka E, Bełtowski J. Hydrogen sulfide (H2S) – the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- 46.Mancardi D, et al. Physiological and pharmacological features of the novel gasotransmitter: hydrogen sulfide. Biochim Biophys Acta. 2009;1787:864–872. doi: 10.1016/j.bbabio.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang G, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimura H, et al. Physiological roles of hydrogen sulfide: synaptic modulation, neuroprotection, and smooth muscle relaxation. Antioxid Redox Signal. 2005;7:795–803. doi: 10.1089/ars.2005.7.795. [DOI] [PubMed] [Google Scholar]
- 50.Nagai Y, et al. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- 51.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 52.Warenycia MW. Monoamine oxidase inhibition as a sequel of hydrogen sulfide intoxication: increases in brain catecholamine and 5-hydroxytryptamine levels. Arch Toxicol. 1989;63:131–136. doi: 10.1007/BF00316435. [DOI] [PubMed] [Google Scholar]
- 53.Han Y, et al. Hydrogen sulfide may improve the hippocampal damage induced by recurrent febrile seizures in rats. Biochem Biophys Res Commun. 2005;327:431–436. doi: 10.1016/j.bbrc.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 54.Hu LF, et al. Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell. 2010;9:135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- 55.Mustafa AK, et al. Signaling by gasotransmitters. Sci Signal. 2009;2:re2. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 57.Johnson DR, et al. Genetic and biochemical studies of protein N-myristoylation. Annu Rev Biochem. 1994;63:869–914. doi: 10.1146/annurev.bi.63.070194.004253. [DOI] [PubMed] [Google Scholar]
- 58.Parpura V, Mohideen U. Molecular form follows function: (un)snaring the SNAREs. Trends Neurosci. 2008;31:435–443. doi: 10.1016/j.tins.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 59.Hess DT, et al. The 25 kDa synaptosomal-associated protein SNAP-25 is the major methionine-rich polypeptide in rapid axonal transport and a major substrate for palmitoylation in adult CNS. J Neurosci. 1992;12:4634–4641. doi: 10.1523/JNEUROSCI.12-12-04634.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang R, et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heindel U, et al. Palmitoylation sites and processing of synaptotagmin I, the putative calcium sensor for neurosecretion. FEBS Lett. 2003;544:57–62. doi: 10.1016/s0014-5793(03)00449-6. [DOI] [PubMed] [Google Scholar]
- 62.Martin BR, Cravatt BF. Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods. 2009;6:135–138. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE. 2006;2006:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 64.Degtyarev MY, et al. Increased palmitoylation of the Gs protein alpha subunit after activation by the beta-adrenergic receptor or cholera toxin. J Biol Chem. 1993;268:23769–23772. [PubMed] [Google Scholar]
- 65.Linder ME, et al. Lipid modifications of G proteins: alpha subunits are palmitoylated. Proc Natl Acad Sci U S A. 1993;90:3675–3679. doi: 10.1073/pnas.90.8.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Dowd BF, et al. Palmitoylation of the human beta 2-adrenergic receptor. Mutation of Cys341 in the carboxyl tail leads to an uncoupled nonpalmitoylated form of the receptor. J Biol Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 67.Loisel TP, et al. Activation of the beta(2)-adrenergic receptor-Galpha(s) complex leads to rapid depalmitoylation and inhibition of repalmitoylation of both the receptor and Galpha(s) J Biol Chem. 1999;274:31014–31019. doi: 10.1074/jbc.274.43.31014. [DOI] [PubMed] [Google Scholar]
- 68.Hayashi T, et al. Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron. 2009;64:213–226. doi: 10.1016/j.neuron.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayashi T, et al. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron. 2005;47:709–723. doi: 10.1016/j.neuron.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 70.Fukata M. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 71.Topinka JR, Bredt DS. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron. 1998;20:125–134. doi: 10.1016/s0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 72.El-Husseini A, et al. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- 73.Kang R, et al. Presynaptic trafficking of synaptotagmin I is regulated by protein palmitoylation. J Biol Chem. 2004;279:50524–50536. doi: 10.1074/jbc.M404981200. [DOI] [PubMed] [Google Scholar]
- 74.Noritake J, et al. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- 76.Fang C, et al. GODZ-mediated palmitoylation of GABA(A) receptors is required for normal assembly and function of GABAergic inhibitory synapses. J Neurosci. 2006;26:12758–12768. doi: 10.1523/JNEUROSCI.4214-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Biljmakers MJ, Marsh M. The on-off story of protein palmitoylation. Trends Cell Biol. 2003;13:32–42. doi: 10.1016/s0962-8924(02)00008-9. [DOI] [PubMed] [Google Scholar]
- 78.Chow EW, et al. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophr Res. 2006;87:270–278. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pulver AE, et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis. 1994;182:476–478. doi: 10.1097/00005053-199408000-00010. [DOI] [PubMed] [Google Scholar]
- 80.Edelmann L, et al. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet. 1999;64:1076–1086. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mukai J, et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang K, et al. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yanai A, et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Halpain S, et al. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heun P. SUMOrganization of the nucleus. Curr Opin Cell Biol. 2007;19:350–355. doi: 10.1016/j.ceb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 86.Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 87.Zhao J. Sumoylation regulates diverse biological processes. Cell Mol Life Sci. 2007;64:3017–3033. doi: 10.1007/s00018-007-7137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mukhopadhyay D, Dasso M. Modification in reverse: the SUMO proteases. Trends Biochem Sci. 2007;32:286–295. doi: 10.1016/j.tibs.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Kahyo T, et al. Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 90.Pichler A, et al. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–120. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- 91.Kagey MH, et al. The polycomb protein Pc2 is a SUMO E3. Cell. 2003;113:127–137. doi: 10.1016/s0092-8674(03)00159-4. [DOI] [PubMed] [Google Scholar]
- 92.Martin S, et al. Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat Rev Neurosci. 2007;8:948–959. doi: 10.1038/nrn2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shalizi AK, Bonni A. brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- 94.Shalizi A, et al. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 95.Wilson VG, Rosas-Acosta G. Wrestling with SUMO in a new arena. Sci STKE. 2005;2005:pe32. doi: 10.1126/stke.2902005pe32. [DOI] [PubMed] [Google Scholar]
- 96.Plant LD, et al. K2P channels and their protein partners. Curr Opin Neurobiol. 2005;15:326–333. doi: 10.1016/j.conb.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 97.Pannasch U, et al. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol Cell Neurosci. 2006;33:401–411. doi: 10.1016/j.mcn.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 98.Benson MD, et al. SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc Natl Acad Sci U S A. 2007;104:1805–1810. doi: 10.1073/pnas.0606702104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martin S, et al. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rosas HD, et al. Complexity and heterogeneity: what drives the ever-changing brain in Huntington’s disease? Ann N Y Acad Sci. 2008;1147:196–205. doi: 10.1196/annals.1427.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Steffan JS, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 102.Subramaniam S, et al. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Subramaniam S, et al. Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between e1 and ubc9. J Biol Chem. 2010;285:20428–20432. doi: 10.1074/jbc.C110.127191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Okamoto S, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Riley BE, et al. SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J Biol Chem. 2005;280:21942–21948. doi: 10.1074/jbc.M501677200. [DOI] [PubMed] [Google Scholar]
- 106.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 107.Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J Biol Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- 108.Um JW, Chung KC. Functional modulation of parkin through physical interaction with SUMO-1. J Neurosci Res. 2006;84:1543–1554. doi: 10.1002/jnr.21041. [DOI] [PubMed] [Google Scholar]
- 109.Um JW, et al. Parkin ubiquitinates and promotes the degradation of RanBP2. J Biol Chem. 2006;281:3595–3603. doi: 10.1074/jbc.M504994200. [DOI] [PubMed] [Google Scholar]
- 110.Abou-Sleiman PM, et al. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 111.Shinbo Y, et al. Proper SUMO-1 conjugation is essential to DJ-1 to exert its full activities. Cell Death Differ. 2006;13:96–108. doi: 10.1038/sj.cdd.4401704. [DOI] [PubMed] [Google Scholar]
- 112.Covington HE, 3rd, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- 114.Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang E, Reichardt L. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nott A, et al. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- 117.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 118.Sen N, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–873. doi: 10.1038/ncb1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jeong H, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li F, et al. Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem. 2002;277:45004–45012. doi: 10.1074/jbc.M205259200. [DOI] [PubMed] [Google Scholar]