

Abstract
Bromoacetoxy-calcidiol (B3CD), a pro-apoptotic and cytotoxic agent in neuroblastoma (NB) cell lines, displayed therapeutic potential in vivo as an anti-cancer drug in a NB xenograft mouse model. Tumors of all animals treated intraperitoneally with B3CD went into regression within 10–30 days of treatment, while tumors in control animals grew aggressively. The response mechanisms of NB cells to B3CD in vitro were studied and included differential targeting of cell cycle key regulators p21 and cyclin D1 on the transcriptional and expression level leading to arrest in G0/G1 phase. In contrast to the effect in ovarian cancer cells, B3CD-induced cell death in SMS-KCNR NB cells was only marginally mediated by the p38 MAPK signaling pathway. Signaling induced by exogenous recombinant EGF lead to a partial restoration of the negative effects of B3CD on SMS-KCNR cell proliferation and survival. Upon combinational treatment of SMS-KCNR cells with B3CD and recombinant EGF, the EGF receptor (EGF-R) was highly activated. We suggest future studies to include analysis of the effects of B3CD in combination therapy with pharmacological inhibitors of cell cycle regulators or with EGF-R targeting inhibitors, -toxins or -antibodies in vitro and their translation into in vivo models of tumor development.
Keywords: Neuroblastoma, Animal Model, Vitamin D3 derivative B3CD, anti-cancer activity
Introduction
Neuroblastoma (NB) is a rare cancer of the peripheral sympathetic nervous system. Peripheral neuroblastic tumors (pNTs) range from benign ganglioneuroma to stroma-rich ganglioneuroblastoma with well differentiated neuroblastic cells to highly malignant NB (1). Three distinct cell types have been isolated from NB cell lines: N-type cells with properties of embryonic sympathoadrenoblasts, S-type cells resembling non-neuronal Schwannian, glial, melanoblastic precursors, and I-type stem cells that can differentiate into either N- or S-type cells(2,3). About 500 new cases of NB are diagnosed in the US each year (4). The majority of NB cases occur in children below the age of five and NB account for 7–10% of all childhood cancers. In the majority of patients older than 1 year of age the disease is fatal. Multimodal treatment methods include surgery, radiation therapy, chemotherapy, autologous stem-cell transplantation (5,6,7) either alone or in combination, depending on the location and biological characteristics of the cancer cells, stage and the risk group to which the patient belongs. However, more than 50% of children with high-risk disease relapse, due to drug-resistant residual disease (8,9,10). Eradication of refractory microscopic disease remains one of the most significant challenges in the treatment of the high-risk NB and innovative treatments need to be designed.
Past studies led to the development of bromoacetoxy-calcidiol (B3CD, Fig. 1), a bromoacetoxy-ester derivative of calcidiol, which exerted potent selective anti-proliferative effects on prostate cancer cells (11,12,13) and NB cell lines (14). Calcidiol is the natural precursor to calcitriol/vitamin D3, is found abundantly in serum and is biologically inactive both in terms of binding to the vitamin D receptor (VDR) and in` transcription regulation (15). B3CD through indirect approaches was suggested to interact directly with the VDR receptor and in prostate cancer cells the authors hypothesize that this drug exerts cellular effects via the VDR signaling pathway (11). Bromoacetoxy analogs such as B3CD generally display an improved pharmacologic profile, exert less toxicity and greater stability compared to their parent compounds (16,17). Previous in vitro studies showed that B3CD at concentrations as low as 1.0 μM displayed strong growth-inhibitory effects in prostate cancer cell lines while other cancer cells such breast cancer cells or primary keratinocytes were significantly less affected (12,13). Previous studies on various neuroblastoma cell lines revealed high cytotoxicity of B3CD at 1 μM and anti-proliferative effects with IC50 concentrations as low as 30–100 nM (14). Cell death of NB cells upon treatment with B3CD is mediated by the intrinsic signaling pathway of apoptosis (14) whereas for prostate cancer cells, in addition to the intrinsic pathway, B3CD-induced apoptosis is mediated by the extrinsic pathway (11). In NB cells (SMS-KCNR) the cytotoxic response to B3CD is correlated with suppression of Akt mediated pro-survival signaling as well as with suppression of the oncogenic transcription factor MYCN (14), which is over-expressed in more than 65% of human NB (18). In ovarian cancer cells (SKOV-3) B3CD induced cell death is directly mediated by p38 MAPK function (19) which is essential for EGF-dependent ovarian cancer invasiveness (20). Interestingly, NB cells lines express a variety of EGF receptors and EGF can stimulate the proliferation of NB cell lines in vitro (21) and induce expression of pro-survival factors including p38 (22).
Figure 1. VDR Expression in Neuroblastoma cell lines after treatment with Calcidiol derivative B3CD.

(A) Structure of B3CD and precursor Calcidiol
(B) Vitamin D receptor expression. SMSK-CNR or SK-N-SH cells were treated with 1 or 3 μM B3CD for 48 h. Western Blot analysis of cell lysates was carried out as described (Material and Methods) using primary antibodies against VDR. As an internal standard for equal loading the blots were probed with an anti-GAPDH antibody.
The objective of the present study was to investigate the therapeutic potential of B3CD to treat NB in vivo in a NB xenograft animal model. Because B3CD was postulated to exert cellular effects via the VDR signaling pathway (11) we analyzed the expression change of the VDR receptor upon B3CD treatment of NB cell lines SMS-KCNR and SK-N-the correlation to the cytotoxicity exerted by the drug. We addressed the hypothesis that B3CD induced cell death, similar to ovarian cancer cells (19), may be mediated by p38 signaling and might be altered by the growth-stimulating effects of growth factor EGF. Because B3CD has previously been reported to affect cell cycle progression in SMS-KCNR cells (14) we studied the expression profile of several cell cycle regulators upon BC3D treatment.
Materials and Methods
Synthesis of B3CD
A procedure described earlier, with suitable modifications was used to synthesize B3CD (23,24). Briefly, equimolar amounts of calcidiol and bromoacetic acid were stirred with excess of dicyclohexylcarbodiimide and dry pryridine in dichloromethane in an ice bath for 2-to-4 hours. Our modifications entail preparative high performance liquid chromatography (HPLC; Waters Milford, MA, USA) using a C18 Luna column (4.6 × 150 mm; 5 μm; Phenomenex) (Torrance, CA, USA) of B3CD followed by 1H NMR and Mass spectroscopy characterization (14).
Cell Culture
SH-SY5Y (human NB) cells were obtained from American Type Culture Collection (Manassas, VA). SMS-KCNR and SK-N-SH (human NB) cell lines were provided by Giselle Saulnier Sholler (University of Vermont, Burlington, VT). The SK-N-SH MYCN deficient cell line displays both neuronal (N)- and stromal (S)-type NB cells and SH-SY5Y (N)-type cells were originally derived from this cell line (25). SMS-KCNR cells feature MYCN amplification and generally exhibit a uniform phenotype with small, round N-type cells that have short neuritic processes (26), yet cells in confluent culture can display stromal morphology. Cells were seeded at 5 X 105/T75 flask (Corning, New York, NY) and cultured to ~80% confluency in RPMI medium (Invitrogen) supplemented according to the suppliers recommendations at 37°C, 5% CO2, in a humidified incubator.
NB Xenograft Model
Animals
In vivo experiments were carried out at the animal facility of Rhode Island Hospital (RIH), Providence, RI, with strict adherence to the guidelines of the Animal Welfare Committee of RIH and Women & Infants Hospital. Four to six week-old immunodeficient nude mice (NU/NU; strain code 088/homozygous) (Charles River Laboratories, Wilmington, MA) were maintained at a temperature of 22±1 °C and a relative humidity of 55±5%, with a 12h light/dark cycle.
Treatment
SK-N-SH cells were cultured to 80% confluence, washed in PBS twice, harvested by trypsinization, pooled in complete medium, washed in PBS twice, and 5×106 cells/inoculate were suspended in 0.1 mL of matrigel and inoculated subcutaneously in the flank of mice. Mice with developing tumors (of max. 8 mm diameter) were randomly assigned to experimental groups. B3CD was prepared as a stock solution of 10 mM in 100% EtOH and diluted 1:100 in PBS for administration. Mice were treated intraperitoneally every other day for 30 days with either vehicle control (control group; 9 animals) or 38 μL (150 μg/kg body weight) of B3CD (treatment group; 8 animals) for 30 days.
Evaluation
Mice were weighed and tumor size calculated using a caliper every 4–6 days.
Western blot analysis
Cells were seeded into 100 mm2 tissue culture dishes (5×105 cells/dish), and treated with 1 μM B3CD for 24 or 48 h. Preparation of cell lysates, PAGE and immunoblotting was carried out as described previously (14). Primary antibodies were diluted 1:1000 in 5% BSA/PBST against P21 (ac-817), P27 (sc-53906) and GAPDH (sc-69778) were purchased from Santa Cruz, antibody (AB-1) against VDR was purchased from Neomarker (Fremont, CA), and antibodies against phosphorylated (#9215S) or inactive (#9212) p38 MAPK, phosphorylated EGF-R (53A5) and β-actin (#1501) were purchased from Cell Signaling Technology Inc. (Beverly, MA). The bands were visualized using horseradish peroxidase-conjugated secondary antibodies (Amersham-Pharmacia Biotech, Piscataway, NJ) diluted 1:2000 in PBST containing 5% non-fat dry milk, followed by enhanced chemiluminescence (Upstate, Waltham, MA) and documented autoradiography (F-Bx810 Film, Phenix, Hayward, CA). As a size standard pre-stained Precision Plus Protein Kaleidoscope (BioRad, Hercules, CA) marker was used.
Cell Viability Assay
Viability of various NB cell lines was determined by the CellTiter 96R AQueous One Solution Assay (Promega Corp, Madison, WI) following the manufacturer’s recommendations. An ELISA plate reader (Thermo Labsystems, Waltham, MA) allowed quantification of this colorimetric assay [conversion of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium “MTS” reagent in the presence of phenazine methosulfate into a soluble formazan product] at 490nm. Briefly, cells (5 X 103/well) were plated into 96 well flat bottom plates (Corning, Inc., Corning, NY) and allowed to attach overnight before treatment with various drugs as indicated (result section) in FCS free medium. Stock solutions of drugs dissolved in EtOH as well as vehicles alone (control) were serially diluted in serum-free medium and added to the wells. Following incubation at 37 °C in a cell-culture incubator for 20 h MTS reagent was added at a 1:10 dilution to the medium. For some assays cells were pre-incubated for 2 h with inhibitors (19) prior to B3CD addition (p38 MAPK inhibitor A: #559389/SB203580, inhibitor B: #559388/SB202190; Calbiochem, La Jolla, CA; inhibitor concentration 0–40 μM; for structure of inhibitors see: http://www.emdchemicals.com/). In some studies recombinant human EGF (GF001; Chemicon, Temecula, CA) at 40 nM concentration was added to the assay. Following incubation at 37°C in a cell culture incubator for 20 h MTS reagent was added at a 1:10 dilution to the medium. The samples were incubated for an additional 4 h before absorbance was measured at 490 nm. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % of absorbance in samples with untreated cells [100%].
Cell Proliferation Assay
Proliferation of various cell lines was determined by a BrdU (5-bromo-2′-deoxyuridine) incorporation assay (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s recommendations. Briefly, cells (5 X 103/well) were plated into 96 well flat bottom plates (Corning, Inc., Corning, NY) and allowed to attach overnight before treatment with B3CD, calcitriol/vitaminD3 or recombinant human EGF (GF001; Chemicon, Temecula, CA; 20 nM) for 18 h in FCS free media. BrdU (10 μM final concentration) was added to the cells grown for a further 6 h. After washing the cells were fixed and incubated for 2 h at 37 °C with an anti-BrdU antibody-peroxidase conjugate. Immune complexes were detected by addition of a tetramethyl-benzidine (TMB) substrate solution according to the manufacturer’s recommendations. The reaction was stopped by adding 50 μl of 1M sulfuric acid, and the absorbance was measured with an ELISA plate reader (Thermo Labsystems, Waltham, MA) at 450 nm. In this assay, the color intensity correlates directly to the amount of BrdU incorporated into the DNA, which in turn represents proliferation. Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % of absorbance of samples with untreated cells [100%].
Semi-quantitative RT-PCR
Total RNA was isolated from cells treated with 0, 1 and 3 μM of B3CD in CM for 48 h by using Charge Switch®Total RNA cell Kit (Invitrogen, Carlsbad, CA). Briefly, 200 ng of total RNA was preincubated at 65°C for 5 min with 50 μM oligo dt, 10 μM dNTP mix. Samples were reverse transcribed for 50 min at 50°C with 200 U SuperScriptTM RT in a total volume of 20 μl. 1 μl template DNA was used in a total 10 μl reaction volume with AccuPrime TM Taq DNA Polymerase System (Invitrogen). 0.2 μM sense and antisense primers were added to the mixture reaction. Amplifications were achieved in 25–35 cycles using MyCycler TM (BIO-RAD, Hercules, CA). For cyclin D1, p21, p27 and GAPDH, each PCR cycle consisted of denaturing for 20 sec at 94°C and annealing for 20 sec at 56°C, with an extension step for 20 sec at 68°C. Sequences of primers used were as follows.
Cyclin D1: forward 5′-GGTGAACAAGCTCAAGTGGA-3′; reverse 5′-GAGGGCGGATTGGAAATGAA-3′
p21: forward 5′-ACTTCGACTTTGTCACCGAG-3′; reverse 5′-AATCTGTCATGCTGGTCTGC-3′
p27: forward 5′-CAGCTTGCCCGAGTTCTA-3′; reverse 5′-TGGGGAACCGTCTGAAAC-3′
GAPDH: forward 5′-AAGGTCGGAGTCAACGGATTTGGT-3′; reverse 5′-ATGGCATGGACTGTGGTCATGAGT-3′
Data Analysis
Means ± standard deviations (SD) were calculated. The Student t-test was used for the biostatistical interpretation of the animal data. Software used for these analyses was STATA 9.0 (StataCorp, College Station, TX).
Results
Bromoacetoxy-calcidiol upregulates VDR expression
Bromoacetoxy-calcidiol (B3CD, Fig 1A), a derivative of calcitriol/vitamin D3, was proposed to be a potential drug to treat certain tumors such as prostate (12) or ovarian cancer (19). Calcitriol/vitamin D3 at higher concentrations inhibits cell proliferation and induces differentiation via binding to the vitamin D receptor (VDR) (27). It was hypothesized that B3CD exerts cellular effects in prostate cancer cells via the VDR signaling pathway (11). In the present study we confirmed that B3CD treatment upregulated VDR expression in NB cells. SMS-KCNR or SK-N-SH cells were treated with 1 or 3 μM B3CD and immunoblotting of cell lysates was performed using primary antibodies against VDR. This experiment revealed a dose-dependent and upregulation of expression of VDR within 48 h of treatment (Fig 1B) which correlated with the cytotoxicity displayed by B3CD in these cell lines (14). Conversely, calcitriol/vitamin D3 also strongly upregulates VDR expression but displays only minor cytotoxicity at similar drug concentrations (14).
Anti-cancer activity of B3CD in a neuroblastoma cancer xenograft model
In a previous in vitro study B3CD, in contrast to hypercalcemic calcitriol/vitamin D3, proved to be cytotoxic against various NB cell lines at concentrations as low as 0.3 μM (14). In addition B3CD, depending on cell line and concentration tested, displayed up to 5-fold greater cytotoxicity than its parent compound calcidiol (data not shown). In addition, calcidiol is hypercalcemic in vivo at the supraphysiologic concentrations required to achieve anti-tumor effect, whereas B3CD did not (11,19). These observations led us to investigate the potential in vivo anti-NB activity of B3CD.
The anti-tumor efficacy of B3CD was studied using human SK-N-SH NB cell derived xenografts in 4–6-week-old nude (NU/NU) mice. Cells suspended in matrigel were inoculated subcutaneously in one flank of each animal. Mice were treated intraperitoneally every other day with either B3CD (150 μg/kg body weight) or vehicle control. Animals were weighed (Fig. 2, right) and tumor size calculated (Fig. 2 left panel) using a caliper every 2 days.
Figure 2. Anti-cancer activity of B3CD in a NB animal model.

SMS-KCNR cells were inoculated subcutaneously in one flank of immunodeficient nude mice. After tumor development, mice were treated intraperitoneally every other day with either B3CD (150 μg/Kg) or vehicle control. Mice were weighed (right panels) and tumor size calculated (left panel) using a caliper every 2 days.
Throughout the study period no significant (≥10%) weight loss or gain was observed in either group and no animals developed ulcers. Tumor size consistently increased in the control animals where tumors measured on average 6.64±1.97 mm in diameter at the beginning of treatment and reached an average of 12.64±2.25 mm within 30 days. In the group treated with B3CD, tumors in all mice regressed from an average of 6.62±1.67 mm in diameter at the beginning of the study to an average of 3.38±1.61 mm at 30 days. Within this time frame complete responses (full tumor regression) to B3CD were not observed. However, in the treatment group the average tumor size stabilized at ~3 mm diameter after 22 days of treatment until the end of study. Due to aggressive tumor growth and increased tumor burden in the controls the study was ended after an observation period of 30 days.
Effect of B3CD on cell cycle regulators of NB cell lines
Previously we reported that blocking cell cycle progression is one of the mechanisms by which B3CD at subcytotoxic or mildly cytotoxic concentrations affects NB cells. Cell cycle analysis revealed a dramatic increase in the apoptotic sub-diploidal population (severe DNA damage) after B3CD treatment for 48 h along with a full block of cell cycle progression (G2/M subpopulation = 0%) with the majority of the cells arrested in G0/G1 phase [(14) and Fig. 3A]. In the present report we analyzed the direct effect of B3CD key regulators of cell-cycle progression such as cyclin D1, p21 and p27 in SMS-KCNR and SH-SY5Y NB cells, since drug targeting of cell cycle checkpoints has been suggested as an alternative approach to anti-cancer therapies (28). In both of these cell lines B3CD (48 h treatment) downregulated p27 expression, as shown by Western blotting of cell lysates (Fig. 3B), and transcription of p27, as shown through RT-PCR (Fig. 3C), at a concentration of 3 μM but not 1 μM. The lower dose of 1 μM B3CD was, however, sufficient to reduce cyclin D1 transcription (Fig. 3C) and expression (Fig 3B). At a higher concentration of B3CD (3 μM) cyclin D1 transcription and translation were completely abrogated. One μM treatment of SMS-KCNR and SK-N-SH cells also led to an upregulation of p21 expression (Fig 3B) even though p21 transcription was not affected (Fig 3C). In summary, B3CD, at concentrations that minimally affect viability of NB cells but dramatically suppresses proliferation (14), targets two key cell cycle regulators p21 and cyclin D1. This drug acts by upregulating p21 and downregulating cyclin D1 expression in NB cells and by affecting the transcription of cyclin D1 but not that of p21.
Figure 3. B3CD effect on cell proliferation and cell cycle progression in NB cells.
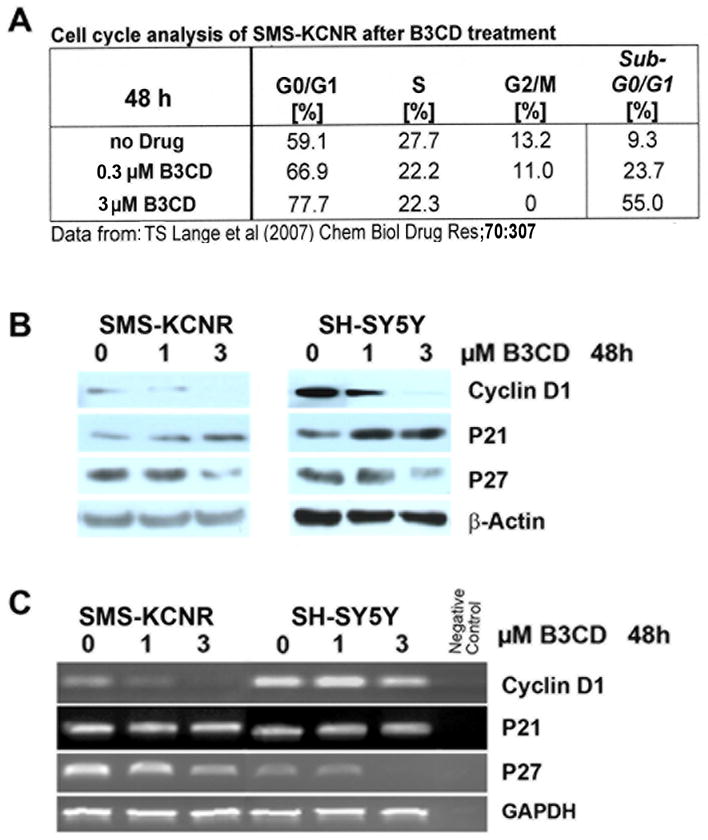
(A) Cell Cycle Analysis by FACS. Summary of previously published data (14).
(B) Expression of cyclin D1 and Cyclin-Dependent Kinase inhibitors in B3CD treated NB cells. Expression of cyclin D1, p21 and p27 cell cycle regulators in B3CD and vehicle treated SMS-KCNR or SH-SY5Y cells were analyzed by western blotting of lysates with the appropriate primary and secondary antibodies in combination with a chemiluminescence detection system as described in (Materials and Methods). As an internal standard for equal loading (50 μg total cell protein/lane) blots were probed with an anti-GAPDH antibody.
(C) Semi-quantitative RT-PCR of cyclin D1 and Cyclin-Dependent Kinase inhibitors in B3CD treated NB cells. Total RNA was isolated from SMSK-CNR or SH-SY5Y cells treated with 0, 1 and 3 μM of B3CD for 48 h. RT PCR was carried out as described (Material and Methods).
Effect of pro-apoptotic p38 MAPK inactivation in NB cells after B3CD treatment
We reported that B3CD treatment led to an activation of p38 MAPK and that inhibition of p38-signaling counteracted B3CD induced death in ovarian cancer cells (SKOV-3) (19). We analyzed the expression of p38 MAPK in SMS-KCNR NB cells before and after B3CD treatment by Western Blot analysis using primary antibodies against pro- and activated/phosphorylated p38. Immunblotting revealed that activation/phosphorylation of p38 in non-treated NB cells or cells treated with B3CD for 12h is barely detectable. P38 activation increased modestly in SMS-KCNR cells within 24 h of treatment with phosphorylation levels maintained throughout 48 h of treatment (Fig. 4A). In contrast, non-activated p38 MAPK expression levels, as seen in non-treated controls, were maintained within 24 h of B3CD treatment, but decreased during longer treatment periods (48 h) (Fig. 3A).
Figure 4. Expression of p38 MAPK in NB cells; effect of p38 inhibition on cell growth.

(A) Activation of p38 MAPK by B3CD. SMS-KCNR cells were treated with 3 μM B3CD for 12 or 24 h. Western Blot analysis of cell lysates was carried out as described (Material and Methods) using primary antibodies against pro- and activated/phosphorylated (P-) p38 MAPK. As an internal standard for equal loading the blots were probed with an anti-GAPDH antibody.
(B) Effect of p38 MAPK inactivation on B3CD mediated cytotoxicity. SMS-KCNR cells were pre-incubated with a specific inhibitors (40 μM) against p38 MAPK for 2 h and treated with B3CD (0, 1 or 3 μM) in the continued presence of the inhibitor for an additional 24 h. As control cell line sensitive to p38 inhibition upon B3CD treatment (19), SKOV-3 ovarian cancer cells were included. The MTS viability assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % cell viability of samples with untreated cells [=100%].
We screened the effect of two different inhibitors (40 μM) against p38 MAPK in a viability assay employing SMS-KCNR cells treated with 1 or 3 μM B3CD. As a positive control (for involvement of the p38 pathway and inhibitor activity) SKOV-3 cells were treated with 1 μM B3CD and p38 inhibitor (Fig. 4A). As shown previously (19), a dramatic suppression of the cytotoxicity of B3CD in SKOV-3 ovarian cancer cells was achieved by interfering with the activity of p38 MAPK. However, in SMS-KCNR cells, p38 inhibitors could only partially counteract the effect of B3CD and restore viability to only 7–14 % of cells depending on drug concentration or inhibitor type (Fig 4B). This observation suggests that B3CD-induced cell death in NB cells is not or only marginally mediated by the p38 MAPK signaling pathway.
Effect EGF on B3CD mediated cytotoxicity in NB cells
SMS-KCNR cells were treated with 20 nM EGF in combination with either B3CD or calcitriol/vitaminD3 and a proliferation assay was carried out. While B3CD at 100 nM concentration over a treatment period of 24 h exerted strong anti-proliferative effects on SMS-KCNR cells (proliferation reduced by ~50 %), calcitriol/vitaminD3 at this concentration was ineffective (Fig. 5A). As described previously for NB cell lines, EGF at 20 nM stimulated proliferation of untreated SMS-KCNR cells by 15–20%. Interestingly, EGF treatment partially prevented the anti-proliferative effect of B3CD in SMS-KCNR cells (91% proliferation at 100 nM B3CD+EGF, 57% at 100 nM B3CD-EGF; 76% proliferation at 1 μM B3CD+EGF, 38% at 1 μM B3CD-EGF). Similarly, EGF completely reversed the mild anti-proliferative effect of calcitriol/vitamin D3 on SMS-KCNR at 1μM (Fig. 5A).
Figure 5. Effect of EGF on B3CD treatment of SMS-KCNR cells.
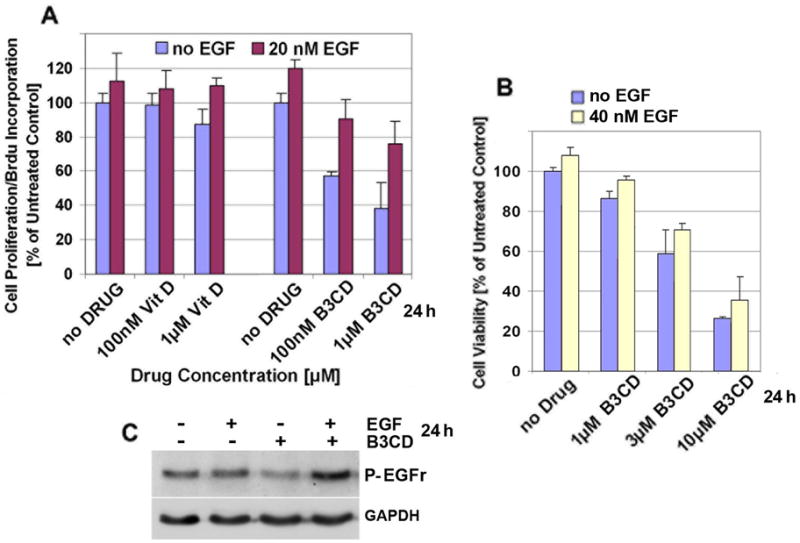
(A) Effect of Vitamin D, B3CD and EGF on cell proliferation. SMS-KCNR cells were treated with or without recombinant EGF (20 nM) and various concentrations (100 nM, 1 μM) of B3CD or calcitriol/vitaminD3 for 24 h. A colorimetric assay measuring cell proliferation (based on BrdU incorporation detected by a BrdU-antibody peroxidase conjugate) was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) in % of absorbance by triplicate samples of untreated cells [=100%].
(B) Cytotoxicity of B3CD during EGF treatment. SMS-KCNR cells were incubated with B3CD (1, 3 or 10 μM) and/or recombinant EGF (40 nM) for a total of 24 h. The MTS viability assay was carried out as described (Materials and Methods). Experiments were performed in triplicates; data are expressed as the mean of the triplicate determinations (X±SD) of a representative experiment in % cell viability of samples with untreated cells [=100%].
(C) Activation of ERF-R. SMS-KCNR treated with a combination of 3 μM B3CD and 50 nM EGF, or with either agent individually for 24 h. Western Blot analysis of cell lysates was carried out as described (Material and Methods) using primary antibodies against phosphorylated EGF-R. As an internal standard for equal loading the blots were probed with an anti-GAPDH antibody.
A viability assay was employed to test if EGF treatment can influence the cytotoxicitiy exerted by B3CD on SMS-KCNR cells (Fig 5B). Cells were incubated with B3CD (1, 3 or 10 μM) and/or recombinant EGF (40 nM) for a total of 24 h. The NB cells treated with EGF as compared to untreated controls displayed a higher OD which is directly proportional to the number of living cells (29). Interestingly, EGF at this concentration, could partially counteract the cytotoxic effect of B3CD (96% viability at 1 μM B3CD+EGF, 86% at 1 μM B3CD-EGF; 71% viability at 3 μM B3CD+EGF, 59% at 3 μM B3CD-EGF; 36% viability at 10 μM B3CD+EGF, 26% at 10 μM B3CD-EGF) (Fig 5B). Western Blot analysis of cell lysates was carried out using a primary antibody recognizing activated EGF-R receptors. SMS-KCNR before treatment revealed a basal level of activated EGF-R which slightly receded upon 24 h exposure to 50 nM of recombinant EGF (Fig. 5C), an adjustment effect typical for growth factor receptors in cells exposed to the ligand. In cells treated with 3 μM B3CD but without EGF supplementation, the activation of EGF-R was reduced to background levels. Interestingly, when cells were treated with the combination of B3CD and EGF, the receptor was highly phosphorylated within 24 h (Fig. 5C). Such receptor activation is correlated with the counteractive effects of EGF on B3CD mediated cytotoxicity (described above).
Discussion
Previous work has shown that B3CD displayed strong growth-inhibitory effects both with respect to proliferation and viability in certain prostate cancer cell lines (11) while other cancer cells such as MCF-7 (breast cancer) or primary keratinocytes were less affected (12,30). Thus, B3CD in contrast to the parent compound calcitriol/vitamin D3, which is highly calcemic in vivo (31–33), was proposed to be a potential anti-cancer drug specific for certain cancer types. However, in a previous study we observed that B3CD, even though highly cytotoxic to defined cell lines including certain ovarian cancer cells (SKOV-3) did not reveal specificity with respect to tumor origin (other ovarian cancer cell lines were less responsive) (19). When B3CD was used as a drug in an ovarian cancer animal model (human SKOV-3 cell derived xenografts in nude mice) mixed results were obtained. We observed that the majority of B3CD treated mice displayed delayed tumor growth or full tumor regression while in a few B3CD treated mice tumor growth accelerated (19). Accordingly, for the treatment of ovarian cancer we proposed further development of non-calcemic bromoacetoxy derivatives of calcitriol/vitaminD3 as potential anti-cancer therapeutics.
In contrast to ovarian cancer cell lines, a variety of NB cell lines studied revealed a consistent cytotoxic response to B3CD treatment (14). B3CD, depending on the cell line and concentration tested, displayed up to 5-fold greater cytotoxicity than the parent compound calcidiol. The structural difference between B3CD and calcidiol is the presence of a bromoacetate functionality which, must be correlated to the efficacy of this drug in NB cells. This observation led us to investigate the potential anti-cancer activity of B3CD in a NB xenograft model for the present study. We choose (NU/NU) mice originated from the NIH, which lack a thymus, are unable to produce T-cells and are used for xenograft and syngeneic tumor studies. Choice of dosage of the drug (non-toxic concentration of 150 μg/kg body weight) was based on previous studies. A recent systemic study in CD-1 mice showed that B3CD did not raise serum-calcium nor exhibit toxicity (166 μg/kg, repeated intraperitoneal administration) unlike other synthetic vitamin D3 derivatives (12), and references therein). In general, bromoacetic acid, a possible metabolite after B3CD administration in clinically relevant doses is unlikely to reach toxic concentrations; the LD50 of bromoacetic acid in male rats is 88 mg/kg (34) which is several hundred-fold higher than concentrations of B3CD used in our in vivo studies.
B3CD treatment of NB tumor cell derived xenografts in mice revealed a chemotherapeutic effect within 6 days of treatment. From day 6 forward the average tumor in treated animals was reduced in diameter as compared to treatment start. In contrast, tumors in control mice grew aggressively. Due to increased tumor burden in the controls the study was ended at 30 days. By study end tumors in untreated mice were 3.75 times larger in diameter than tumors in B3CD treated mice. Within this time frame complete responses (full tumor regression) to B3CD were not observed. However, in the treatment group tumor size stabilized within 22 days of treatment at ~3 mm diameter. In summary, B3CD displayed anti-cancer activities in this NB-xenograft animal model. We did not observe ulcerations around NB-cell derived tumor sites or acceleration of tumor growth, unlike our previous study using ovarian cancer cell xenografts in the same mouse strain (19). Given the fact that B3CD 1) did not cause adverse effects and 2) revealed activity against neuroblastoma xenografts, we propose to continue the evaluation of this bromoacetoxy-ester derivative of calcidiol in neuroblastoma cancer model systems. We also initiated studies to develop other non-calcemic vitamin D derivatives as potential anti-cancer agents (35). In addition, we suggest studies on the effects of B3CD in combination therapy with other anti-cancer drugs, antibodies or cytokines in tumor cells in vitro and their translation into in vivo models of tumor development.
Like various other cytotoxic agents B3CD displays cell-cycle regulatory effects in NB cells at the IC50 as well as at sub-cytotoxic concentrations (14). After B3CD treatment the sub-diploidal apoptotic population increases and this correlates with the onset of apoptotic signaling and DNA fragmentation (14). With respect to the cycling cells B3CD causes an increase, or depending on the concentration, an arrest of the G0/G1 population along with decrease of cells in S-phase and G2/M phase.
In the present report we analyzed the direct effect of B3CD on regulators of cell-cycle progression such as cyclin D1, or p21 and p27 (both are regulators of cyclin D- dependent kinases; CDK) in SMS-KCNR and SH-SY5Y NB cells. In these cell lines B3CD at a concentration of 1 μM, which mildly suppresses viability of NB cells, targets two key cell cycle regulators, p21 and cyclin D1. It acts by upregulating p21, downregulating cyclin D1 expression and by downregulating the transcription of cyclin D1 but not of p21. Drug targeting of cell cycle checkpoints and key regulators which are frequently altered in human cancer, such as cyclin D1 and p21, has been suggested as an alternative approach to anti-cancer therapies (28,36,37). We also observed that at the cytotoxic dosage of 3 μM B3CD, p27 expression and transcription was downregulated. It has been reported that depending on the experimental parameters, either p27 up- or down-regulation can be associated with a G1 arrest in NB cells (38,39). P27 sequestration by cyclinD-cdk complexes is a key factor in establishing a balance between cell proliferation and cycle arrest even though p27 knockout animals are relatively free of malignancy (36; and references therein). Given the multi-faceted role displayed by p27 and the fact that p27 expression in NB cells changes only upon high dosage of B3CD we suggest that p27 regulation is not a major regulator of specific cell cycle regulation exerted by B3CD. In contrast, cyclin D1 and p21 are modulated by B3CD at lower dosage. Cyclin D1 downregulation and p21 induction leading to inhibition of cyclin D dependent kinases is a prerequisite for the arrest of cells in G1 phase in general (28,37) as well as in NB cell lines (38). However, modulation of a regulator alone depending on the experimental setting and cell system does not necessarily lead to cell cycle arrest as was shown for NB cells where p21 (as well as p53) induction lead to apoptosis, yet G1 cell cycle arrest was attenuated (40). Similarly, inhibition of CDK activity by itself does not necessarily lead to the arrest of cycling cancer cells (28). In contrast, B3CD interferes with the progression of cells through G1 phase by regulation of cyclin D1 and p21 expression. Therefore, the use of drugs such as B3CD alone or in combination with pharmacological CDK inhibitors may specifically prevent the progression of NB.
In a previous study in ovarian cancer cells we observed a dramatic suppression of the cytotoxicity of B3CD by interfering with the activity of p38 MAPK while inhibition of other MAPKs such as Erk 1, Erk2, JNK 1, 2 and 3, Jun/JNK, or MEK did not significantly alter B3CD mediated cell death (19). P38 and other MAPKs such as JNK, MEK, Erk1/2 mediate signaling pathways in cancer and control cell lines responding to inflammatory cytokines, UV light, cytotoxic drugs, and diverse other pro-apoptotic stimuli (41,42,43). Activation of p38 generally is a pro-apoptotic trigger and is a key determinant for drug-induced apoptosis, such as cisplatin in ovarian cancer cells (44). Similarly, sustained activation of the p38 and JNK MAPK pathways by different drugs (e.g. Fenretinide) initiated cell-death of NB cell lines (45) and certain apoptotic stimuli in NB cells act mainly via the p38 MAPK pathway (46). B3CD caused an up-regulation of the activity of this pro-apoptotic signaling factor in SMS-KCNR NB cells (19). However, in SMS-KCNR cells p38 inhibitors could only partially counteract the effect of B3CD and restore cell viability by only 5–10%. This observation suggests that the p38 MAPK signaling pathway in NB cells, unlike that of ovarian cancer cells, does not play a pivotal role in the response to B3CD.
We observed a stimulating effect of exogenously added recombinant EGF on SMS-KCNR NB cell proliferation. It has been reported that EGF can stimulate the growth of a variety of NB cell lines in vitro along with activation of both mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/AKT pathways (21). It is known that NB primary tumors and cells lines express a variety of growth factor receptors including epidermal growth factor receptors (EGF-R; HER1-4) (21). Moreover, resistance of NB to standard drugs such as cisplatin, has been directly linked to enhanced levels of EGF-R expression. Such cells are sensitive to treatment with specific toxins and antibodies targeting EGF-R (47). Consequently, an alternative treatment option for NB might be combination treatment with EGF-R targeting agents as well as standard (e.g. cisplatin) or newly developed drugs such as B3CD. We analyzed whether the cytotoxic effect of B3CD on NB cells might be altered by exposure to exogenous recombinant EGF and whether these treatments change the expression levels of the EGF-R.
Recombinant EGF, added at 20 nM concentration, prevented almost completely the anti-proliferative effect of 1 μM B3CD on these NB cells. Similarly, EGF reversed the mild anti-proliferative effect of calcitriol/vitamin D3 on SMS-KCNR cells. In addition, recombinant EGF at 40 nM could partially counteract the cytotoxic effect of B3CD on this NB cell line. Therefore, we hypothesize that signaling induced by EGF leads to a restoration of both the negative effects of subcytotoxic concentrations of B3CD on cell cycle progression and that of cytotoxic concentrations on NB cell death. EGF signaling is primarily induced by EGF receptors (HER1-4). Upon combination treatment of SMS-KCNR cells with B3CD and recombinant EGF, EGF receptors were highly activated. Apparently, access to exogenous EGF allowed NB to counteract B3CD induced cell death by an increase in EGF-R signaling as an effort to survive drug treatment. Apart from specific toxins or antibodies clinically established EGF-R-specific tyrosine kinase inhibitors such as Gefitinib (also known as Iressa or ZD1839) could be evaluated in their effect on NB in combinational treatment with B3CD. Even though effective in vitro concentrations cannot be clinically reached Gefitinib also displayed chemosensitizing effects when used with other drugs (e.g. topotecan, vincristine) in NB cell lines (48). In summary, we postulate that the chemotherapeutic properties of B3CD might be enhanced by co-treatment with EGF-R-targeting agents (e.g. toxins, antibodies, tyrosine kinase inhibitors) and suggest further animal models to substantiate the findings. Future studies, beyond the scope of the present manuscript, might investigate if in NB cells B3CD targets in a reverse manner the same MAPK and PI3K/AKT pathways as EGF (21).
Conclusion
The present study reveals the therapeutic potential of B3CD to treat tumors in vivo in a NB xenograft mice model. The mechanisms of anti-proliferative and cytotoxic activity of B3CD in vitro include differential regulation of cyclin D1 and p21 cell cycle regulators on the transcriptional as well as expressional level. B3CD targets pathways that mediate the response of NB cells to growth factor EGF via the EGF-R. We suggest future studies to include analysis of the effects of B3CD in combination therapy with other anti-cancer drugs, EGF-R inhibitors, pharmacological inhibitors of cell cycle regulators (e.g. CDK), and their translation into in vivo models of tumor development.
Acknowledgments
This work was supported by a NINDS/NIH R21NS051408 grant to Dr. Brard and Penelope and Sam Fund at the University of Vermont to Dr. Sholler. The authors thank NIH COBRE Grant 1-P20RR018728 for providing instrumentation support.
Footnotes
Competing Interests: The authors declare that there are no conflicts of interest.
References
- 1.Brodeur GM, Pizzo PA, Poplack DG. Neuroblastoma. 4. Lippencott-Raven; Philadelphia, USA: 2001. Principles and Practice of Pediatric Oncology; pp. 895–937. [Google Scholar]
- 2.Berthold F. Biology of Neuroblastoma. In: Pochedly C, editor. Tumor Biology and Therapy. CRC Press; Boca Raton: 1990. pp. 1–27. [Google Scholar]
- 3.Ross RA, Hein AM, Braca JA, Spengler BA, Biedler JL, Scammel JG. Glucocorticoids induce neuroendocrine cell differentiation and increase expression of N-myc in N-type human Neuroblastoma cells. Oncology Res. 2002;13:87–94. [PubMed] [Google Scholar]
- 4.Ater JL. Neuroblastoma. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BMD, editors. Nelson Textbook of Pediatrics. 18. chapter 498 Saunders Elsvier; St. Louis, USA: 2007. [Google Scholar]
- 5.Matthay KK, Perez C, Seeger RC, Brodeur GM, Shimada H, Atkinson JB, Black CT, Gerbing R, Haase GM, Stram DO, Swift P, Lukens JN. Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children’s Cancer Group study. J Clin Oncol. 1998;16:1256–1264. doi: 10.1200/JCO.1998.16.4.1256. [DOI] [PubMed] [Google Scholar]
- 6.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeu GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 7.Perez CA, Matthay KK, Atkinson JB, Seeger RC, Shimada H, Haase GM, Stram DO, Gerbing RB, Lukens JN. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: a children’s cancer group study. J Clin Oncol. 2000;18:18–26. doi: 10.1200/JCO.2000.18.1.18. [DOI] [PubMed] [Google Scholar]
- 8.Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;1999:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- 9.Goldsby RE, Matthay KK. NEUROBLASTOMA: evolving therapies for a disease with many faces. Paediatr Drugs. 2004;6:107–122. doi: 10.2165/00148581-200406020-00004. [DOI] [PubMed] [Google Scholar]
- 10.Matthay KK, Atkinson JB, Stram DO, Selch M, Reynolds CP, Seeger RC. Patterns of relapse after autologous purged bone marrow transplantation for neuroblastoma: a Childrens Cancer Group pilot study. J Clin Oncol. 1993;11:2226–2233. doi: 10.1200/JCO.1993.11.11.2226. [DOI] [PubMed] [Google Scholar]
- 11.Swamy N, Chen TC, Peleg S, Dhawan P, Christakos S, Stewart LV, Weigel NL, Mehta RG, Holick MF, Ray R. Inhibition of proliferation and induction of apoptosis by 25-hydroxyvitamin D3–3beta-(2)-Bromoacetate, a nontoxic and vitamin D receptor-alkylating analog of 25-hydroxyvitamin D3 in prostate cancer cells. Clin Cancer Res. 2004;10:8018–8027. doi: 10.1158/1078-0432.CCR-04-0881. [DOI] [PubMed] [Google Scholar]
- 12.Swamy N, Persons KS, Chen TC, Ray R. 1α,25-Dihydroxyvitamin D3–3beta-(2)-bromoacetate, an affinity labeling derivative of 1 α,25-dihydroxy-vitamin D3 displays strong antiproliferative and cytotoxic behavior in prostate cancer cells. J Cell Biochem. 2003;89:909–916. doi: 10.1002/jcb.10585. [DOI] [PubMed] [Google Scholar]
- 13.Lambert JR, Young CD, Persons KS, Ray R. Mechanistic and pharmacodynamic studies of a 25-hydroxyvitamin D(3) derivative in prostate cancer cells. Biochem Biophys Res Commun. 2007;361:189–195. doi: 10.1016/j.bbrc.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 14.Lange TS, Singh RK, Kim KK, Zou Y, Kalkunte SS, Sholler GL, Swamy N, Brard L. Anti-proliferative and pro-apoptotic properties of 3-Bromoacetoxy Calcidiol (B3CD) in high-Risk neuroblastoma. Chem Biol Drug Design. 2007;70:302–310. doi: 10.1111/j.1747-0285.2007.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt-Gayk H, Bouillon R, Roth HJ. Measurement of vitamin D and its metabolites (calcidiol and calcitriol/vitamin D3) and their clinical significance. Scand J Clin Lab Invest Suppl. 1997;227:35–45. [PubMed] [Google Scholar]
- 16.Cottens S, Sedrani R. O-alkylated rapamycin derivatives and their use. 6440990 United States Patent. 2002
- 17.Driedger PE, Quick J. Protein kinase C modulators, E. 5886017 United States Patent. 1999
- 18.Pession A, Tonelli R. The MYCN oncogene as a specific and selective drug target for peripheral and central nervous system tumors. Curr Cancer Drug Targets. 2005;5:273–283. doi: 10.2174/1568009054064606. [DOI] [PubMed] [Google Scholar]
- 19.Lange TS, Robison K, Stuckey AR, Kim KK, Singh RK, Raker CA, Brard L. Effect of a Vitamin D3 derivative (B3CD) with postulated anti-cancer activity in an ovarian cancer animal model. Invest New Drugs. 2009 doi: 10.1007/s10637-009-9284-y. [epub ahead of print, July 7 2009] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou HY, Pon YL, Wong AS. Synergistic effects of epidermal growth factor and hepatocyte growth factor on human ovarian cancer cell invasion and migration: role of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Endocrinology. 2007;148:5195–5208. doi: 10.1210/en.2007-0361. [DOI] [PubMed] [Google Scholar]
- 21.Ho R, Minturn JE, Hishiki T, Zhao H, Wang Q, Cnaan A, Maris J, Evans AE, Brodeur GM. Proliferation of human neuroblastomas mediated by the epidermal growth factor receptor. Cancer Res. 2005;65:9868–9875. doi: 10.1158/0008-5472.CAN-04-2426. [DOI] [PubMed] [Google Scholar]
- 22.Chiu B, Mirkin B, Madonna MB. Mitogenic and apoptotic actions of epidermal growth factor on Neuroblastoma cells are concentration-dependent. J Surg Res. 2006;135:209–212. doi: 10.1016/j.jss.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Swamy N, Ray R. Affinity labeling of rat serum vitamin D binding protein. Arch Biochem Biophys. 1996;333:139–144. doi: 10.1006/abbi.1996.0374. [DOI] [PubMed] [Google Scholar]
- 24.Warren JC, Sweet F. Synthesis and use of affinity labeling steroids for analysis of macromolecular steroid-binding sites. Methods Enzymol. 1975;36:374–410. doi: 10.1016/s0076-6879(75)36036-9. [DOI] [PubMed] [Google Scholar]
- 25.Ross RA, Spengler BA, Biedler JL. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst. 1983;71:741–747. [PubMed] [Google Scholar]
- 26.Hettmer S, McCarter R, Ladisch S, Kaucic K. Alterations in neuroblastoma ganglioside synthesis by induction of GD1b synthase by retinoic acid. British Journal of Cancer. 2004;91:389–397. doi: 10.1038/sj.bjc.6601914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar R. 1Alpha,25-dihydroxyvitamin D(3) - not just a calciotropic hormone. Nephron. 2002;91:576–581. doi: 10.1159/000065015. [DOI] [PubMed] [Google Scholar]
- 28.Shapiro GI, Harper JW. Anticancer drug targets: cell-cycle and checkpoint control. J Clin Invest. 1999;104:1645–1653. doi: 10.1172/JCI9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malich G, Markovic B, Winder C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicol. 1997;124:179–192. doi: 10.1016/s0300-483x(97)00151-0. [DOI] [PubMed] [Google Scholar]
- 30.Lambert JR, Young CD, Persons KS, Ray R. Mechanistic and pharmacodynamic studies of a 25-hydroxyvitamin D(3) derivative in prostate cancer cells. Biochem Biophys Res Commun. 2007;361:189–195. doi: 10.1016/j.bbrc.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 31.DeLuca HF, Zierold C. Mechanisms and functions of vitamin D. Nutr Rev. 1998;56:4–10. 54–75. doi: 10.1111/j.1753-4887.1998.tb01686.x. [DOI] [PubMed] [Google Scholar]
- 32.Dusso AS, Brown AJ. Mechanism of vitamin D action and its regulation. Am J Kidney Dis. 1998;32(S2):13–24. doi: 10.1053/ajkd.1998.v32.pm9808140. [DOI] [PubMed] [Google Scholar]
- 33.Olick MF. Vitamin D: the underappreciated D-lightful hormone that is important for skeletal and cellular health. Curr Opin Endocrinol Diabetes. 2002;9:87–98. [Google Scholar]
- 34.United Sates Environmental Protection Agency (USEPA) report (2005) Bromoacetic acid - Identification, toxicity, use, water pollution potential, ecological toxicity and regulatory information. CAS number 79–08–3. http://yosemite.epa.gov
- 35.Brard L, Robison K, Singh RK, Kim KK, Lange TS. A novel non-hypercalcemic vitamin D derivative in the treatment of ovarian cancer. J Wom Health. 2007;16:1098–1099. [Google Scholar]
- 36.Blain S, Scher H, Cordon-Cardo C, Koff A. p27 as a target for cancer therapeutics. Cancer Cell. 2003;3:111–115. doi: 10.1016/s1535-6108(03)00026-6. [DOI] [PubMed] [Google Scholar]
- 37.Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV, Bates SE. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer. 2000;83:817–825. doi: 10.1054/bjoc.2000.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouwehand K, de Ruijtera AJM, van Breeb C, Caron HN, van Kuilenburg ABP. Histone deacetylase inhibitor BL1521 induces a G1-phase arrest in neuroblastoma cells through altered expression of cell cycle proteins. FEBS letters. 2005;579:1523–1528. doi: 10.1016/j.febslet.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 39.Matsuo T, Seth P, Thiele CJ. Increased expression of p27Kip1 arrests neuroblastoma cell growth. Med Pediatr Oncol. 2001;36:97–99. doi: 10.1002/1096-911X(20010101)36:1<97::AID-MPO1022>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 40.McKenzie PP, Guichard SM, Middlemas DS, Ashmun RA, Danks MK, Harris LC. Wild-Type p53 Can Induce p21 and Apoptosis in Neuroblastoma Cells But the DNA Damage-induced G1 Checkpoint Function Is Attenuated1. Clin Canc Res. 1999;5:4199–4207. [PubMed] [Google Scholar]
- 41.Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 42.Birkenkamp KU, Dokter WH, Esselink MT, Jonk LJ, Kruijer W, Vellenga E. A dual function for p38 MAP kinase in hematopoietic cells: involvement in apoptosis and cell activation. Leukemia. 1999;13:1037–1045. doi: 10.1038/sj.leu.2401447. [DOI] [PubMed] [Google Scholar]
- 43.Ahmed-Choudhury J, Williams KT, Young LS, Adams DH, Afford SC. SCCD40 mediated human cholangiocyte apoptosis requires JAK2 dependent activation of STAT3 in addition to activation of JNK1/2 and ERK1/2. Cell Signal. 2006;18:456–468. doi: 10.1016/j.cellsig.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 44.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 45.Osone S, Hosoi H, Kuwahara Y, Matsumoto Y, Lehara T, Sugimoto T. Fenretinide induces sustained-activation of JNK/p38 MAPK and apoptosis in a reactive oxygen species-dependent manner in neuroblastoma cells. Int J Canc. 2004;112:219–224. doi: 10.1002/ijc.20412. [DOI] [PubMed] [Google Scholar]
- 46.Arvidsson Y, Hamazaki TS, Ichijo H, Funa K. ASK1 resistant neuroblastoma is deficient in activation of p38 kinase. Cell Death Diff. 2001;8:1029–1037. doi: 10.1038/sj.cdd.4400922. [DOI] [PubMed] [Google Scholar]
- 47.Michaelis M, Bliss J, Arnold SC, Hinsch N, Rothweiler F, Deubzer HE, Witt O, Langer K, Doerr HW, Wels WS, Cinatl J. Cisplatin-Resistant Neuroblastoma Cells Express Enhanced Levels of Epidermal Growth Factor Receptor (EGF-R) and Are Sensitive to Treatment with EGFR-Specific Toxins. Clin Canc Res. 2008;14:6531–6537. doi: 10.1158/1078-0432.CCR-08-0821. [DOI] [PubMed] [Google Scholar]
- 48.Rosseler J, Odenthal E, Geoerger A, Gerstenmeyer A, Lagodny J, Niemmeyer CM, Vassal G. EGFR Inhibition Using Gefitinib Is Not Active in Neuroblastoma Cell Lines. Anticancer Res. 2009;29:1327–1333. [PubMed] [Google Scholar]