

Abstract
It has been confirmed that stress plays an important role in the induction and development of cardiovascular diseases, but its mechanism and molecular basis remain unknown. In the present study, a myocardial injury model induced by restraint stress was established in rat. To screen for the related proteins involved in stress-induced myocardial injury, proteomic techniques based on 2-DE and mass spectrometry were used. In our results, ten proteins were found to be altered. The expression of eight of these proteins was increased after restraint stress, including cardiac myosin heavy chain, dihydrolipoamide succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial aldehyde dehydrogenase, H+-transporting ATP synthase, albumin, and apolipoprotein A-I precursor. The expression of uncoupling protein 3 (UCP3) and mitochondrial aconitase was decreased. Most of the proteins were related to energy metabolism. Further research indicated that UCP3 may mediate the myocardial cell response induced by restraint stress.
Keywords: Myocardial injury, Mitochondria, Proteomic, Restraint stress, UCP3
Introduction
Stress is defined as a physiological response to the disruption of homeostasis and contributes to diseases, including diabetes, gastric ulcer, obesity, cancer, and Parkinson's disease (Rasola and Bernardi 2007). In particular, the relationship between stress and the risk of cardiovascular disease has been confirmed (Feuerstein and Young 2000). Restraint is considered to be a nonspecific stressor; hence, the animal model of restraint stress is often used to study the influence of stress on physiological function and pathological processes (Liu et al. 2004; Zhao et al. 2007). In our previous study, pathological alterations in electrocardiograms and disruption of cardiovascular function were observed in chronic restraint stressed rat (Zhao et al. 2007).
Mitochondria are important subcellular organelles, which play a crucial role in diverse cellular functions, such as energy production, modulation of redox status, osmotic regulation, Ca2+ homeostasis, inter-organelle communication, cell proliferation and senescence, and cell response to a multiplicity of physiological stresses (Marcil et al. 2006; Mattson and Liu 2003; Nicholls 2004). Several publications have shown that stress induces impaired mitochondrial function (Ott et al. 2007). Under stress conditions, the permeability of mitochondria membranes will be increased, which leads to the loss of mitochondria membrane potential and to the uncoupling of oxidative phosphorylation, and several factors such as cytochrome C and apoptosis-induced factor were released by the opening of a proteinaceous channel, commonly known as the permeability transition pore (PTP).
Another factor associated with mitochondria is reactive oxygen species (ROS), a group of reactive and short-lived oxygen free radicals that includes superoxide (O2·−), singlet oxygen1 (O2·), hydroxyl radical (·OH), and hydrogen peroxide (H2O2). The destructive capability of ROS depends on their concentration in cell. Mitochondria are considered one of the sources of ROS. Batandier et al. reported that under stress conditions, the opening of the mitochondria PTP induces ROS production at the level of respiratory chain complex I and complex II (Ott et al. 2007; Batandier et al. 2004; Muller et al. 2004). In a further study, Perier et al. (2005) found that mitochondria-generated ROS play an important role in the release of cytochrome C and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis. Whereas cardiomyocyte death is considered an important cellular basis for stress-induced cardiovascular injury and disease, the detailed molecular basis of stress-induced cardiovascular injury remains unclear.
In the present study, a proteomic technique based on 2-DE and MS was used to explore novel significant proteins which correlate with chronic stress and cardiomyocyte injury. We focused on mitochondrial protein alteration as a result of ROS concentration in cardiomyocyte and found that the expression of ten proteins was altered in cardiomyocyte mitochondria of restrained rat. Further functional study revealed a dramatical change in UCP3 in mitochondria. Our results indicated that proper expression of UCP3 in mitochondria protects against stress-induced myocardial injury.
Materials and methods
Materials
Acrylamide, methylenebis-acrylamide, glycine, Tris, SDS, urea, glycerol, bromophenol blue, Triton X-100, IPG buffers, IPG strips, and 2-D Quant kit were purchased from Amersham Biosciences (Uppsala, Sweden). Pharmalyte, DCFH-DA, Rh123, TEMED, CHAPS, thiourea, iodoacetamide, and ammonium persulfate were obtained from Sigma (St. Louis, MO, USA). DTT was purchased Promega (Madison, WI, USA). The in situ cell death detection kit was purchased from Boehringer Mannheim (Mannheim, Germany). The (125I-) cortisol radioactivity immunoassay kit was purchased from North (Beijing, China). Other reagents were of the highest purity commercially available.
Experimental animal model of chronic restraint stress
Adult male Wistar rats (180–200 g in weight) from the same parents were divided into stress and control groups randomly as previously described (Galea et al. 1997). The animals in the stress group were placed into an adjustable restraint cage separately 6 for hours per day (from 09:00 am to 15:00 pm) for 4 weeks. The control group was kept under the same living conditions as the stress groups except for the restraint treatment. The investigation conformed to the guide for the care and use of laboratory animals published by the US National Institutes of Health.
Isolation and culture of cardiomyocytes
Cardiomyocytes were isolated from 3-day-old neonatal Wistar rats according to the previously described method (Qian et al. 2004). The animals were given heparin, 100 units, hypodermically. The rat hearts were quickly removed and washed with chilled PBS, then cut into 1- to 2-mm cubes and dissociated by blowing five times with 0.25% trypsin and 0.02% EDTA plus 0.1% sodium citrate. Cardiomyocytes from the first dissociation were discarded, and the rest were collected in ice-cold MEM with 10% FBS containing penicillin 50 U/ml and streptomycin 50 μg/ml, and centrifuged (1,000 rpm, 10 min). The precipitate was washed twice in MEM, then incubated in the same medium containing 10% FBS in culture dishes at 37°C in humidified air with 5% CO2. A single 30-min preplating period provided the best separation for >95% cardiomyocytes. An aliquot of the non-attached cells was counted with a hemocytometer in quadruplicate in 0.4% trypan blue, and 1 ml of the cells in culture medium with 10% FBS and 0.1 mM Brdu was plated in 35-mm culture dishes (at 220–300 trypan blue-negative cells per mm2). After 24 h, the cardiomyocytes were washed with PBS. The culture medium with 10% FBS was renewed at this time. Cardiomyocytes exceeding 100 beats/min were selected for the following experiments.
Cardiomyocyte model of stress
Stress can activate the hypothalamic–pituitary–adrenocortical axis and the sympathetic–adrenomedullary system, and the increased content of glucocorticoid (GC) and catecholamine in plasma is considered as a significant biological basis for evaluating stress load. The major component of GC in rodents is corticosterone (CORT). To establish a cell model of stress, the cardiomyocytes were cultured in serum-free minimum essential medium (MEM) for 2 h before stress and then were incubated with different doses of corticosterone and norepinephrine (Fluka) (10−5 M norepinephrine and 10−4 M corticosterone) for 2 h, 6 h, and 12 h, respectively.
Preparation for mitochondria
For isolation and preparation of mitochondria, the animals were decapitated. The hearts were excised, and the left ventricles were separated on an ice dish. A half ventricular wall was selected for isolating mitochondria, and another half was used for analyzing adenosine triphosphate (ATP) content in myocardium. For isolation of mitochondria, the myocardium was minced and washed three times with 37°C physiological saline (0.9% NaCl). The homogenate of myocardium was prepared in ice-cold medium (1:8 wt/vol) containing 250 mM sucrose, 50 mM Tris, and 1 mM EDTA, pH 7.4. The mitochondria were purified by differential centrifugation (700 ×g for 10 min, 9000 ×g for 30 min). The mitochondria were then suspended in sucrose buffer (250 mM sucrose, 20 mM N-2-hydroxyethylpiperazine-N9-2-ethanesulfonicacid [HEPES], 1 mM EDTA, 1 mM dithiothreitol [DTT], pH 7.4) for up to 2 h at 4°C for functional examination. The precipitation of mitochondria was schizolysis by 1× SDS gel loading buffer and stored at −70°C for use (Wang et al. 2009).
Measurement of mitochondrial ROS
Measurement of mitochondrial ROS was performed with flow cytometry (Liu et al. 2005). After mixing 200 µg mitochondria and the measurement buffer (250 mM sucrose, 20 mM MOPS, 10 mM Tris, 100 μM K2HPO4, 0.5 mM MgCl2, pH7.4), 1 μM cyclosporine A and 5 mM succinate were added, then the mixture was incubated 10 min at room temperature. The fluorescence intensity was recorded by flow cytometry with FL2 passage (FSC/SSC gate) as the background. Subsequently, DCFH-DA (10 μg/ml) was added and the fluorescence intensity was recorded again.
Determination of mitochondrial membrane potential
Mitochondrial membrane potential was determined by flow cytometry (Raquel et al. 2008). 100 μg mitochondria were put into the determination buffer (100 mM KCl, 10 mM Tris, 2 mM MgCl2, 4 mM K2HPO4, 1 mM EDTA, pH7.4) and incubated for 10 min at room temperature to determine the fluorescence intensity of FL2 passage (10,000 cells/second, FSC/SSC gate). The fluorescence intensity of FL2 passage was recorded as the background, then substrate (20 mM succinate 6 μl, 0.8 μM Rh123 6 μl) was added, and incubated for 10 min at room temperature to determine the fluorescence intensity of FL2 passage again, then FCCP 4 μl was added to record the fluorescence intensity. The difference between the two fluorescence intensity levels is the mitochondria membrane potential.
Preparation of myocardial proteins
Cardiomyocyte whole-cell lysates were prepared. Briefly, the ventricle muscle of stressed rat was separated, and neonatal rat cardiomyocytes digested by 0.25% trypsin were homogenized in precooled 2× SDS gel loading buffer containing protease inhibitors, PMSF, and leupeptin. The homogenate was centrifuged (10,000 ×g for 10 min) after heating in boiling water for 10 min. The supernatant was used as cardiomyocyte whole-cell lysates. The protein content was assayed using the 2-D Quant Kit.
2-DE
The sample was dissolved in lysis buffer (8 M urea, 5% w/v CHAPS, 65 mM DTT and proteinase inhibitor) at room temperature. IEF was carried out on Ettan IPGphor isoelectric focusing system (Pharmacia Biotech, Uppsala, Sweden) using 18 cm IPG strips (pH 3–10). Protein (1.5 mg) was added to the rehydration solution (8 M urea, 2% w/v CHAPS, 65 mM DTT, 0.5% IPG buffer and a trace of bromophenol blue) to a total volume of 340 ml. Strips were rehydrated for 6 h and then IEF was performed for a total of 56,000 Vh. The second dimension electrophoresis was carried out on 1.5 mm and 10%T SDS-PAGE gels (Bio-Rad vertical system; Bio-Rad, Hercules, CA, USA). The parameters were a constant current of 20 mA/gel for 30 min and 30 mA/gel until the bromophenol blue dye reached the bottom of the gel. After electrophoresis, gels were stained with CBB R-250. 2-DE gels were scanned with an ImageScanner (Amersham Biosciences) and analyzed using ImageMaster 2D Elite software (Amersham Biosciences). The difference in the abundance of differential protein spots was analyzed with the Student's t test (p < 0.005 was considered significant).
In-gel digestion and MALDI-TOF MS
The significant protein spots were excised from the gel and then digested with trypsin as described by Rosenfeld et al. (1992). Briefly, protein spots were destained with 50% ACN and dried in a vacuum concentrator. The dried gel was then rehydrated in trypsin solution and incubated overnight at 37°C. After the peptides were eluted in turn with TFA of different concentrations, the peptide mixture was measured on a Micomas Tof Spec MALDI-TOF mass spectrometer (Manchester, UK) according to the method of Jungblut and Thiede (1997). The data from MALDI-TOF MS were analyzed by searching an NCBInr database using the MASCOT search engine. One possible missed cleavage for trypsin digestion was selected. Errors of peptide mass were set to 20 ppm.
Western blotting
Protein lysate was resolved by 10% SDS-PAGE and transferred onto Immobilon-P transfer membrane (Millipore) using a semidry transfer apparatus (Bio-Rad Laboratories). The membrane was blocked with nonfat milk in PBST and incubated with rabbit polyclonal UCP3 antibody (Santa Cruz Biotechnology Incorporation, 1:2,000), followed by secondary horseradish peroxidase-linked antibody (Beijing Zhongshan Corporation, 1:1,000). The bound antibody was visualized using the ECL Western blotting detection system (Amersham).
Statistical analysis
Experimental data were expressed as mean ± SD. The significance of the differences was determined by multiple comparison test post ANOVA and expressed as a probability value.
Results
Restraint stress induced ROS in cardiomyocytes
Mitochondria are the key position of cellular oxidative stress and the major site to produce ROS. Our results showed that restraint stress had different effects on mitochondrial ROS content in the rat myocardium (Fig. 1a). Compared with the control, the mitochondrial ROS content showed no significant change after 1 and 2 weeks of restraint stress, but its content gradually increased with restraint-time prolongation in chronic stress groups to about 16% and 25%, respectively (Fig. 1b). Additionally, treating cardiomyocytes with CORT and NE in vitro significantly increased the content of mitochondrial ROS in a dose-dependent manner. These results showed that the content of ROS increased approximately to 120%, 132%, and 146% in treated cardiomyocytes by NE in 2, 6, and 12 h, respectively (Fig. 1c). The results of GC-treated cardiomyocytes are most similar to NE.
Fig. 1.

Effect of stress on ROS production in the mitochondria. a Detection of fluorescence intensity of mitochondria by dyeing with oxygen free radical fluorescent probe (DCFH-DA). b Changes of ROS content in cardiomyocytes under stress. The mitochondrial ROS content showed no significant change after 1 and 2 weeks of restraint stress, but gradually increased in the chronic stress groups to about 16, and 25% of content, after 3 and 4 weeks respectively (p < 0.05). c Changes of ROS content in cardiomyocyte treatment with GC and NE. The content increased approximately to 120, 132, and 146% in treated cardiomyocytes by NE in 2, 6, and 12 h, respectively (p < 0.05). The results of GC-treated cardiomyocytes are most similar to NE
Changes of mitochondria PTP in cardiomyocytes under stress
The integrality of mitochondria is important for cell survival, which is associated with oxidative phosphorylation. Damage to mitochondria would result in the disturbance of the intracellular homeostasis such as calcium overload, oxidative stress, and acidosis, which has been identified in our previous and present studies. Here, we analyzed the mitochondrial membrane potential with mitochondria proton gradient (ΔμH+) by flow cytometry (Fig. 2a). The result showed that mitochondrial PTP alternated clearly after rats were exposed to restraint stress for 3 and 4 weeks, or cardiomyocytes were exposed to NE and GC for 6 and 12 h, respectively (Fig. 2b and c). The influence of stress on mitochondrial PTP was significant if a higher intensity was given; as the ΔμH+ of mitochondria was increased, the alteration of mitochondrial PTP was also increased. The results suggested that cell stress may result in injury of mitochondrial PTP. Mitochondrial PTP is an important structure for maintaining mitochondrial homeostasis and membrane potential. Although there are still no direct methods to detect the opening of the mitochondrial PTP, the swelling of mitochondria and the decrease of mitochondrial membrane potential undoubtedly suggest an abnormal opening of the mitochondrial PTP.
Fig. 2.

Changes of mitochondrial MPT in stressed cardiomyocytes. a The mitochondrial membrane potential with mitochondrial proton gradient (ΔμH+) was analyzed by flow cytometry. b MPT alternated evidently after rats were exposed to restraint stress for 3 and 4 weeks, respectively (p < 0.05). c Cardiocytes exposed to NE and GC for 6 and 12 h (p < 0.05)
Analysis of differentially expressed proteins
The higher stability and reproducibility of 2-DE are the important bases for proteomic analysis. The match rate of protein spots among different 2-DE maps is an effective index of reproducibility. In the present study, both the protein preparation and 2-DE protocol were performed using a standardized procedure. Based on ImageMaster 2D Elite software analysis, 1,301 ± 59 and 1,325 ± 42 spots were detected in the control- and stress-group gels, respectively (CBB R-250 staining). The match rate of control- and stress-group gels was 87.3% ± 2.0%. The expression patterns of mitochondrial proteins are shown in Fig. 3. Most of the protein spots were distributed in the region of pH 4–8 and had MW between 15 and 55 kDa. The differences in protein profiles between the control and stressed groups were detected by image analysis. Compared with the control group, ten protein spots with differential abundance were found in the stressed group as shown in Fig. 4, and among them, eight protein spots were increased and two were decreased.
Fig. 3.

Representative 2D map of rat heart from control and stress groups. Proteins (1.5 mg) were loaded on pH 3–10 linear IPG strips (18 cm), with 10%T vertical SDS-PAGE as the second dimension. The gel was visualized by CBB R-250 staining. Spots differentially expressed with p < 0.05 calculated with Student's t test, are indicated
Fig. 4.

Ten differentially expressed proteins from rat heart. Each pair of gel maps is from control and stress groups, respectively. The arrowed and circled spots differ in average ratios (t-test value was P < 0.05; C control group, S stress group)
Identification of differentially expressed myocardial proteins
The differentially expressed proteins were analyzed by MALDI-TOF MS. After database searching, ten protein spots were successfully identified. The proteins are listed in Table 1, including cardiac myosin heavy chain, dihydrolipoamide succinyltransferase component of 2-oxoglutarate dehydrogenase complex, similar to dihydrolipoamide S-succinyltransferase (E2 component of 2-oxoglutarate complex), mitochondrial aldehyde dehydrogenase, mitochondrial aconitase (nuclear aco2 gene), albumin myosin heavy chain, apolipoprotein A-I precursor (Apo-AI), H(+)-transporting ATP synthase, and uncoupling protein 3.
Table 1.
Protein alternations induced by stress (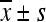 , n = 3)
, n = 3)
| Spot | Protein name | PI | MW | Norm. vol. | |
|---|---|---|---|---|---|
| Control | Stress | ||||
| 1 | Cardiac myosin heavy chain | 7.67 | 76.19 | 0.75 ± 0.24 | 1.54 ± 0.32* |
| 2 | Dihydrolipoamide succinyltransferase component (E2) (E2K) | 7.27 | 62.40 | 0.16 ± 0.08 | 0.57 ± 0.12** |
| 3 | Similar to dihydrolipoamide S-succinyltransferase (E2 component) | 7.09 | 62.64 | 0.32 ± 0.11 | 0.50 ± 0.21* |
| 4 | Mitochondrial aldehyde dehydrogenase | 7.09 | 61.22 | 0.14 ± 0.09 | 0.36 ± 0.10** |
| 5 | Mitochondrial aconitase (nuclear aco2 gene) | 6.93 | 55.61 | 1.29 ± 0.21 | 0.55 ± 0.23* |
| 6 | Albumin | 7.46 | 56.05 | 0.31 ± 0.10 | 1.04 ± 0.21** |
| 7 | Myosin heavy chain, polypeptide 6 | 8.06 | 44.90 | 0.86 ± 0.23 | 1.68 ± 0.48* |
| 8 | Apolipoprotein A-I precursor (Apo-AI) | 7.53 | 30.30 | 0.65 ± 0.22 | 1.39 ± 0.35* |
| 9 | H(+)-transporting ATP synthase | 6.70 | 26.94 | 0.97 ± 0.32 | 1.38 ± 0.46* |
| 10 | Uncoupling protein 3 | 4.35 | 19.02 | 2.15 ± 0.89 | 0.25 ± 0.12** |
*P < 0.05, **P < 0.01 vs control
Protein validation by Western blot
To verify 2DE results, the stressed cardiomyocytes were further analyzed by Western blot. The antibody against UCP3 was used to test whole cell and mitochondrial cardiomyocytes, respectively (Fig. 5a and b). The results from Western blotting showed that restraint stress had varying effects on UCP3 expression in the rat myocardium. Compared with the control, the UCP3 level was increased after 1 week and 2 weeks of restraint stress, but its expression gradually declined with restraint-time prolongation in chronic stress groups with about a 16% and a 40% inhibition of expression, respectively in the 3 weeks and 4 weeks restraint groups (Fig. 5b), which was consistent with the 2DE result (Fig. 4). As an additional evidence, the expression of UCP3 in cardiomyocytes treated with CORT and NE in vitro was significantly degraded (Fig. 5c, d).
Fig. 5.

Changes in UCP3 expression in myocardium and cardiomyocytes under stress. Compared with the control, the results from Western blotting showed a that the UCP3 level declined by 40% after 4 weeks of restraint stress in the rat myocardium (p < 0.05). b The UCP3 levels of mitochondria gradually declined with restraint-time prolongation in chronic stress groups to about 12%, 13%, and 48% inhibition of expression, respectively in the 2 weeks, 3 weeks and 4 weeks restraint groups (p < 0.05). c The UCP3 levels declined approximately to 82% in norepinephrine treated for 12 h (p < 0.05). d The UCP3 level significantly declined to 72% in cardiomyocytes treated with corticosterone (CORT) at 10−4 mol/L concentration for 12 h in vitro (p < 0.05). In all Western blots, uniformity of sample loading was confirmed by probing for tubulin or Cox-2
Discussion
Restraint stress stimulates the generation of ROS and changes mitochondrial membrane potential
Cellular homeostasis can be disrupted by changes in the extra-cellular environment. Such disruption is often accompanied by the undesirable accumulation of metabolic intermediates and the most studied of these are reactive oxygen species produced by disruption of metabolism and the electron transport chain (Korshunov et al. 1997). Mitochondria are considered one of the sources as well as the target organ of ROS, hence, the nature of oxidative damage to mitochondria is being investigated in a variety of organisms (Duchen 2004). In the present study, restraint, a nonspecial stressor, has been used to construct an animal model. We directly measured the ROS and membrane potential of mitochondria by flow cytometry, and the ROS “break out” was observed after 3 weeks of stress treatment (Fig. 1a and b). As oxidative stress markedly sensitizes mitochondria towards PTP induction, it was proposed that mitochondrially generated ROS are directly involved in PTP induction. Hence, we also analyzed the mitochondrial membrane potential (Fig. 2a and b), which putatively reflects the mitochondrial PTP state. Under stress conditions, the level of glucocorticoid will be up-regulated (Zhao et al. 2007). Thus, the cell model of stress stimulation was constructed by treating cardiomyocytes with corticosterone and norepinephrine, respectively. These treatments clearly affected membrane potential and caused ROS accumulation in cells (Figs. 1c and 2c) as treatment was prolonged. The results confirmed that ROS mediated the cell response to restraint stress.
Up-regulation proteins are related to myocardial damage and energy metabolism
In this study, two proteins related to myocardial hypertrophy and four proteins related to energy metabolism were up-regulated in rat myocardia under stress (Table 1). Cardiac myosin heavy chain is the major constituent of myocardial thick myofilament, and the up-regulation of this protein is an adaptive reaction to stress, but it is still the basis of myocardial damage, especially in myocardial hypertrophy (Laredo et al. 2006). Accumulation of apolipoprotein A-I precursor is another basis of myocardial hypertrophy (Hamidi et al. 1999). Dihydrolipoamide succinyltransferase component of 2-oxoglutarate dehydrogenase complex, dihydrolipoamide S-succinyltransferase (E2 component of 2-oxoglutarate complex), and H(+)-transporting ATP synthase are the enzymes of energy metabolism. Energy metabolism in mitochondria is a multistep process which includes the transport of energy substance into the mitochondria, the TCA cycle, electron transport, and ATP synthesis. Mitochondria aldehyde dehydrogenase participates in the deintoxication of acetaldehyde, which can get rid of oxygen free radical production in stress. The up-regulation of energy metabolism protein may be a compensatory mechanism in the face of stress.
Aconitase may be a potential target of ROS attack
Mitochondria appear to be the most powerful intracellular source of ROS. Therefore, mitochondria might also be a primary target for the damaging effects of ROS (Cadenas and Davies 2000). An important mechanism of ROS toxicity is the direct oxidation and inactivation of iron–sulfur (Fe–S) proteins (Fridovich 1997; Iñarrea et al. 2007). In the present study, we found that aconitase was down-regulated more than 2-fold compared with the control. Mitochondrial aconitase was a putative iron–sulfur (Fe–S) protein, which plays a key role in the Krebs cycle, catalyzing the conversion of citrate to isocitrate (Tórtora et al. 2007). Inhibition of mitochondrial aconitase could result in Krebs cycle dysfunction and has an impact on energy production. A recent study further documents that mitochondrial aconitase is associated with protein–mtDNA complexes, called nucleoids. In this novel context, aconitase functions to stabilize mtDNA, perhaps by reversibly remodeling nucleoids to directly influence mitochondrial gene expression in response to changing cellular metabolism (Shadel 2005).
UCP3 protects against myocardial injury induced by restraint stress
Uncoupling proteins (UCPs), made up of four members, are thought to be a family of mitochondrial H+/fatty acid transporters that are expressed in a tissue-specific manner (Ricquier and Bouillaud 2000). UCP1 is present exclusively in brown adipose tissue (BAT). UCP2 is expressed in heart and skeletal muscles. UCP3 expression is largely confined to skeletal muscle and heart, and UCP4 is expressed predominantly in neural tissues (Choi et al. 2007). Previously, UCPs were thought to participate in energy metabolism and in the maintenance of temperature (Lowell 1993). Recently, researchers have become aware of a variety of functions for this protein. Mice made null either for Ucp2 or Ucp3 maintain their body temperature in a cold environment (Vidalpuig et al. 2000). Therefore, unlike UCP1, UCP2 and UCP3 are not involved in cold-induced thermogenesis. In addition, UCP3 has been postulated to dissipate the mitochondrial proton gradient and cause metabolic inefficiency (Barger et al. 2006). These data may be connected to the fact that a basal proton leak in the mitochondrial inner membrane is associated with the resting metabolic rate in most tissues. In this study, we found that the level of UCP3 under stress was about 20-fold lower than in normal conditions. Importantly, the negative relationship between ROS and membrane potential and the level of UCP3 was observed. The ROS decreased as the level of UCP3 increased (data not shown). This indicated that UCP3 may participate in the oxidation stress pathway. It is known that mild uncoupling of respiration diminishes mitochondrial ROS formation by complexes I and III (Ott et al. 2007; Batandier et al. 2004; Muller et al. 2004). The explanation for the control of ROS production by respiration uncoupling is that ROS formation depends on the mitochondrial proton gradient and the mitochondrial potential. In particular, Skulachev (1998) postulated that fatty acids may prevent an increase in the mitochondrial electrochemical gradient and thus decrease ROS generation. In other words, a mild uncoupling of respiration may participate in antioxidant defense and the UCPs may be the effectors of such a defense mechanism. This may explain why UCP3 have double acting in cardiocyte as we have shown in the present study.
Acknowledgment
This work was supported in part by grants from the National Natural Science Foundation of China (No. 30770843, No. 30430590).
Conflict of interest None declared.
Abbreviations
- ROS
reactive oxygen species
- NE
norepinephrine
- MEM
minimum essential medium
- PTP
permeability transition pore
- 2-DE
two-dimensional gel electrophoresis
- UCP3
uncoupling protein 3
Footnotes
Xinxing Wang and Jingbo Gong contributed equally to this work.
References
- Barger JL, Barnes BM, Boyer BB. Regulation of UCP1 and UCP3 in arctic ground squirrels and relation with mitochondrial proton leak. J Appl Physiol. 2006;101:339–347. doi: 10.1152/japplphysiol.01260.2005. [DOI] [PubMed] [Google Scholar]
- Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol chem. 2004;279(17):17197–17204. doi: 10.1074/jbc.M310329200. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/S0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Choi CS, Fillmore JJ, Kim JK. Overexpression of uncoupling protein 3 in skeletal muscle protects against fat-induced insulin resistance. J Clin Invest. 2007;117:1995–2003. doi: 10.1172/JCI13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria in health and disease: perspectives on new mitochondrial biology. Mol Aspects Med. 2004;25:365–451. doi: 10.1016/j.mam.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Young PR. Apoptosis in cardiac diseases: stress- and mitogen-activated signaling pathway. Cardiovasc Res. 2000;45:560–569. doi: 10.1016/S0008-6363(99)00372-7. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Superoxide anion radical (O2·−), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- Galea LAM, McEwen BS, Tanapat P. Sex differences in dendritic atrophy of CA3 pyramidal neurons in response to chronic restraint stress. Neurosci. 1997;81:689–697. doi: 10.1016/S0306-4522(97)00233-9. [DOI] [PubMed] [Google Scholar]
- Hamidi AL, Liepnieks JJ, Hamidi AK. Hereditary amyloid cardiomyopathy caused by a variant apolipoprotein A1. Am J Pathol. 1999;154(1):221–227. doi: 10.1016/S0002-9440(10)65268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñarrea P, Moini H, Han D, Cadenas E. Mitochondrial respiratory chain and thioredoxin reductase regulate intermembrane Cu, Zn-superoxide dismutase activity: implications for mitochondrial energy metabolism and apoptosis. Biochem J. 2007;405(1):173–179. doi: 10.1042/BJ20061809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut P, Thiede B. Protein identification from 2-DE gels by MALDI mass spectrometry. Mass Spectrom Rev. 1997;16(3):145–162. doi: 10.1002/(SICI)1098-2787(1997)16:3<145::AID-MAS2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/S0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- Laredo R, Monserrat L, Hermida-Prieto M. Beta-myosin heavy-chain gene mutations in patients with hypertrophic cardiomyopathy. Rev Esp Cardiol. 2006;59(10):1008–1018. doi: 10.1157/13093977. [DOI] [PubMed] [Google Scholar]
- Liu X, Qian L, Gong J, Shen J, Zhang X, Qian X. Proteomic analysis of mitochondrial proteins in cardiomyocytes from chronic stressed rat. Proteomics. 2004;4(10):3167–3176. doi: 10.1002/pmic.200300845. [DOI] [PubMed] [Google Scholar]
- Liu F, Jiang YY, Liu SY, Wang K, Cao J, Zhao YF. Inducement effect of (DIPP-L-Trp)2-L-Lys-OCH3 on apoptosis of K562 cells through mitochondrial-dependent pathway. Ai Zheng. 2005;24(4):448–453. [PubMed] [Google Scholar]
- Lowell BB. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- Marcil M, Bourduas K, Ascah A, Burelle Y. Exercise training induces respiratory substrate-specific decrease in Ca2+-induced permeability transition pore opening in heart mitochondria. Am J Physiol Heart Circ Physiol. 2006;290:1549–1557. doi: 10.1152/ajpheart.00913.2005. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Liu D. Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem Biophys Res Commun. 2003;304(3):539–549. doi: 10.1016/S0006-291X(03)00627-2. [DOI] [PubMed] [Google Scholar]
- Muller FL, Liu Y, Van RH. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial membrane potential and aging. Aging Cell. 2004;3(1):35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- Perier C, Tieu K, Guegan C. Complex I deficiency primes Bax dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Song X, Ren H, Gong J, Suqi C. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones. 2004;9:281–293. doi: 10.1379/CSC-20R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raquel P, María GF, Matías DS, Juan EP, Gloria D, Marian C, Jordi M, Inma CC. Mitochondrial protection by low doses of insulin-like growth factor-Iin experimental cirrhosis. World J Gastroenterol. 2008;14(17):2731–2739. doi: 10.3748/wjg.14.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000;2:161–179. doi: 10.1042/0264-6021:3450161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem. 1992;203(1):173–179. doi: 10.1016/0003-2697(92)90061-B. [DOI] [PubMed] [Google Scholar]
- Shadel GS. Mitochondrial DNA, aconitase ‘wraps’ it up. Trends Biochem Sci. 2005;30(6):294–296. doi: 10.1016/j.tibs.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- Tórtora V, Quijano C, Freeman B. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med. 2007;42(7):1075–1088. doi: 10.1016/j.freeradbiomed.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Vidalpuig AJ, Grujic D, Zhang CY. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 2000;275:16258–16266. doi: 10.1074/jbc.M910179199. [DOI] [PubMed] [Google Scholar]
- Wang XX, Liu XH, Kong RR, Zhan R, Wang XM, Leng X, Gong JB, Duan M, Wang LQ, Wu L, Qian LJ. NGFI-B targets mitochondria and induces cardiomyocyte apoptosis in restraint-stressed rats by mediating energy metabolism disorder. Cell Stress Chaperones. 2009;14(6):639–648. doi: 10.1007/s12192-009-0116-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang W, Qian L. Hsp70 may protect cardiomyocytes from stress-induced injury by inhibiting Fas-mediated apoptosis. Cell Stress Chaperones. 2007;12(1):83–95. doi: 10.1379/CSC-231R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]