

Abstract
Rationale and objectives
The observed relaxivity of gadolinium based contrast agents has contributions from the water molecule(s) that bind directly to the gadolinium ion (inner-sphere water), long lived water molecules and exchangeable protons that make up the second-sphere of coordination, and water molecules that diffuse near the contrast agent (outer-sphere). Inner- and second-sphere relaxivity can both be increased by optimization of the lifetimes of the water molecules and protons in these coordination spheres, the rotational motion of the complex, and the electronic relaxation of the gadolinium ion. We sought to identify new high relaxivity contrast agents by systematically varying the donor atoms that bind directly to gadolinium to increase inner-sphere relaxivity and concurrently including substituents that influence the second-sphere relaxivity.
Methods
Twenty GdDOTA derivatives were prepared and their relaxivity determined in presence and absence of human serum albumin as a function of temperature and magnetic field. Data was analyzed to extract the underlying molecular parameters influencing relaxivity. Each compound had a common albumin-binding group and an inner-sphere donor set comprising the 4 tertiary amine N atoms from cyclen, an α-substituted acetate oxygen atom, two amide oxygen atoms, an inner-sphere water oxygen atom, and a variable donor group. Each amide nitrogen was substituted with different groups to promote hydrogen bonding with second-sphere water molecules.
Results
Relaxivites at 0.47T and 1.4T, 37 °C, in serum albumin ranged from 16.0 to 58.1 mM−1s−1 and from 12.3 to 34.8 mM−1s−1 respectively. The reduction of inner-sphere water exchange typical of amide donor groups could be offset by incorporating a phosphonate or phenolate oxygen atom donor in the first coordination sphere resulting in higher relaxivity. Amide nitrogen substitution with pendant phosphonate or carboxylate groups increased relaxivity by as much as 88% compared to the N-methyl amide analog. Second-sphere relaxivity contributed as much as 24 mM−1s−1 and 14 mM−1s−1 at 0.47 and 1.4T respectively.
Conclusions
Water/proton exchange dynamics in the inner- and second-coordination sphere can be predictably tuned by choice of donor atoms and second-sphere substituents resulting in high relaxivity agents.
Keywords: Relaxivity, gadolinium, second-sphere, protein binding, water exchange, NMRD
INTRODUCTION
Magnetic resonance (MR) imaging contrast agents are characterized by their relaxivity. Relaxivity is the degree to which the agent can enhance the longitudinal or transverse water relaxation rate constant (R1 = 1/T1 or R2 = 1/T2, respectively) normalized to concentration of the contrast agent. Longitudinal and transverse relaxivity are denoted r1 and r2, respectively. Gadolinium-based contrast agents are generally referred to as T1 agents, because on a percentage basis they have a larger effect on tissue T1 than on T2. Reducing tissue T1 results in positive image contrast in T1-weighted images and gadolinium (Gd) contrast agents are widely used in clinical practice to enhance tumors, the blood pool, and myocardial infarcts, to list a few applications. Because Gd agents are most often used for T1 relaxation, longitudinal relaxivity is often simply referred to as “relaxivity” and this will be used here. Equation 1 shows the relationship between Gd concentration and relaxivity on tissue relaxation rate constants, where [CA] is the concentration of the contrast agent and Ti0 is the relaxation time of the tissue in the absence of contrast agent.
| (1) |
Relaxivity is a measure of the sensitivity of the contrast agent. For compounds with similar distribution, a compound with higher relaxivity would provide equivalent contrast at a lower dose compared to a low relaxivity compound. A lower dose may lower the risk of gadolinium (Gd)-induced toxicity, which can manifest itself in nephrogenic systemic fibrosis (NSF).1–3 In addition to safety, molecular imaging is driving improvements in relaxivity.4–7} For targeted contrast agents, higher relaxivity would allow lower concentration targets to be detected by MRI. Indeed, this quest for higher relaxivity contrast agents was recognized in the 1980’s as the utility of the first contrast agents became apparent.8–10 Another driver for identification of high relaxivity chelates is cost of production. Newer agents with complex molecular structures, e.g. peptides conjugated to multiple gadolinium chelates,11–16 require more synthetic steps to prepare compared to first generation agents like GdDTPA; a lower dose enabled by high relaxivity may make such agents more cost effective to manufacture.
For gadolinium (Gd)-based contrast agents, relaxivity depends on extrinsic factors like applied field and temperature and also on the molecular properties of the contrast agent.8,10,17–20 At common clinical field strengths (0.5 – 3T), the relaxivity of rapidly tumbling, small molecules like GdDTPA or GdDOTA depends on the hydration state of the complex and how fast the complex tumbles. Hydration refers to the number of water molecules in the inner-coordination sphere (i.e. the number directly bonded to the Gd and denoted q), as well as the number of water molecules or exchangeable hydrogen atoms in the second and subsequent coordination spheres. The tumbling of the complex creates a fluctuating magnetic field and this fluctuating field induces relaxation of water molecules. For compounds tumbling isotropically this motion is described by a characteristic rotational correlation time, τR. Relaxation will be most effective when the frequency of the fluctuating field (1/τR for small molecules) becomes close to the Larmor frequency. Small molecules tumble with gigahertz rates while the Larmor frequency of clinical scanners is in the 20 – 130 MHz range. It has long been recognized that slowing down rotation is an effective way to increase relaxivity.10,21 This can be done by increasing the size of the contrast agent by incorporating it into a dendrimer22 or polymer, 23,24 or by binding covalently9,25,26 or non-covalently to a protein.27,28
When the tumbling rate is slowed, other molecular factors may begin to limit relaxivity. For example the rate of water exchange (kex = 1/τm, where τm is the lifetime of the water in the inner-sphere) may be too slow to affect a large fraction of bulk water. Alternately the water molecule may not spend enough time in the inner- or second-coordination sphere to have a high probability of being relaxed (τm is too short). Another limiting factor is the rate of electronic relaxation of the Gd3+ ion. Electronic relaxation also creates a fluctuating field and this fluctuation may be too fast so as to limit relaxivity. Because limiting effects of inner- and second-sphere water exchange and electronic relaxation are only observed when the complex is tumbling slowly, it is very difficult to identify new gadolinium chelates that may provide high relaxivity. Synthesizing and screening small molecules for relaxivity will not reflect the relaxivities that may be achieved if these new chelates were to be conjugated to a protein or polymer.
In a companion paper,29 we reported a series of GdDOTA derivatives. Those compounds all had a common functional group that provided high affinity to serum albumin and this allowed relaxivity to be determined in a fast tumbling (no albumin) and slow tumbling (albumin bound) state. By changing one of the donor groups that bind to gadolinium, the relaxivity at 37 °C, 0.47T in albumin solution ranged from 12.3 to 55.6 mM−1s−1. Further analysis showed that these differences were largely a result of differences in the inner-sphere water exchange rate. This single donor group modification resulted in water exchange rates that differed over 3 orders of magnitude. Donor groups increased water exchange rate in the order: phosphonate ~ phenolate > α-substituted acetate > acetate > hydroxamate ~ sulfonamide > amide ~ pyridyl ~ imidazole. In that paper, the optimal exchange rate was observed when the donor groups were the 4 cyclen nitrogen atoms, two acetate oxygen atoms, and two α-substituted acetate oxygen atoms.
We noted that when the donor group was an amide oxygen, the relaxivity strongly depended on the amide substituent. For instance the amide with a CH2COOH substituent had a 30% higher relaxivity than the amide with a CH3 substituent. Chain length was also important since the CH2(CH2)nCOOH (n=1,2) did not show this increased relaxivity. We also noted an increased relaxivity when the amide substituent was CH2PO3H2. This is most likely a result of the carboxylate or phosphonate forming strong hydrogen bonding interactions with water molecule(s) in the second hydration sphere. Indeed, this interaction increases the lifetime of water molecule(s) in the second sphere thereby increasing the probability that they will be relaxed. Phosphonates are known from the literature to promote these second-sphere effects.30–33 The GdDOTA derivative, Gd(DOTA-4AMP), where there are four amide donors with pendant methylphosphonate substituents shows relatively high relaxivity that is almost solely due to a second-sphere effect.34–36 Notable second-sphere effects have also been reported with pendant carboxylate groups.37
The drawback of using substituted amide donors to increase second-sphere relaxivity is that the amide group slows down the water exchange rate of the inner-sphere water molecule.38,39 So although relaxivity may be increased through second-sphere effects, this is offset by a lower contribution to relaxivity from the inner-sphere water. In this work, we sought to test the hypothesis that inner-sphere water exchange at GdDOTA derivatives was an additive effect of the donor atom types. That is, if replacement of a carboxylate donor group with an amide donor group is known to slow water exchange, but replacement with a phenolate donor group increases water exchange, would incorporation of a water exchange promoting phenolate group offset the water exchange retarding effect of an amide? In this way, could the inner-sphere water exchange rate of a complex containing amide donors be increased?
Based on this hypothesis, we prepared a series of complexes based on the thermodynamically stable GdDOTA complex. Each of the complexes has a common, high affinity serum albumin binding group attached via a substituted acetate (Figure 1). Two amide donor groups are positioned at N4 and N10 on the cyclen ring, and the amide substituents are systematically varied to increase second-sphere relaxivity. One donor group at N7 in the cyclen ring is systematically varied to explore the effect of donor group on inner-sphere water exchange kinetics. By varying the N7 donor group and the amide substituents we sought to optimize both inner- and second-sphere relaxivity. The compounds were screened by measuring relaxivity at 0.47 and 1.4T in the presence (immobilized) and absence of human serum albumin. A subset of the complexes were further studied by variable temperature proton nuclear magnetic relaxation dispersion (NMRD) to extract the molecular determinants of relaxivity for this class of compounds.
Figure 1.

General approach to identifying high relaxivity complexes with enhanced inner- and second-sphere relaxivity.
MATERIALS AND METHODS
Compound synthesis
Compounds used in this report have been described in a patent application.40 The synthetic procedure was similar for all compounds and a representative synthesis of Gd3-asp is described in detail (see Figures 2 and 3 for scheme and numbering).
Figure 2.

General synthetic scheme for the compounds described in this report.
Figure 3.

Chemical structures of the compounds studied in this report arranged by different donor groups.
Synthesis of (S)-2-(2-bromoacetylamino)succinic acid di-tert-butyl ester, 6
Bromoacetyl bromide (2.57 g, 12.8 mmol, 1.2 eq.) was added to a solution of L-aspartic acid β-t-butyl α-t-butyl ester hydrochloride (3.00 g, 10.6 mmol, 1 eq.) and triethylamine (2.5 g, 24.7 mmol, 2.3 eq.) in 50 mL CH2Cl2 at −40°C over 5 minutes. The brown solution was warmed up to room temperature (RT) and stirred for 2 hours. The solvent was evaporated and the residue was partitioned between 0.2N KHSO4 and ether. The aqueous layer was extracted with ether and the organic layers were combined and washed successively with water (2 × 50 mL), saturated NaHCO3 (50 mL) and brine (20 mL). The organic layer was dried over Na2SO4 in the presence of decolorizing activated charcoal. Filtration and evaporation of solvent gave 4.10 g of the crude desired product as a deep brown oil, which was purified by flash chromatography on silica gel (hexanes/EtOAc 75:25 to 70:30 to 65:35) to give 2.92 g (63%) of bromoacetamide 6 as a brown oil. 1H NMR (300 MHz, CDCl3): δ 7.42 (br d, J = 8.1 Hz, 1H), 4.6–4.7 (m, 1H), 3.82–3.95 (m, 2H), 2.92 (dd, J1 = 4.2 Hz, J2 = 17.1 Hz, 1H), 2.73 (dd, J1 = 4.2 Hz, J2 = 17.1 Hz, 1H), 1.47 (s, 9 H), 1.45 (s, 9 H).
Synthesis of (S)-2-({7-[(2-(S)-tert-butyl aspartate-carbamoyl)-methyl]-1,4,7,10-tetraazacyclododec-1-yl}-acetylamino)-succinic acid di-tert-butyl ester 8
1,7-Bis-Cbz cyclen (1.0 g, 2.3 mmol, 1 eq.) and (S)-2-(2-bromo-acetylamino)-succinic acid di-tert-butyl ester 6 (1.92 g, 5.2 mmol, 2.3 eq.) were dissolved in 15 mL acetonitrile and Na2CO3 (2.8 g, 26.4 mmol, 11.7 eq.) was added. The reaction mixture was microwaved for 5 minutes at 120°C. The solvent was evaporated. The residue was dissolved in ether (150 mL) and washed successively with 0.2N KHSO4 and with brine. The organic layer was dried over Na2SO4. Evaporation of solvent gave 3.01 g of the crude desired bis-amide 7 which was purified by flash chromatography on silica gel (CH2Cl2/Hex/MeOH: 50:50:0 to 100:0:0 to 97:0:3 to 96:0:4) to give 1.95 g (85%). MS [M+1] = 1011.7
Removal of Cbz groups of protected bis-amide 7 (1.95g, 1.9 mmol) was performed in EtOH (100 mL) using 20% Pd(OH)2/C (0.4 g) under 45 psi H2. Filtration of catalyst and evaporation of solvent gave 1.49 g the desired product 8 as an oil (crude yield 104%). MS: [M+1] = 743.5
Synthesis of (R)-2-{4,10-bis-[(S)-(tert-butyl aspartate-carbamoyl)-methyl]-1,4,7,10-tetraazacyclododec-1-yl}-5-[tert-butoxy-(2,5,2′,4′,6′-pentamethyl-biphenyl-4-yloxy)-phosphoryloxy]-pentanoic acid tert-butyl ester 9
Tri-substituted macrocycle 9 was prepared by stirring the corresponding bis-amide 8 (0.41 g, 0.55 mmol, 1 eq.) with (S)-5-[tert-butoxy-(2,5,2′,4′,6′-pentamethyl-biphenyl-4-yloxy)-phosphoryloxy]-2-(4-nitrobenzenesulfonyloxy)-pentanoic acid tert-butyl ester (“nosylate” from ref 29, 0.21 g, 0.29 mmol, 0.53 eq.) and Na2CO3 (1.20 g, 11.3 mmol, 20 eq.) for 15 hours at RT and at 50°C for 24 hours. The solvent was evaporated and the residue was dissolved in EtOAc (15 mL) and washed with saturated NaHCO3 and brine. The organic layer was dried over Na2SO4 and evaporated to give 0.33 g of the desired product 9 (crude yield: 90%). MS: [M+1] = 1274.9
Synthesis of (R)-2-{7-(2-methoxymethoxy-5-nitrobenzyl)-4,10-bis-[(tert-butyl aspartate-carbamoyl)-methyl]-1,4,7,10-tetraazacyclododec-1-yl}-5-[tert-butoxy-(2,5,2′,4′,6′-pentamethyl-biphenyl-4-yloxy)-phosphoryloxy]-pentanoic acid tert-butyl ester 10
The trisubstituted macrocycle 9 (160 mg, 0.12 mmol, 1 eq.), 2-methoxymethoxy-5-nitrobenzaldehyde (116 mg, 0.55 mmol, 4.6 eq.) and NaBH(OAc)3 (116 mg, 0.55 mmol, 4.6 eq.) were dissolved in 0.6 mL of dichloroethane in a 20 mL vial and the mixture was shaken on a orbital shaker for 24 hours. The crude reaction mixture was purified by flash chromatography on silica gel (CH2Cl2/MeOH/Et3N 100:0:0 to 95:5:0.5 to 90:10:0.5) to give 40 mg of the desired product 10 (22%). MS: [M+1] = 1470.9.
Synthesis of (R)-2-{4,10-bis-[(aspartate-carbamoyl)-methyl]-7-(2-methoxymethoxy-5-nitrobenzyl)-1,4,7,10-tetraazacyclododec-1-yl}-5-[hydroxy-(2,5,2′,4′,6′-pentamethyl-biphenyl-4-yloxy)-phosphoryloxy]-pentanoic acid 3-asp
Protected ligand 10 (40 mg, 0.027 mmol) was dissolved in 10 mL of a deprotection cocktail (95 parts trifluoroacetic acid (TFA): 2.5 parts methanesulfonic acid: 2.5 parts phenol) and the reaction mixture was stirred at RT for 3 hours. After precipitation from ether 40 mg of crude ligand 3-asp was obtained. MS: [M+1] = 1089.3.
Synthesis of Gd3-asp
The ligand 3-asp was dissolved in H2O and the pH adjusted to 6 with a 1N NaOH solution. A 245 mM solution of GdCl3 was added (1 eq based on ligand weight) and the pH adjusted to 6. The reaction mixture was stirred at 45°C for 5 hours and the chelation monitored by LC-MS. EDTA solution was added to chelate the excess Gd3+ and the product was purified by preparative-HPLC on a C-18 column (reversed-phase HPLC, RP-HPLC) using a gradient of 50 mM aqueous ammonium formate and acetonitrile. After lyophilization of the pure fractions, Gd3-asp (7 mg, 95% purity by HPLC) was obtained. MS: [M+1] = 1242.3 with expected isotopic mass distribution.
Sample preparation and concentration determination
Human serum albumin (Fraction V, Sigma) was dissolved in 50 mM HEPES buffered saline (HBS) of 150 mM sodium chloride content (pH 7.4) to reach a final protein solution of ~6% (w/v). Protein concentration was estimated from absorbance at 280 nm compared to a series of albumin standards (Sigma). This stock solution was used to prepare 4.5% (w/v) HSA solutions (0.67 mM) by appropriate dilution with a gadolinium chelate stock solution (5 – 10 mM) and HBS. For relaxivity determinations, the Gd concentration ranged from 0 – 0.1 mM and for nuclear magnetic relaxation dispersion (NMRD) and protein binding studies the Gd concentration was 0.1 mM. All gadolinium concentrations were determined by inductively coupled plasma mass spectroscopy (ICP-MS) on an Agilent 7500a system.
Relaxivity
Water proton relaxation rates (1/T1 and 1/T2) were determined at 20 MHz (0.47 T) and 60 MHz (1.41 T) using a Bruker NMS 120 Minispec and a Bruker mq60 Minispec, respectively. Samples, 200 μL, were pipetted into glass vials and were equilibrated for at least 20 minutes at 37°C before being measured at 37°C. T1 was measured with an inversion recovery pulse sequence using 10 different TI times; the pre-acquisition delay was always set to at least 5T1. T2 was measured using a Carr Purcell Meiboom Gill (CPMG) sequence with 300 to 500 echoes collected. The 1H NMRD profiles were recorded at 5°, 15°, 25°, and 35°C on a field cycling relaxometer41 at NY Medical College over the frequency range 0.01–50 MHz (0.0002 – 1.2 tesla). Each dispersion curve consisted of a total of 22 data points.
RESULTS
Synthesis
Synthesis of this two-variable library of compounds followed a common route as outlined in Figure 2. First, if it was not commercially available, the substituted amide “arm” was prepared by acylation of the appropriate amine with 2-bromoacetylbromide. A slight excess of the amide arm was used to dialkylate 1,7-bis(Cbz)-protected cyclen. The Cbz groups were readily removed by hydrogenation with a palladium catalyst. The synthesis of the biphenyl phosphate albumin-binding group was described previously.42 The nosylate of this albumin-binding moiety was prepared,29 and used to alkylate a 3rd cyclen nitrogen. At this stage, the donor group (D1) was introduced. To prepare the phenolate, we used reductive amination. When D1 was a phosphonate, acetate, or amide, these groups were introduced via alkylation reactions from a precursor with a triflate or bromide leaving group (see D1 precursors in Figure 2). The methylphosphinate was prepared by reaction of the secondary amine with diethyl methylphosphonite and paraformaldehyde. Further conversion to the gadolinium complex followed three common steps: acid deprotection of the tert-butyl groups, neutralization, metallation with gadolinium chloride, then preparative HPLC to give the final purified compound. Compounds were assessed for purity by LC-MS and stock solutions then prepared for biophysical studies with accurate gadolinium concentration determined by ICP-MS.
A range of compounds were prepared and analyzed and are shown in Figure 3. For D1 donor groups we focused on groups that were previously shown29 to increase inner-sphere water exchange at GdDOTA derivatives: phosphonates, phenolates, phosphinates, and alpha-substituted acetates. As controls we also prepared examples where D1 was an unsubstituted acetate (Gd1a-gly) or an amide (Gd4-gly) where the inner-sphere water exchange rate may be expected to be slow. To promote second-sphere relaxivity, we built on our original observation that the CH2COOH substitution gave enhanced relaxivity. Such an amide is derived from glycine (NH2CH2COOH) and these are denoted “gly”. We formed amides from iminodiacetic acid (ida) and from aspartic acid (asp) in order to assess whether additional pendant carboxylates could further enhance relaxivity. Methyl (me) and dimethylamides (dme) were prepared as controls. We also assessed other amide substituents like trifluoromethyl (CF3) and methylphosphonate esters (PEtOH, PEt2).
Relaxivity
Longitudinal proton relaxation rates (R1) were measured in HEPES buffer, pH 7.4, 37 °C at two proton Larmor frequencies, 20 MHz and 60 MHz corresponding to 0.47 and 1.41T, respectively. Measurements were made with gadolinium concentrations ranging from 0 to 0.1 mM and either no HSA or with 0.67 mM HSA. Relaxivity (r1) was calculated from the slope of the R1 dependence on concentration. Relaxivities in HSA reported are all “observed” relaxivities, i.e. the relaxivity obtained from the R1 dependence and not calculated to reflect the relaxivity of the protein-bound species. However this protein binding group results in a very high fraction bound to HSA under the conditions used (>98%) and the observed relaxivity is essentially that of the protein-bound species.
Figure 4 shows the relaxivity of these compounds in HEPES buffer at both 0.47 and 1.4T. The relaxivities range from 5.6 to 11.3 mM−1s−1. As expected for fast tumbling complexes, the relaxivities at both fields for a given compound were similar. In the fast tumbling regime, relaxivity will be unaffected by differences in inner-sphere water residency times (τm), unless τm is very long (>1 μs). This data set should be sensitive to differences in τR, q, or second-sphere effects. Inspection of Figure 4 reveals some obvious trends. Complexes with two pendant carboxylates per amide (ida and asp) show considerably higher relaxivity than compounds with the same D1 group but a different amide substituent. For instance Gd2-asp has a 96% higher relaxivity at 1.4T than the dimethylamide complex Gd2-dme. The much higher relaxivities for the asp and ida derivatives cannot be rationalized by slight differences in molecular weights. These differences are likely due to second-sphere relaxation wherein the carboxylates form strong hydrogen bonds with hydrating water molecules. For common amide substituents, there is a clear influence on D1 donor group and relaxivities increase as D1 = phosphonate > phenolate > alpha-substituted acetate. Phosphonate donors are known to enhance second-sphere relaxivity32,33 and that is likely that the phosphonate donor contributes to increased second-sphere relaxivity in these compounds as well.
Figure 4.
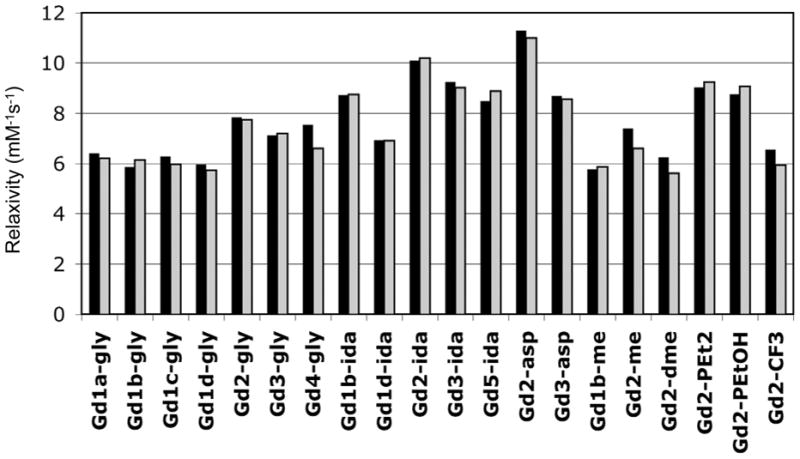
Relaxivities determined in HEPES buffer, pH 7.4, 37 °C at 0.47T (black bars) and 1.4T (grey bars).
Figure 5 shows the relaxivity of these compounds in HSA solution at 0.47 and 1.4T grouped by amide substituent to illustrate the effect of the D1 donor group. The highest relaxivity was observed for Gd3-ida which had a relaxivity of 58.1 mM−1s−1 at 0.47T, 37 °C. For a given amide substituent, D1 = phenolate gave the highest relaxivity at 0.47T followed by D1 = phosphonate, but at 1.4T the relaxivities were similar for the phenolate and phosphonate derivatives. The acetate derivatives gave lower relaxivities and, as expected, when D1 = amide the relaxivity was quite poor likely due to very slow inner-sphere water exchange. Only one phosphinate derivative was prepared, Gd5-ida, but its relaxivity was comparable to the substituted acetates. Among the substituted acetate donors tested, the highest relaxivities were seen when the α-substituent was a carboxylate (Gd1d). The relaxivities of Gd1d-gly and Gd1d-ida were 30% and 20% higher at 0.47T than those of Gd1b-gly and Gd1b-ida (α-substituent = isopropyl) respectively. We speculate that the higher relaxivity observed for the Gd1d derivatives may also be due to a second-sphere effect.
Figure 5.

Relaxivities determined in HSA solution (0 < Gd < 0.1 mM; 0.67 mM HSA) at 37 °C at 0.47T (black bars) and 1.4T (grey bars) grouped by: A) glycyl amide substituent (gly); B) iminodiacetate amide substituent (ida) or aspartyl amide substituent (asp); C) other amide substituents.
Figure 6 shows the relaxivities measured in HSA grouped according to donor type to illustrate the impact of the amide substituent on relaxivity. Similar to what was observed in HEPES buffer, the gly, ida, and asp substitutions have a very positive effect on relaxivity. For Gd1b and Gd2 methyl amides were synthesized giving a clear view of the impact of the second-sphere effect provided by the pendant carboxylates. At 0.47T, relaxivities of Gd1b-gly and Gd1b-ida were 12% and 38% higher than that of Gd1b-me, respectively. For the phosphonate derivatives (Gd2), the relaxivities of the gly, ida, asp, PEt2, and PEtOH derivatives were 45 – 88% higher than Gd2-me at 0.47T and 46 – 69% higher at 1.4T. For the phenolate derivatives (Gd3), a methylamide control was not prepared, but it is likely that the very high relaxivities observed for Gd3-gly, Gd3-ida, and Gd3-asp are partially due to a large second-sphere effect.
Figure 6.

Relaxivities determined in HSA solution (0 < Gd < 0.1 mM; 0.67 mM HSA) at 37 °C at 0.47T (black bars) and 1.4T (grey bars) grouped by: A) acetate or substituted acetate donor group; B) phosphonate donor group; C) phenolate (3), amide (4) or phosphinate (5) donor group.
Nuclear Magnetic Relaxation Dispersion (NMRD)
Nuclear magnetic relaxation dispersion (NMRD) is a measure of relaxivity (or relaxation rate) as a function of magnetic field or Larmor frequency. To better understand the differences in relaxivity among these compounds, NMRD profiles of 9 of these compounds (0.1 mM) in HSA solution (0.66 mM, pH 7.4) were recorded each at 4 temperatures: 5, 15, 25, and 35 °C. Since the HSA solutions alone also show a field dependence on relaxation rate,41 the NMRD relaxivity was calculated for each magnetic field and temperature using equation 1, and subtracting the appropriate diamagnetic relaxation rate for HSA solution at that same field and temperature.
Relaxivity can be factored into contributions arising from the water molecule in the inner-sphere (IS, directly bonded to Gd3+), long-lived water molecules in the second-sphere (SS, lifetime greater than diffusion time constant), and from water molecules diffusing near the complex (OS, outer-sphere), as shown in equation 2. Here q represents the number of water molecules in the inner-sphere and q′ is the number of water molecules in the second sphere. These water hydrogen atoms in the inner- and second-sphere have relaxation times T1m and T′1m, respectively, and residency times denoted τm and τ′m respectively. For slow tumbling complexes such as those described here, relaxation of the inner-sphere water is fairly efficient and T1m is comparable to τm. When the temperature is lowered, τm will increase but T1m will decrease; depending on which is longer, relaxivity will either increase or decrease. Analysis of variable temperature NMRD of slow tumbling complexes can result in an estimation of the water residency time, τm.
| (2) |
At higher fields (ν ≥ 6 MHz) the relaxation time of the inner-sphere water hydrogen atoms is given by equation 3. At these fields, the contribution to electronic relaxation from the static zero-field splitting (ZFS) is negligible and use of modified Solomon-Bloembergen-Morgan theory is appropriate.17,43 For albumin-bound complexes it is necessary to apply the Lipari-Szabo modification of the spectral density term to account for internal motion.44–46 At these proton frequencies (6 MHz and up), the contribution to relaxation dependent on the electronic Larmor frequency, ωs, has dispersed and the spectral density term is simplified. The relevant correlation times, τc and τf, contain contributions from electronic relaxation (T1e), rotation, and exchange (equations 4 and 5). In the Lipari-Szabo model, τc takes into account the global motion of the albumin-bound complex and τf is a correlation time that takes into account fast local motion (τl is a correlation time for fast motion).
| (3) |
| (4) |
| (5) |
F2 is an order parameter (often denoted S2, but here F is used to avoid confusion with the spin quantum number S) that can range from 0 (local motion completely decoupled from global motion) to 1 (isotropic global motion with no contribution from local motion). ωH is the Larmor frequency of the proton (rad/s), γH is the proton magnetogyric ratio, ge is the electronic g-factor (ge = 2 for Gd(III)), μB is the Bohr magneton, and μ0 is the permittivity of vacuum. The field dependence on T1e is given by equation 6.
| (6) |
The relaxation of the second-sphere water hydrogen atoms is also described by equation 3, except the Gd-H distance will be different and the correlation time τ′m replaces τm. The various correlation times should all be temperature dependent. The NMRD data has strong temperature and field dependencies but there are too many molecular parameters to model this data exactly. However, in order to gain some insight into the differences in relaxivity observed among these compounds, we modeled the data in two ways.
In the first analysis, we fit the data using the smallest number of parameters that would provide an adequate fit. To estimate the outer-sphere contribution to relaxivity at each temperature and frequency we assumed it was the same as that of a q=0 GdDOTA derivative bound to albumin reported previously.47 The distance rGdH for the inner-sphere water was set to 3.1Å based on numerous ENDOR studies on frozen solutions of gadolinium complexes that showed that this distance did not vary among 8 and 9 coordinate Gd(III) complexes.47–49 Because rotation is already slow due to protein binding, the data could be well fit with a single, temperature independent τR (i.e. the temperature dependence on τR was not well defined). Similarly it is known from previous work that τv has a weak temperature dependence38,44 and we found that all the variable temperature NMRD data could be well fit with a single τv. We also found that the data was insensitive to the fast internal motion correlation time τl, which was always very short (≤100 ps). Under these conditions the (1−F2)τf/(1+ωH2τf2) term tends to zero and introducing a term for the temperature dependence of τl is meaningless. The data are however very sensitive to τm and its temperature dependence is given by equation 7 where τm310 is the water residency time at 37 °C and ΔH‡ is the activation energy for water exchange. We did not include additional parameters to account for second-sphere relaxivity in this first analysis. Second-sphere effects would be reflected in a larger value of the order parameter, F2.
| (7) |
Altogether the variable temperature NMRD data (4 temperatures, 60 data points) could be well fit using 6 parameters: τR, τv, τm310, ΔH‡, Δ2, and F2. NMRD data for each compound was simultaneously fit to equations 2 – 7. Examples of the observed NMRD relaxivities and the fitted data are shown in Figure 7. Figure 7 shows the impact of the donor group, D1, on relaxivity for 3 compounds where the amide substituent is CH2COOH (gly). Figure 7A shows relaxivity of Gd1b-gly (α-substituted acetate donor) where the relaxivity is decreasing slightly as temperature decreases. Second- and outer-sphere relaxivity should increase at lower temperatures, so this indicates that slow exchange of the inner-sphere water is limiting relaxivity. This is also suggested by the rather flat NMRD profile. Figure 7B shows NMRD data for the phosphonate donor Gd2-gly where relaxivity is increasing markedly as the temperature is decreased. This indicates that the system is in fast exchange (T1m > τm). At 5 °C very high relaxivities were observed with this compound but at 35 °C, relaxivity appears limited by water exchange that is too fast. Figure 7C shows data for the phenolate donor Gd3-gly where relaxivity is also increasing with decreasing temperature, although the temperature dependence is not as marked as that observed for Gd2-gly with a phosphonate donor. This also suggests that the system is in fast exchange and that relaxivity at 35 °C is limited by water exchange that is slightly too fast.
Figure 7.
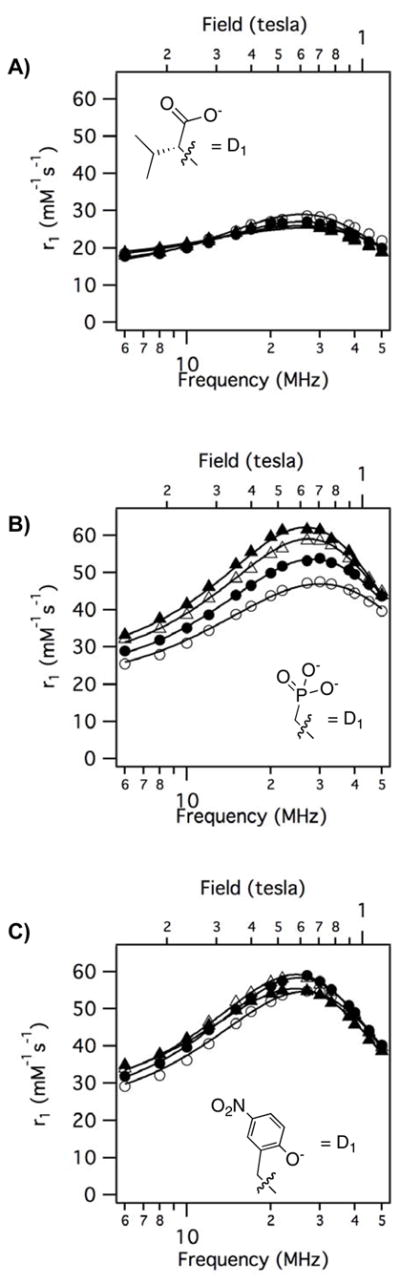
Representative variable temperature (t = 35 °C (open circles); t = 25 °C (filled circles); t = 15 °C (open triangles); t = 5 °C (filled triangles)) NMRD showing effect of donor group on relaxivity with solid lines as fits to the data as described in the text. A) Gd1b-gly; B) Gd2-gly; C) Gd3-gly.
Figure 8 shows the effect of the amide substituent on relaxivity when D1 is phosphonate. For all four phosphonate compounds, relaxivity increases as temperature is decreased. This suggests that all 4 compounds are in fast exchange and that relaxivity is limited by inner-sphere water exchange that is too fast. Replacing the amide methyl substituent with a group containing a pendant carboxylate results in a large relaxivity increase that must be due to increased second-sphere relaxivity.
Figure 8.

Variable temperature (t = 35 °C (open circles); t = 25 °C (filled circles); t = 15 °C (open triangles); t = 5 °C (filled triangles)) NMRD showing effect of amide substituent on relaxivity when D1 = phosphonate with solid lines as fits to the data as described in the text. A) Gd2-me; B) Gd2-gly; C) Gd2-ida; D) Gd2-asp.
In total, NMRD data for 9 compounds was analyzed. The results of these six parameter NMRD analyses are given in Table 1 along with the estimated standard deviation for each parameter in parentheses. The water residency times at 37 °C (τm310) were very similar for a given donor group and the analysis supports the qualitative observation that fast inner-sphere water exchange limits relaxivity for the phosphonate class (mean τm = 6.4 ns) while inner-sphere water exchange is too slow for α-substituted acetate class (mean τm = 290 ns). The phenolate class had inner-sphere exchange rates that were approaching the ideal range (mean τm = 18.4 ns; ideal τm ~ 30 ns). Table 1 also shows that other parameters (τR, ΔH‡, τv, Δ2) were similar for a given D1 group; differences in relaxivity for compounds with the same D1 were reflected in different values of the order parameter F2. The values of τR are in the range reported for other complexes non-covalently bound to serum albumin.28,44,46 The values of τv are also similar to reported numbers for these types of gadolinium complexes.32,33,38 The Δ2 numbers are much smaller by a factor of 2–3 than what is typically reported for fast tumbling GdDOTA derivatives, i.e. electronic relaxation of the protein-bound compounds is slower than may have been anticipated from the literature. This inconsistency may arise from the use of approximate theories for the relaxivity of small complexes where the effect of the static ZFS is neglected. It has been shown that when the fast rotational motion is included as source of modulation of static ZFS, smaller Δ2 values are always obtained.50 Furthermore, more thorough analyses of multifrequency EPR and NMRD of [Gd(DOTA)(H2O)]− which take static ZFS into account show lower values of Δ2.51–53
Table 1.
Molecular parameters derived from simultaneous fitting of variable temperature NMRD of albumin-bound gadolinium complexes. See text for details. Numbers in parentheses represent one standard deviation. RSD = relative standard deviation.
| τR310 (ns) | τm310 (ns) | τv310 (ps) | ΔH‡ (kJ/mol) | Δ2 (1018 s−2) | F2 | |
|---|---|---|---|---|---|---|
| D1=phosphonate | ||||||
| Gd2-me | 5.0 (0.5) | 5.2 (0.3) | 12 (1) | 45.0 (4.8) | 6.48 (0.14) | 0.38 (0.01) |
| Gd2-gly | 5.6 (0.2) | 4.5 (0.1) | 17 (1) | 43.1 (1.6) | 6.40 (0.12) | 0.69 (0.01) |
| Gd2-ida | 5.4 (0.3) | 8.9 (0.5) | 16 (1) | 45.5 (1.9) | 6.01 (0.09) | 0.71 (0.01) |
| Gd2-asp | 5.5 (0.2) | 6.8 (0.3) | 14 (1) | 46.5 (1.5) | 5.95 (0.07) | 0.75 (0.01) |
| mean | 5.4 (0.3) | 6.4 (2.0) | 15 (2) | 45.0 (1.4) | 6.20 (0.30) | 0.60 (0.20) |
| D1=α-acetate | ||||||
| Gd1b-gly | 9.2 (2.7) | 290 (100) | 17 (3) | 26.1 (5.9) | 8.24 (1.15) | 0.40 (0.08) |
| Gd1b-me | 6.5 (2.5) | 287 (200) | 18 (1) | 17.1 (9.4) | 9.81 (2.11) | 0.36 (0.12) |
| mean | 7.9 (1.9) | 289 (2) | 18 (1) | 21.6 (6.4) | 9.0 (1.1) | 0.38 (0.03) |
| D1=phenolate | ||||||
| Gd3-gly | 6.0 (0.2) | 9.5 (0.4) | 17 (1) | 53.9 (1.1) | 5.38 (0.07) | 0.67 (0.01) |
| Gd3-ida | 5.7 (0.1) | 25.7(1.5) | 18 (1) | 45.8 (1.3) | 5.12 (0.06) | 0.72 (0.04) |
| Gd3-asp | 5.4 (0.1) | 20.0 (1.2) | 18 (1) | 51.5 (1.4) | 6.02 (0.07) | 0.51 (0.01) |
| mean | 5.7 (0.3) | 18.4 (8.2) | 18 (1) | 50.4 (4.2) | 5.51 (0.46) | 0.63 (0.11) |
In a second analysis, we sought a better understanding of the second-sphere effect. The phosphonate series (Figure 8) offers the possibility to estimate the lifetime of these second-sphere water molecules. Considering the structures of Gd2-me, Gd2-gly, Gd2-asp, and Gd2-ida it is reasonable to assume that the increased relaxivity of the later 3 compounds compared to Gd2-me is due to the different amide substituents. This is illustrated in Figure 9A where the NMRD profiles of Gd2-me and Gd2-ida are plotted together. To estimate this second-sphere effect, we subtracted the measured relaxivity of Gd2-me from the relaxivity of each of Gd2-gly, Gd2-ida, and Gd2-asp. This analysis assumes that the contributions from inner- and outer-sphere water to relaxivity are the same, which seems reasonable based on the similarity in structures and because all 4 phosphonates showed a similar temperature dependence on relaxivity. The NMRD curves for r1SS of Gd2-gly, Gd2-ida, and Gd2-asp at 35 °C are shown in Figure 9B. The second-sphere obviously contributes considerably to the relaxivity of these compounds, as much as 24 mM−1s−1.
Figure 9.

A) NMRD for Gd2-me (filled circles) and Gd2-ida (open circles) at 35 °C in HSA solution showing the large enhancement in relaxivity due to the pendant carboxylate groups in Gd2-ida. This difference can be attributed to second-sphere relaxivity, r1SS. B) r1SS at 35 °C in HSA solution for Gd2-gly (filled triangles), Gd2-ida (open circles), Gd2-asp (open triangles) calculated by subtracting the relaxivity of Gd2-me from the relaxivity of each compound (Gd2-gly, Gd2-ida, Gd2-asp) at each frequency. Solid lines are from the global fit to the data as described in the text.
The three data sets in Figure 9B were simultaneously fit to equations 2–4. Since the inner-sphere is the same for all three compounds, we assume that the electronic relaxation parameters Δ2 and τv would be the same for all three compounds, and these were treated as global parameters. We also treated τR as a global parameter since the compounds have such similar structures and a common HSA binding group. For each individual compound, two local parameters were also included: the ratio of number of second-sphere water to the gadolinium - hydrogen distance (q′/rGdH) and the residency time in the second-sphere (τ′m). This analysis described the data quite well (see solid lines in Figure 9B) and returned τR = 5.5 ± 0.2 ns, Δ2 = (7.37 ± 0.17) × 1018 s2, τv = 15 ± 1 ps. These values are in good agreement with the values found in the first analysis (Table 1). For Gd2-gly, the mean residency time of water in the second sphere, τ′m = 7.8 ± 1.4 ns; for Gd2-ida, τ ′m = 20.6 ± 4.6 ns; for Gd2-asp, τ ′m = 11.2 ± 1.7 ns. The number of water molecules is strongly correlated with the distance between the Gd ion and the hydrogen atom and cannot be independently determined. For one water molecule (2 exchangeable protons) in the second sphere, the Gd-H distance would be 3.82 ± 0.02 Å for Gd2-gly, 3.68 ± 0.01 Å for Gd2-ida, and 3.64 ± 0.01 Å for Gd2-asp. For two water molecules in the second sphere the Gd-H distance for Gd2-gly would be 4.30 Å, and if there were three waters, the distance would be 4.59 Å (other values are easily calculated from the q′/(rGdH)6 dependence). The distances for one or two water molecules are physically quite reasonable.
DISCUSSION
The purpose of this study was to identify new high-relaxivity Gd chelates and was based upon the hypothesis that the rate of inner-sphere water exchange could be rationally optimized and further, that large gains in relaxivity could be realized through second-sphere effects. Based on this limited data set with 20 compounds, it appears that the inner-sphere water exchange rate can be tuned. It is established that changing carboxylate donor groups to amides results in a decrease in water exchange, and that water exchange gets increasingly slower when more carboxylates are converted to amides.39,54,55 However using amide donors opens up a great deal of synthetic flexibility to try and improve relaxivity through the second hydration sphere. Amides with different substituents are readily prepared and preparation of these DOTA-bis(amide) derivatives is straightforward.56 In order to offset the impact of two amide donor groups on inner-sphere water exchange, we introduced donor groups that had been previously shown to accelerate this water exchange compared to the carboxylate donor group. While it was known that amide substitution of carboxylate decreased exchange54 and phosphonate substitution increased exchange,32 it was not clear at the outset whether these effects would be additive. Based on the relaxivity data and especially the variable temperature NMRD, it is clear that a phosphonate or phenolate donor group offsets the water exchange reducing effect of the amides. This is a powerful finding that offers many possibilities for tuning water exchange dynamics and relaxivity in GdDOTA-like complexes.
In a related study, we found the τm value at 37 °C for a DOTA derivative with 4 cyclen nitrogen donor atoms, 3 carboxylate oxygen donor atoms, and a phosphonate oxygen donor atom to be 0.7 ns.29 When 2 of those carboxylate groups are converted to amide groups as in this study, the mean τm value at 37 °C increased to 6.4 ns or an order of magnitude slower exchange rate. Similarly, compounds with a phenolate donor group and three carboxylate donor groups had a mean τm value at 37 °C of 1.7 ns;29 in this study, when 2 carboxylates were converted to amides, the mean τm value at 37 °C increased by an order of magnitude to 18.4 ns. The same effect was seen when D1 was the isopropyl substituted acetate donor: here τm at 37 °C increased from 34 ns to 290 ns when two carboxylates were converted to amides. This same order of magnitude increase in τm has been reported for simple bis(amides) of GdDOTA compared to GdDOTA itself,38,55,57 and is also seen with GdDTPA and its bis(amides).38,54
For the types of compounds described here, the phenolate donor produced the best water exchange rate. The substituted acetates gave exchange rates that were too slow and the phosphonates gave exchange rates that were too fast, although it should be noted that the fast inner-sphere water exchange rate in the phosphonate series mainly limited the peak low field relaxivity between 0.5–0.7T and relaxivities at 1.4T were quite high with this class of compounds.
The second goal of this work was to exploit amide substituents with hydrogen bond acceptors to increase the residency time of water in the second coordination sphere and thus increase relaxivity. Pendant hydrogen bond acceptors like carboxylates and phosphonates have previously been shown to enhance relaxivity.35,37 Increasing the rotational correlation time increases this second-sphere relaxivity effect further.31,36 For the complexes described here, we found that an amide substituted with a methylene group followed by a carboxylate (i.e. CH2COOH) or phosphonate or phosphonate ester resulted in a large second-sphere effect compared to simple methyl group substitutions. At pH 7.4, the carboxylic acid will be in its conjugate base form and can act as a H-bond acceptor through either of its oxygen atoms. Larger second sphere effects were observed when there were two pendant carboxylate groups per amide, i.e. the ida and asp substituents had higher relaxivity than the analogous gly substituent. Analysis of NMRD data indicated that the mean water residency time in the second sphere was longer in the Gd2-ida and Gd2-asp complexes than in Gd2-gly. This suggests that having two pendant carboxylate groups per amide doubles the probability of forming a stable H-bond with water, and thus increases the mean residency time of second-sphere water. If there were twice as many second-sphere water molecules associated with Gd2-ida compared to Gd2-gly, then one might expect to see similar mean residency times.
An alternate hypothesis for the higher relaxivities observed with the carboxylate-substituted amides is that these pendant groups could interact with HSA and serve to further rigidify the protein-bound complex. This would reduce internal motion and increase relaxivity. While this hypothesis is difficult to disprove with the available data, we note that the relaxivities of the compounds that gave high relaxivity when bound to albumin also gave higher relaxivities in the absence of protein. Figure 4 shows that the gly, ida, asp, PEt2, and PEtOH complexes all have substantially higher relaxivity than analogous compounds with amide methyl substituents. This data suggests that the second-sphere effect is present in the absence of protein binding, and it is reasonable to expect that the second-sphere effect should be present when the complex is immobilized by albumin binding. Of course, one cannot rule out simultaneous effects of differences in rotational dynamics due to pendant arm interactions with the protein and second-sphere effects.
The mean residency time of water in the second-sphere for the gly, ida, and asp derivatives was quite long (>5 ns) and this offers great potential in using this class of gadolinium chelates as high field agents. At fields higher than 3T there is little contribution to the overall correlation time from T1e. Most gadolinium-based contrast agents suffer from lower relaxivity at high field.58 By modifying the rotational dynamics to provide an intermediate rotational correlation time (0.5 < τR < 2 ns) it is possible to improve relaxivities at 3T and higher fields.20,59–61 The data from this report indicate that it should also be possible to engineer significant contributions from second-sphere relaxivity at high field.
The amide “arms” were prepared using 2-bromoacetylbromide and an amine. This approach lends itself readily to a wide range of substituted amides and it may be possible to improve relaxivity even further. For instance, other amino acids like serine or arginine could be used to assess the impact of pendant hydroxyl or guanidinium groups on second sphere hydration and relaxivity.
There are several limitations to this study. The mechanistic conclusions regarding inner-sphere water exchange rates and second-sphere contributions to relaxivity are based on variable temperature NMRD measurements. For slow tumbling protein-bound complexes such as these, the VT NMRD profiles are quite sensitive to water exchange. Unfortunately the traditional O-17 method of determining water exchange rates requires gadolinium concentrations in excess of 5 mM and this precludes making measurements under conditions where there is primarily a single protein-bound complex in solution, as was the case for the NMRD measurements. While protein binding may change the water exchange rate,62 it would be worthwhile to measure water exchange independently on the complexes in the absence of protein. Another limitation is that the second-sphere effect is difficult to measure directly and is mainly implied by the data. The results described here suggest some useful compounds to explore this effect by way of molecular dynamics simulations.63 Finally, the NMRD analysis was not sensitive enough to identify increases in τR with decreasing temperature, but such increases must occur. We used an analysis that treated the VT NMRD data with the smallest number of adjustable parameters and assumed that outer-sphere relaxivity could be described by a model compound and that the inner-sphere hydration number did not change when the complexes was protein bound. Clearly the rotational and hydration dynamics are more complex than described by our basic analysis, and discrepancies between assumed values and true values would obviously result in differences in the parameters in Table 1. Further NMR, ENDOR, EPR, and time-resolved luminescence studies could be performed on these complexes or other lanthanide surrogates to better understand the rotational and hydration dynamics of these complexes, and to further test the validity of the assumptions used in the NMRD analyses.47,48,51,62,64
The underlying design tenet for these studies was to use the macrocylic cyclen core to create octadentate ligands. The thermodynamic stability of these types of complexes is generally quite high and more importantly, this class of complexes are usually very kinetically inert to Gd dissociation or transmetallation. High stability and inertness are key criteria for any new gadolinium-based contrast agent development, especially with the advent of NSF. The goal of this work was to identify new high relaxivity Gd-chelates, and several were identified. Future work with this class of molecules should include measures of thermodynamic stability, gadolinium dissociation and transmetallation kinetics, as well as rodent biodistribution data to understand in vivo stability.
CONCLUSION
Overall this study demonstrates that the inner-sphere water exchange rate of GdDOTA derivatives depends on the choice of donor groups and can be rationally tailored to reach an optimal value for relaxivity. Incorporation of a phosphonate or phenolate donor group offsets the water exchange retarding effect of two amide donor groups. The presence of the amide groups enables large contributions to relaxivity to be made via stabilization of second-sphere hydration. Several promising new gadolinium chelates were identified that may be useful in the design of new molecular MR imaging agents.
Acknowledgments
PC acknowledges financial support from the National Institutes of Health through grants R01EB009062 and R21EB009738.
References
- 1.Grobner T, Prischl FC. Gadolinium and nephrogenic systemic fibrosis. Kidney Int. 2007;72:260–264. doi: 10.1038/sj.ki.5002338. [DOI] [PubMed] [Google Scholar]
- 2.Rydahl C, Thomsen HS, Marckmann P. High Prevalence of Nephrogenic Systemic Fibrosis in Chronic Renal Failure Patients Exposed to Gadodiamide, a Gadolinium-Containing Magnetic Resonance Contrast Agent. Invest Radiol. 2008;43:141–144. doi: 10.1097/RLI.0b013e31815a3407. [DOI] [PubMed] [Google Scholar]
- 3.Haylor J, Dencausse A, Vickers M, et al. Nephrogenic Gadolinium Biodistribution and Skin Cellularity Following a Single Injection of Omniscan in the Rat. Invest Radiol. 2010;45:XXX–XXX. doi: 10.1097/RLI.0b013e3181eb51f2. [DOI] [PubMed] [Google Scholar]
- 4.Aime S, Castelli DD, Crich SG, et al. Pushing the sensitivity envelope of lanthanide-based magnetic resonance imaging (MRI) contrast agents for molecular imaging applications. Acc Chem Res. 2009;42:822–831. doi: 10.1021/ar800192p. [DOI] [PubMed] [Google Scholar]
- 5.Major JL, Meade TJ. Bioresponsive, cell-penetrating, and multimeric MR contrast agents. Acc Chem Res. 2009;42:893–903. doi: 10.1021/ar800245h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta A, Raymond KN. Gd-hydroxypyridinone (HOPO)-based high-relaxivity magnetic resonance imaging (MRI) contrast agents. Acc Chem Res. 2009;42:938–947. doi: 10.1021/ar800250h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bazeli R, Coutard M, Daumas-Duport B, et al. In Vivo Evaluation of a New MRI Contrast Agent (P947) to Target Matrix Metalloproteinases in Expanding Experimental Abdominal Aortic Aneurysms. Invest Radiol. 2010;45:XXX–XXX. doi: 10.1097/RLI.0b013e3181ee5bbf. [DOI] [PubMed] [Google Scholar]
- 8.Koenig SH, Brown RD., III Relaxation of solvent protons by paramagnetic ions and its dependence on magnetic field and chemical environment: implications for NMR imaging. Magn Reson Med. 1984;1:478–495. doi: 10.1002/mrm.1910010407. [DOI] [PubMed] [Google Scholar]
- 9.Lauffer RB, Brady TJ. Preparation and water relaxation properties of proteins labeled with paramagnetic metal chelates. Magn Reson Imaging. 1985;3:11–16. doi: 10.1016/0730-725x(85)90004-9. [DOI] [PubMed] [Google Scholar]
- 10.Lauffer RB. Paramagnetic Metal Complexes as Water Proton Relaxation Agents for NMR Imaging: Theory and Design. Chem Rev. 1987;87:901–927. [Google Scholar]
- 11.Spuentrup E, Katoh M, Buecker A, et al. Molecular MR imaging of human thrombi in a swine model of pulmonary embolism using a fibrin-specific contrast agent. Invest Radiol. 2007;42:586–595. doi: 10.1097/RLI.0b013e31804fa154. [DOI] [PubMed] [Google Scholar]
- 12.Spuentrup E, Ruhl KM, Botnar RM, et al. Molecular magnetic resonance imaging of myocardial perfusion with EP-3600, a collagen-specific contrast agent: initial feasibility study in a swine model. Circulation. 2009;119:1768–1775. doi: 10.1161/CIRCULATIONAHA.108.826388. [DOI] [PubMed] [Google Scholar]
- 13.Vymazal J, Spuentrup E, Cardenas-Molina G, et al. Thrombus imaging with fibrin-specific gadolinium-based MR contrast agent EP-2104R: results of a phase II clinical study of feasibility. Invest Radiol. 2009;44:697–704. doi: 10.1097/RLI.0b013e3181b092a7. [DOI] [PubMed] [Google Scholar]
- 14.Katoh M, Haage P, Wiethoff AJ, et al. Molecular Magnetic Resonance Imaging of Deep Vein Thrombosis Using a Fibrin-Targeted Contrast Agent: A Feasibility Study. Invest Radiol. 2009 doi: 10.1097/RLI.0b013e318195886d. [DOI] [PubMed] [Google Scholar]
- 15.Caravan P, Das B, Dumas S, et al. Collagen-Targeted MRI Contrast Agent for Molecular Imaging of Fibrosis. Angew Chem Int Ed Engl. 2007;46:8171–8173. doi: 10.1002/anie.200700700. [DOI] [PubMed] [Google Scholar]
- 16.Overoye-Chan K, Koerner S, Looby RJ, et al. EP-2104R: a fibrin-specific gadolinium-based MRI contrast agent for detection of thrombus. J Am Chem Soc. 2008;130:6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- 17.Bloembergen N, Morgan LO. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 18.Dwek RA, Richards RE, Morallee KG, et al. Lanthanide cations as probes in biological systems. Proton relaxation enhancement studies for model systems and lysozyme. Eur J Biochem. 1971;21:204–209. doi: 10.1111/j.1432-1033.1971.tb01457.x. [DOI] [PubMed] [Google Scholar]
- 19.Caravan P, Ellison JJ, McMurry TJ, et al. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 20.Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]
- 21.Lauffer RB. Targeted Relaxation Enhancement Agents for MRI. Magn Reson Med. 1991;22:339. doi: 10.1002/mrm.1910220237. [DOI] [PubMed] [Google Scholar]
- 22.Wiener EC, Brechbiel MW, Brothers H, et al. Dendrimer-based metal chelates: a new class of magnetic resonance imaging contrast agents. Magn Reson Med. 1994;31:1–8. doi: 10.1002/mrm.1910310102. [DOI] [PubMed] [Google Scholar]
- 23.Schuhmann-Giampieri G, Schmitt-Willich H, Frenzel T, et al. In vivo and in vitro evaluation of gadolinium-DTPA-polylysine as a macromolecular contrast agent for magnetic resonance imaging. Invest Radiol. 1991;26:969–974. doi: 10.1097/00004424-199111000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Bogdanov AA, Weissleder R, Frank HW, et al. A new macromolecule as a contrast agent for MR angiography: preparation, properties, and animal studies. Radiology. 1993;187:701–706. doi: 10.1148/radiology.187.3.8497616. [DOI] [PubMed] [Google Scholar]
- 25.Manabe Y, Longley C, Furmanski P. High-level conjugation of chelating agents onto immunoglobulins: use of an intermediary poly(L-lysine)-diethylenetriaminepentaacetic acid carrier. Biochim Biophys Acta. 1986;883:460–467. doi: 10.1016/0304-4165(86)90285-0. [DOI] [PubMed] [Google Scholar]
- 26.Ogan MD, Schmiedl U, Moseley ME, et al. Albumin labeled with gadolinium-DTPA an intravascular contrast-enhancing agent for magnetic resonance blood pool imaging: Preparation and characterization. Invest Radiol. 1987;22:665–671. [PubMed] [Google Scholar]
- 27.Jenkins BG, Armstrong E, Lauffer RB. Site-Specific Water Proton Relaxation Enhancement of Iron(III) Chelates Noncovalently Bound to Human Serum Albumin. Magn Reson Med. 1991;17:164–178. doi: 10.1002/mrm.1910170120. [DOI] [PubMed] [Google Scholar]
- 28.Caravan P, Cloutier NJ, Greenfield MT, et al. The interaction of MS-325 with human serum albumin and its effect on proton relaxation rates. J Am Chem Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 29.Dumas S, Jacques V, Sun W-C, et al. High relaxivity MRI contrast agents part 1: Impact of single donor atom substitution on relaxivity of serum albumin-bound gadolinium complexes. Invest Radiol. 2010;45:XXX–XXX. doi: 10.1097/RLI.0b013e3181ee5a9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aime S, Botta M, Terreno E, et al. Gd(DOTP)5-outer-sphere relaxation enhancement promoted by nitrogen bases. Magn Reson Med. 1993;30:583–591. doi: 10.1002/mrm.1910300509. [DOI] [PubMed] [Google Scholar]
- 31.Caravan P, Greenfield MT, Li X, et al. The Gd(3+) complex of a fatty acid analogue of DOTP binds to multiple albumin sites with variable water relaxivities. Inorg Chem. 2001;40:6580–6587. doi: 10.1021/ic0102900. [DOI] [PubMed] [Google Scholar]
- 32.Rudovsky J, Cigler P, Kotek J, et al. Lanthanide(III) complexes of a mono(methylphosphonate) analogue of H4dota: The influence of protonation of the phosphonate moiety on the TSAP/SAP isomer ratio and the water exchange rate. Chem Eur J. 2005;11:2373–2384. doi: 10.1002/chem.200400367. [DOI] [PubMed] [Google Scholar]
- 33.Lebduskova P, Hermann P, Helm L, et al. Gadolinium(III) complexes of mono- and diethyl esters of monophosphonic acid analogue of DOTA as potential MRI contrast agents: solution structures and relaxometric studies. Dalton Trans. 2007:493–501. doi: 10.1039/b612876a. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Wu K, Sherry AD. A novel pH-sensitive MRI contrast agent. Angew Chem Int Ed Engl. 1999;38:3192–3194. [PubMed] [Google Scholar]
- 35.Kalman FK, Woods M, Caravan P, et al. Potentiometric and relaxometric properties of a gadolinium-based MRI contrast agent for sensing tissue pH. Inorg Chem. 2007;46:5260–5270. doi: 10.1021/ic0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ali MM, Woods M, Caravan P, et al. Synthesis and relaxometric studies of a dendrimer-based pH-responsive MRI contrast agent. Chem Eur J. 2008;14:7250–7258. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowe MP, Parker D, Reany O, et al. pH-dependent modulation of relaxivity and luminescence in macrocyclic gadolinium and europium complexes based on reversible intramolecular sulfonamide ligation. J Am Chem Soc. 2001;123:7601–7609. doi: 10.1021/ja0103647. [DOI] [PubMed] [Google Scholar]
- 38.Powell DH, Ni Dhubhghaill OM, Pubanz D, et al. Structural and Dynamic Parameters Obtained from 17O NMR, EPR, and NMRD Studies of Monomeric and Dimeric Gd3+ Complexes of Interest in Magnetic Resonance Imaging: An Integrated and Theoretically Self-Consistent Approach. J Am Chem Soc. 1996;118:9333–9346. [Google Scholar]
- 39.Zhang S, Wu K, Sherry AD. Gd3+ complexes with slowly exchanging bound-water molecules may offer advantages in the design of responsive MR agents. Invest Radiol. 2001;36:82–86. doi: 10.1097/00004424-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Caravan P, Jacques V, Dumas S, et al. PCT WO2008098056 Synthesis of high relaxivity chelates for potential use as MRI contrast agents. 2008
- 41.Koenig SH, Brown RD., III Field-cycling relaxometry of protein solutions and tissue: immplications for MRI. Prog Nucl Magn Reson Spectrosc. 1990;22:487–567. [Google Scholar]
- 42.Dumas S, Troughton JS, Cloutier NJ, et al. A High Relaxivity Magnetic Resonance Imaging Contrast Agent Targeted to Serum Albumin. Aus J Chem. 2008;61:682–686. [Google Scholar]
- 43.Solomon I. Relaxation Processes in a System of Two Spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 44.Caravan P, Parigi G, Chasse JM, et al. Albumin Binding, Relaxivity, and Water Exchange Kinetics of the Diastereoisomers of MS-325, a Gadolinium(III)-Based Magnetic Resonance Angiography Contrast Agent. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 45.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 46.Troughton JS, Greenfield MT, Greenwood JM, et al. Synthesis and evaluation of a high relaxivity manganese(II)-based MRI contrast agent. Inorg Chem. 2004;43:6313–6323. doi: 10.1021/ic049559g. [DOI] [PubMed] [Google Scholar]
- 47.Zech S, Sun W-C, Jacques V, et al. Probing the Water Coordination of Protein-Targeted MRI Contrast Agents by Pulsed ENDOR Spectroscopy. ChemPhysChem. 2005;6:2570–2577. doi: 10.1002/cphc.200500250. [DOI] [PubMed] [Google Scholar]
- 48.Astashkin AV, Raitsimring AM, Caravan P. Pulsed ENDOR Study of Water Coordination to Gd3+ Complexes in Orientationally Disordered Systems. J Phys Chem A. 2004;108:1900–2001. [Google Scholar]
- 49.Raitsimring AM, Astashkin AV, Poluektov OG, et al. High field pulsed EPR and ENDOR of Gd3+ complexes in glassy solutions. Appl Magn Reson. 2005;28:281–295. [Google Scholar]
- 50.Kruk D, Kowalewski J. Nuclear spin relaxation in paramagnetic systems (S≥1) under fast rotation conditions. J Magn Reson. 2003;162:229–240. doi: 10.1016/s1090-7807(03)00011-9. [DOI] [PubMed] [Google Scholar]
- 51.Benmelouka M, Van Tol J, Borel A, et al. A high-frequency EPR study of frozen solutions of Gd(III) complexes: straightforward determination of the zero-field splitting parameters and simulation of the NMRD profiles. J Am Chem Soc. 2006;128:7807–7816. doi: 10.1021/ja0583261. [DOI] [PubMed] [Google Scholar]
- 52.Borel A, Helm L, Merbach AE, et al. T1e in Four Gd3+ Chelates: LODEPR Measurements and Models for Electron Spin Relaxation. J Phys Chem A. 2002;106:6229–6231. [Google Scholar]
- 53.Rast S, Fries PH, Belorizky E, et al. A general approach to the electronic spin relaxation of Gd(III) complexes in solutions. Monte Carlo simulations beyond the Redfield limit. J Chem Phys. 2001;115:7554–7563. [Google Scholar]
- 54.Helm L, Merbach AE. Inorganic and bioinorganic solvent exchange mechanisms. Chem Rev. 2005;105:1923–1959. doi: 10.1021/cr030726o. [DOI] [PubMed] [Google Scholar]
- 55.Zhang S, Merritt M, Woessner DE, et al. PARACEST agents: modulating MRI contrast via water proton exchange. Acc Chem Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 56.De Leon-Rodriguez LM, Kovacs Z, Esqueda-Oliva AC, et al. Highly regioselective N-trans symmetrical diprotection of cyclen. Tetrahedron Lett. 2006;47:6937–6940. [Google Scholar]
- 57.Zhang S, Kovacs Z, Burgess S, et al. [DOTA-bis(amide)]lanthanide complexes: NMR evidence for differences in water-molecule exchange rates for coordination isomers. Chem Eur J. 2001;7:288–296. doi: 10.1002/1521-3765(20010105)7:1<288::aid-chem288>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Noebauer-Huhmann IM, Szomolanyi P, Juras V, et al. Gadolinium-based MR Contrast Agents at 7 Tesla: In Vitro T1 Relaxivities in Human Blood Plasma. Invest Radiol. 2010;45:XXX–XXX. doi: 10.1097/RLI.0b013e3181ebd4e3. [DOI] [PubMed] [Google Scholar]
- 59.Caravan P, Farrar CT, Frullano L, et al. Influence of molecular parameters and increasing magnetic field strength on relaxivity of gadolinium- and manganese-based T(1) contrast agents. Contrast Media Mol Imaging. 2009;4:89–100. doi: 10.1002/cmmi.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Livramento JB, Toth E, Sour A, et al. High relaxivity confined to a small molecular space: a metallostar-based, potential MRI contrast agent. Angew Chem Int Ed Engl. 2005;44:1480–1484. doi: 10.1002/anie.200461875. [DOI] [PubMed] [Google Scholar]
- 61.Livramento JB, Weidensteiner C, Prata MI, et al. First in vivo MRI assessment of a self-assembled metallostar compound endowed with a remarkable high field relaxivity. Contrast Media Mol Imaging. 2006;1:30–39. doi: 10.1002/cmmi.92. [DOI] [PubMed] [Google Scholar]
- 62.Zech SG, Eldredge HB, Lowe MP, et al. Protein binding to lanthanide(III) complexes can reduce the water exchange rate at the lanthanide. Inorg Chem. 2007;46:3576–3584. doi: 10.1021/ic070011u. [DOI] [PubMed] [Google Scholar]
- 63.Borel A, Helm L, Merbach AE. Molecular dynamics simulations of MRI-relevant GdIII chelates: direct access to outer-sphere relaxivity. Chem Eur J. 2001;7:600–610. doi: 10.1002/1521-3765(20010202)7:3<600::aid-chem600>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 64.Raitsimring AM, Astashkin AV, Baute D, et al. Determination of the hydration number of gadolinium(III) complexes by high-field pulsed (17)O ENDOR spectroscopy. ChePphysChem. 2006;7:1590–1597. doi: 10.1002/cphc.200600138. [DOI] [PubMed] [Google Scholar]