

Abstract
Cytochromes P450 (CYPs or P450s) contain a highly conserved threonine residue in the active site, which is referred to as Thr302 in the amino acid sequence of CYP2B4. Extensive biochemical and crystallographic studies have established that this Thr302 plays a critical role in activating molecular oxygen to generate Compound I, a putative iron(IV)-oxo porphyrin cation radical, that carries out the preliminary oxygenation of CYP substrates. Because of its proximity to the center of the P450 active site, this Thr302 is susceptible to mechanism-based inactivation under certain conditions. In this article, we review recent studies on the mechanism-based inactivation of three mammalian P450s in the 2B family, CYP2B1 (rat), 2B4 (rabbit) and 2B6 (human) by tert-butylphenylacetylene (tBPA). These studies showed that tBPA is a potent mechanism-based inactivator of CYP2B1, 2B4 and 2B6 with high kinact/KI ratios (0.23-2.3 min-1 μM-1) and low partition ratios (0-5). Furthermore, mechanistic studies revealed that tBPA inactivates these three CYP2B enzymes through the formation of a single ester adduct with the Thr302 in the active site. These inhibitory properties of tBPA allowed the preparation of a modified CYP2B4 where the Thr302 was covalently and stoichiometrically labeled by a reactive intermediate of tBPA in quantities large enough to permit spectroscopic and crystallographic studies of the consequences of covalent modification of Thr302. Molecular modeling studies revealed a unique binding mode of tBPA in the active site that may shed light on the potency of this inhibition. The results from these studies may serve as a basis for designing more specific and potent inhibitors for P450s by targeting this highly conserved threonine residue which is present in the active sites of most mammalian P450s.
Keywords: Mechanism-based inactivation, Thr302, tert-butylphenylacetylene, specific inhibitor, cytochrome P450, active site
INTRODUCTION
The cytochromes P450 (CYPs or P450s) are members of a superfamily of hemoproteins that catalyze the oxidation of a vast number of organic compounds including many endogenous compounds such as fatty acids, steroid hormones, and eicosanoids, and numerous xenobiotic compounds including drugs, chemical carcinogens, and environmental pollutants. The P450s are one of the most versatile enzymes in nature. The general reaction for P450-catalyzed reactions is expressed as:
| (1) |
where R-H is substrate. From the perspective of chemical synthesis, P450s are extraordinary in their ability to activate relatively inert hydrocarbons (C-H) at physiological temperatures. This catalytic prowess is ascribed to the delicate molecular machinery of the P450s that is capable of activating dioxygen to generate a highly reactive iron-oxo species often referred to as Compound I. This molecular machinery comprises a heme cofactor, critical amino acid residues, and protein architecture that are fine-tuned to control substrate binding, electron and proton flow, and the activation of molecular oxygen during the overall catalytic cycle (for reviews, see [1-3]).
Because of the highly oxidizing nature of Compound I, P450s may become the victim of this remarkable catalytic reaction through a process known as mechanism-based inactivation or “suicide” inhibition. Mechanism-based inactivation of P450s often occurs when P450-catalyzed reactions generate reactive intermediates that can covalently modify critical components of the P450s required for catalysis such as the heme and critical amino acid residues. Several classes of chemicals are known to act as mechanism-based inactivators of P450s including acetylenes, organosulfur compounds, arylamines, cyclic tertiary amines, and furanocoumarins [4, 5]. Mechanism-based inactivation of human P450s may have significant ramifications for the pharmacokinetics of drugs and other xenobiotics and this inactivation has the potential to result in significant, sometimes lethal adverse drug-drug interactions and toxicity [6].
On the other hand, inhibition of P450 activity may have therapeutic potential if the targeted P450 is involved in the pathogenesis of diseases and toxicity. For instance, human aromatase (CYP19A1) inhibitors such as letrozole, anastrozole, and exemestane, are currently being used to treat hormone-dependent breast cancer. Exemestane is, in fact, a potent mechanism-based inhibitor of CYP19A1 that causes time-dependent and irreversible loss of aromatase activity [7]. Because the mechanism-based inhibitors like exemestane bind to the active site of CYP19A1, they are specific and have lasting inhibitory effects in vivo. It was reported that a single dose of 25 mg of exemestane results in a long-lasting reduction (4-5 days) in plasma and urinary estrogens [8]. A number of studies have also demonstrated that dietary and synthetic isothiocyanates have cancer chemopreventive activity and that this activity may correlate with the abilities of isothiocyanates to act as mechanism-based inhibitors of a number of CYP isozymes [9-12].
In the past four decades it has been of great interest to pharmacologists, chemists, and toxicologists to understand the structure-function relationships of the P450s since these monooxygenases play a central role in the metabolism of drugs in humans. A better understanding of the mechanism(s) by which mechanism-based inhibitors inactivate P450s will provide a molecular basis for designing newer, safer drugs to minimize adverse drug-drug interactions and toxicity. In this article, we review recent studies on the mechanism-based inactivation of mammalian CYP2B1, 2B4 and 2B6 by tert-butylphenylacetylene (tBPA). tBPA offers unique advantages to investigate the mechanism by which aryl acetylenes inactivate members of the CYP2B family because of its high kinact, low partition ratio, and 1:1 stoichiometry leading to the formation of a single ester adduct with the critical active site Thr302 residue. In addition, we have been able to obtain Thr302-modified CYP2B4 in large quantities for biophysical characterization using resonance Raman (RR), EPR, and X-ray crystallography. Through the combined use of biochemical analysis, site-directed mutagenesis, biophysical spectroscopy, and molecular modeling, we have been able to obtain structural and functional details for the mechanism-based inactivation of mammalian CYP2Bs by tBPA. Our results show that the mechanism-based inactivation of these three CYP2B enzymes is due to the covalent modification of the highly conserved Thr302 residue and this modification causes significant alterations in the properties of both the heme and the substrate binding site. These findings may aid in designing more potent and specific CYP inhibitors for therapeutic applications.
ROLE OF THR302 IN P450 CATALYSIS
Thr302 is highly conserved among many different families of P450s. The exact residue number of this conserved threonine varies between various P450s due to variations in their amino acid sequences. For convenience, we will refer to this threonine residue as Thr302 based on the amino acid sequence of CYP2B4, unless specified otherwise. In humans, the residue corresponding to Thr302 is conserved in ~86% of the 57 human CYP genes (http://drnelson.uthsc.edu/CytochromeP450.html). Much of our understanding of the role of Thr302 in mammalian P450s stems from extensive studies of bacterial P450cam (CYP101). In this article, we provide only a brief review on the role of Thr302 in P450 catalysis. For more details, readers are referred to a few recent review articles [1, 3, 13].
The first crystal structure of P450s, which was obtained for CYP101, showed that Thr252, which is equivalent to Thr302 in CYP2B4, is located in the I-helix and that the Oγ atom of Thr252 is 5.8 Å away from the heme iron [14]. Adjacent to Thr252 in the I-helix is Asp251, another catalytically important residue. These two residues comprise an important acid-alcohol pair that was later proven to be critical for dioxygen activation. Replacement of Asp251 with Asn by site-directed mutagenesis led to a sharp decrease in the NADH consumption rate by ~99% and this mutation shifted the rate-limiting step to O-O bond scission [15, 16]. In addition, a large kinetic solvent isotope effect was observed for D251N under steady-state turnover conditions [17]. These results suggest that Asp251 plays an essential role in the proton delivery required for O-O bond scission. In contrast, replacement of Thr252 with Ala did not affect the NADH consumption rate, but did lead to a large decrease in product formation. As a result, the reducing equivalents from the NADH consumed were uncoupled from product formation to produce H2O2, with only <10% of the NADH being used to produce hydroxylated product [18, 19]. Replacement of Thr252 with Ala also led to destabilization of the oxyferrous intermediate, indicating that the Thr252 may also interact with the iron-bound oxygen to stabilize the oxyferrous intermediate [19]. These studies provided unequivocal evidence that Thr252 and Asp251 are part of the proton delivery network involved in activating O-O bond scission en route to the generation of Compound I in CYP101.
The elaborate proton delivery network of P450s was best illustrated by the elegant work of Schlichting and coworkers using trapping techniques and cryocrystallography [20]. The crystal structures of both ferric and ferrous CYP101 with d-camphor bound showed no H2O molecules in the active sites, consistent with a previous crystallographic study of CYP101 [21]. The crystal structure of the dioxygen complex of CYP101 showed that dioxygen is bound end-on (η1) to the heme iron with the distal oxygen atom pointing toward Thr252. Associated with dioxygen binding, two H2O molecules, WAT901 and WAT902, appear in the active site, and Asp251 and Thr252 undergo conformational changes such that the carbonyl oxygen of Asp251 flips by 90° toward Asn255 and the amide nitrogen of Thr252 rotates toward the heme pocket. These changes yield a hydrogen bond network consisting of Asp251, Thr252, and H2O molecules that are requisite for O-O bond scission. The hydrogen bond network is extended to the side chain of Glu366 via a chain of H2O molecules.
Subsequent crystallographic studies on the dioxygen complexes of the D251N and T252A variants of CYP101 led to re-consideration of the role of Thr252 [22]. This study demonstrated that the two active site H2O molecules are absent in the D251N, but remain in the T252A variant with almost exactly the same geometry as in the wild type (WT) CYP101. This result suggests that Thr252 is not required to hold the H2O molecules in place and that the uncoupling is not due to disordering of the active site H2O molecules. As illustrated in Figure 1, Nagano and Poulos hypothesized that Thr252 functions as a hydrogen acceptor to stabilize the hydroperoxy intermediate rather than as a hydrogen bond donor. This hypothesis gained strong support from a site-directed mutagenesis study of an unnatural Thr252 variant. To examine the role of the hydroxyl group of Thr252, a methoxy group was substituted for the hydroxyl group of Thr252 through unnatural amino acid mutagenesis [23]. The methoxy-containing variant OMe-Thr252 catalyzes the hydroxylation of d-camphor at one-third of the turnover rate of CYP101 WT, but the reaction is 100% coupled to the NADH consumption. This observation is in agreement with the notion that the Thr252 may function as a hydrogen bond acceptor [22].
Figure 1.
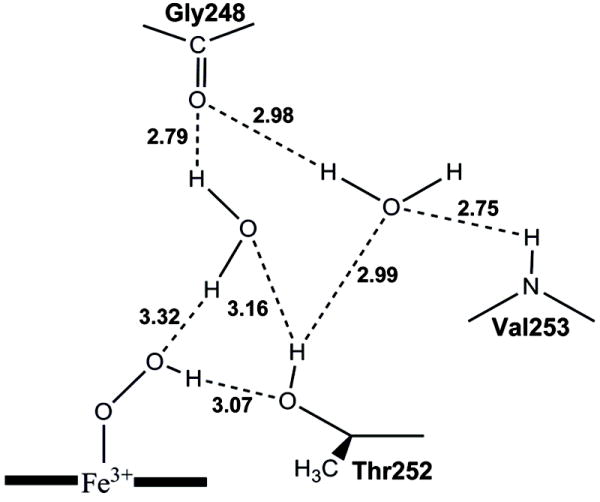
Possible hydrogen bond network for the P450cam hydroperoxy intermediate as proposed by Nagano and Poulos [22]. The distances between the heteroatoms are taken from the crystal structure of the P450cam WT dioxygen complex. This figure is reproduced from Figure 4 of ref. [22].
The importance of this highly conserved threonine in the active site has also been demonstrated in mammalian P450s. As reported by Vaz et al [24], the T302A variant of CYP2B4 exhibited a two- to three-fold increase in H2O2 formation and an 80-90% loss in hydroxylase activity for benzphetamine and cyclohexane. Of particular interest, the T302A variant showed a 10-fold increase in the deformylation of cyclohexane carboxaldehyde, which led to the conclusion that the Thr302→Ala mutation disrupts the formation of Compound I and hence other oxygenated species such as the peroxy or hydroperoxy species may function as alternative oxidants to oxidize cyclohexane carboxaldehyde. An EPR study showed that the T302A variant of CYP2B4 exhibited a higher percentage of high-spin heme (40%) than the CYP2B4 WT [25], indicating that the heme is perturbed in the variant. Mutation of Thr303 (equivalent to Thr302 in CYP2B4) to valine in CYP2E1 also led to an increase in high-spin heme as observed in CYP2B4 and lower hydroxylase activity for fatty acids [26]. Interestingly, the Thr303→Ser mutation did not alter the turnover rate but did alter the regioselectivity of the fatty acid hydroxylase activity as additional hydroxylated products of the C11 to C17 fatty acids were formed by the T303S variant. This change in the regioselectivity indicates that the Thr303 may play an important role in the orientation of substrate. This is consistent with the notion that the I-helix is part of the substrate recognition site (SRS4) as proposed by Gotoh [27]. As discussed in the next section, mutation of the Thr302 also affects the mechanism-based inactivation of P450s by a number of compounds including several acetylenic compounds.
MECHANISM-BASED INACTIVATION OF P450S BY ACETYLENIC COMPOUNDS
Acetylenes represent one of the most informative and effective chemical functional groups for our studies aimed at understanding the mechanism-based inactivation of P450s. Many different acetylene-containing compounds have been used over the years to investigate the topology of the heme active site and the catalytic mechanism of P450s, including 2-ethynylnaphthalene (2EN), 9-ethynylphenanthrene (9EPh), 5-phenyl-1-pentyne (5P1P), 17α-ethynylestradiol (17EE), tert-butylacetylene (tBA), tert-butyl 1-methyl-2-propynyl ether (tBMP), and tert-butylphenylacetylene (tBPA). The kinetic constants for the inactivation of several different P450s by a variety of acetylenic compounds that have been reported in the literature are summarized in Table 1.
Table 1.
Summary of kinetic inactivation constants for acetylenic mechanism-based inhibitors of P450s.
| MBIa | CYPsb | KI (μM) | kinact (min-1) | kinact/KI (min-1μM-1) | Modified Sitec | Ref. |
|---|---|---|---|---|---|---|
| 2EN | 2B1 | 0.08 | 0.83 | 10.4 | Protein | [28] |
| 9EPh | 2B1 | 0.14 | 0.45 | 3.2 | Protein | [31] |
| 5-P1P | 2B1 | 100 | 0.92 | 0.0092 | Heme | [33] |
| 2E1 | 50 | 1.31 | 0.026 | Heme | [33] | |
| Δ2E1 | 54 | 0.78 | 0.014 | Heme | [33] | |
| 4-P1B | 2E1 (LM) | ND | 0.21 | ND | [33] | |
| 2E1 | ND | 0.55 | ND | [33] | ||
| 2-P3B2 | 2E1 (LM) | ND | 0.04 | ND | [33] | |
| 2E1 | ND | 0.13 | ND | [33] | ||
| 1-P2P1 | 2E1 (LM) | ND | 0.3 | ND | [33] | |
| 2E1 (LM) | ND | 0.16 | ND | [33] | ||
| 17-EE | 2B1 | 11 | 0.2 | 0.018 | Protein | [36] |
| 2B6 | 0.8 | 0.03 | 0.038 | Protein | [36] | |
| 3A4 | 18 | 0.04 | 0.0022 | Protein+Heme | [35] | |
| tBA | 2B4 | 75 | 0.23 | 0.0031 | Heme | [42] |
| 2E1 | 1000 | 0.2 | 2×10-4 | Protein+Heme | [41] | |
| tBMP | 2E1 | 100 | 0.12 | 1.2×10-4 | Heme | [41] |
| 2B1 | 17 | 0.56 | 0.33 | Heme | [44] | |
| tBPA | 2B4 | 0.44 | 0.12 | 0.27 | Protein | [45] |
| 2B1 | 0.7 | 1.64 | 2.3 | Protein | [44] | |
| 2B6 | 2.8 | 0.7 | 0.25 | Protein+Heme | unpublished data | |
| DMPB | 2B6 (LM) | 5.1 | 0.09 | 0.018 | Heme | [54] |
Abbreviations: 2EN, 2-ethynylnaphthalene; 9-EPh, 9-ethynylphenanthrene; 5-P1P, 5-phenyl-1-pentyne; 17EE, 17α-ethynylestradiol; tBA, tert-butyl acetylene; tBMP, tert-butyl-1-methyl-2-propynyl ether; tBPA, tert-butylphenylacetylene; 4-P1B, 4-phenyl-1-butyne; 2-P3B2, 2-phenyl-3-buty-2-ol; 1-P2P1, 1-phenyl-2-propyn-1-ol; DMPB, N-(3,5-Dichloro-4-pyridyl)-4-methoxy-3-(prop-2-ynyloxy)benzamide. The chemical structures of the listed MBIs are included in Table 1S in the Supplemental Materials.
LM, liver microsomes
ND, not determined.
One of the most well characterized aryl acetylenes is 2-ethynylnaphthalene (2EN). In 1993, Roberts et al. demonstrated in a reconstituted system that 2EN inactivates the 7-ethoxycoumarin O-deethylase activity of CYP2B1 in a time-, concentration- and NADPH-dependent manner [28]. HPLC analysis revealed that the [3H]-labeled 2EN ([3H]2EN) was covalently bound to the apoprotein with a stoichiometry of ~1.3 molecules of 2EN per molecule of CYP2B1 inactivated (1.3:1). Amino acid sequencing of the [3H]2EN-labeled CYP2B1 peptides following cleavage of the protein by cyanogen bromide (CNBr) led to the identification of a radiolabeled peptide 290-314 of CYP2B1 that contains the Thr302 residue [29]. Subsequent studies showed that [3H]2EN was also an efficient mechanism-based inactivator of CYP2B4 and that the inactivation was the result of the formation of protein adduct with a stoichiometry of ~0.8 [29]. Peptide sequencing following CNBr cleavage of the protein revealed two radiolabeled peptides, one of which was identified as peptide 273-283. Roberts et al also reported that 9-ethynylphenanthrene (9EPh) is a mechanism-based inactivator of CYP2B1 with a kinact of 0.45 min-1 and KI of 0.14 μM [30]. MALDI-MS analyses of the [3H]9EPh-labeled peptides following cleavage with CNBr indicated the addition of phenanthrylacetyl group to the peptide. Peptide mapping of the pepsin-digest demonstrated that the radiolabeled peptide 294-307 was the site of covalent modification [30, 31]. The functional importance of Thr302 in the inactivation of CYP2B4 by 2EN was confirmed when the T302A variant was shown to exhibit a significantly slower rate of inactivation by 2EN compared to CYP2B4 WT [32].
However, not all acetelynic compounds target the apoprotein for covalent modification. In some cases the heme is the target. Examples include the inactivation of CYP2E1 and 2B1 by 5-phenyl-1-pentyne, which has been identified as the most effective acetylenic mechanism-based inhibitor of CYP2E1 investigated so far [33]. HPLC analyses of samples incubated in the presence of NADPH and 5-phenyl-1-pentyne revealed formation of a heme adduct. MALDI-MS data obtained for 5-phenyl-1-pentyne inactivated CYP 2E1 showed the addition of a 2-oxo-5-phenylpentyl group to the heme moiety and this modification was responsible for the inactivation [33].
Mechanism-based inhibitors containing acetylenic groups also include a number of clinically used compounds, one of which is 17α-ethynylestradiol (17EE). 17EE was developed in 1938 and still remains the primary estrogenic component in most oral contraceptives. When 17EE was incubated with human liver microsomes in the presence of NADPH, there was both a loss in spectrally detectable P450 and a decrease in the 17EE 2-hydroxylase activity [34]. It was subsequently demonstrated that the mechanism-based inactivation of CYP3A4 by 17EE results in both heme destruction and covalent binding of 17EE to protein [35]. 17EE also inactivates CYP2B1 and 2B6 as reported by Kent et al [36]. In contrast to CYP3A4, inactivation of CYP2B1 and 2B6 by 17EE led to very little decrease in the P450-CO spectra, nor did it cause heme alkylation. Clearly the inactivations by 17EE were due to protein modification. Mass spectral analysis of the 17EE-inactivated CYP2B1 revealed an increase in the molecular mass of the apoprotein corresponding to the mass of 17EE plus one oxygen atom [37]. In order to identify the modified peptides, CYP2B1 and 2B6 were inactivated with [3H]17EE and digested with CNBr [37]. The results from N-terminal sequencing of the digested samples indicated that the amino acid sequences that correspond to the peptides 347-376 and 347-365 in CYPs 2B1 and 2B6, respectively, were modified by 17EE. Peptide mapping of the tryptic digests of the 17EE-labeled and unlabeled CYP2B6 and 2B1 by LC-MS/MS identified S360 as the site of modification [38]. CYP2B2 and 2B4 which have a alanine and glycine, respectively, at position 360 rather than a serine, were not inactivated by 17EE. The functional significance of S360 in the mechanism-based inactivation of CYP 2B1 was investigated by generating a S360A variant of CYP2B1. Interestingly, this mutation did not prevent the inactivation by 17EE. It is possible that an alternate residue in the S360A variant becomes the target for the inactivation by 17EE [38].
Inactivation of CYP 2B4 and CYP2E1 by structurally smaller acetylene analogues such as tBA and tBMP was investigated by Blobaum et al. [39-41]. It was found that both tBA and tBMP inactivated CYP2E1 WT and the T303A variant in a time- and NADPH-dependent manner and the inactivation led to the formation of two modified heme products with concomitant loss of the P450-CO spectra [41]. The inhibitory efficiency of tBA and tBMP is substantially lower than that of aryl acetylenes like 2EN and tBPA (see Table 1), indicative of the importance of the aromatic ring in determining inhibitory potency, at least for CYP2B1 and 2E1. Surprisingly, the mechanism-based inactivation of the T303A variant of CYP2E1 by tBA was completely reversible. The catalytic activity of the tBA-inactivated T303A was restored after overnight dialysis. Subsequent studies identified a novel reversible acetylene-heme adduct in the tBA-inactivated CYP2E1 T303A and provided support for a role of the Thr303 in the proton delivery process [39, 40]. Similarly, tBA and tBMP inactivate CYP2B4 WT and T302A as well and the inactivation results in heme alkylation [42]. HPLC and ESI-LC/MS analysis detected two different tBA-modified heme products with masses of 661 and 705 Da, respectively. However, inactivation of CYP2B4 T302A by tBA was partially reversible. Molecular dynamics studies of both CYP2B4 WT and T302A suggested that the Thr302 may play a role in the stabilization of the heme adducts.
Recent work by Wright and Cravatt has extended the applications of acetylenic inhibitors to in vivo studies of P450s [43]. These authors harnessed the specificity of the reactivity of 2-EN with P450 to create a chemical probe that reports on P450 drug induction and inhibition. Specifically, 2EN was derivatized with an alkyl acetylene group whose addition provides a moiety for click chemistry-based conjugation to azide-modified rhodamine or biotin reporter groups (2EN-ABP). In a series of experiments, 2EN-ABP was shown to label a number of P450s including CYP1A2, 3A11, 2C29 and 2D9/2D10 in a concentration- and NADPH- dependent manner and the majority of the labeling was exclusively to P450s found in mouse liver microsomes. This approach has been used to monitor drug-drug interaction involving CYP1A2 in mice treated with β-naphthoflavone (CYP1A2 inducer) and α-naphthoflavone (CYP1A2 inhibitor).
Even though many acetylenic compounds have been shown to be mechanism-based inhibitors of P450s, details are still missing at the atomic level with respect to the precise sites of modification, and mechanism(s) of inactivation, and the effects of the inactivations on P450 catalysis. As described below, we have attempted to address these by a combination of biophysical and kinetic studies using tBPA as a mechanistic probe.
MECHANISM-BASED INACTIVATION OF CYP2BS BY tBPA
Kinetics and partition ratios for the mechanism-based inactivation by tBPA
The results from our initial investigation of the mechanism-based inactivation of CYP2B1 by tBPA in a reconstituted system were somewhat surprising because the NADPH- and time-dependent inactivation of CYP2B1 by tBPA was too fast to be determined accurately at 30 °C and we had to lower the incubation temperature to ~20 °C. At this temperature, the mechanism-based inactivation of CYP2B1 by tBPA gives a KI of 0.7 μM and a kinact of 1.6 min-1 [44]. The partition ratio for the mechanism-based inactivation of CYP2B1 is 1, indicating that tBPA is highly efficient in inactivating CYP2B1. When tBMP is substituted for tBPA as the mechanism-based inhibitor, the efficiency (kinact/KI) decreases by more than 70-fold (see Table 1); tBMP inactivates CYP2B1 with a KI of 17 μM, kinact of 0.56 min-1 and partition ratio of 10. This result underscores the importance of the aromatic ring of acetylenic compounds for the mechanism-based inactivation of CYP2Bs.
The amino acid sequences of CYP2B4 and CYP2B6 exhibit approximately 77% homology to that of CYP2B1. Thus, we anticipated that tBPA would inactivate CYP2B4 and CYP2B6. In the case of CYP2B6, the KI and kinact were 2.8 μM and 0.7 min-1, respectively, and the partition ratio is ~5 (unpublished data). For CYP2B4, a KI of 0.44 μM and a kinact of 0.12 min-1 were obtained at 30 °C and the partition ratio is close to zero [45]. It appears that tBPA is most efficient in inactivating CYP2B1 with a kinact/KI ratio of 2.3, which is approximately one order of magnitude higher than those of CYP2B4 and CYP2B6.
Identification of the specific amino acid residue modified by tBPA
The potent inactivation of CYP2B1, 2B4 and 2B6 by tBPA prompted us to investigate the underlying mechanism. The primary targets for covalent modification by mechanism-based inhibitors are the heme and active-site amino acid residue(s). Analyses of the molecular masses of CYP2B1, 2B4 and 2B6 by LC-MS following the inactivation reactions showed that all three CYP2B enzymes inactivated by tBPA exhibit a mass increase of 174±5 Da compared with the unmodified enzymes. This mass increase corresponds to the addition of one molecule of tBPA (158.2 Da) plus one oxygen atom. As shown in Figure 2, the 174 Da increase in mass of CYP2B4 is NADPH- and time-dependent. After a 15 min of incubation, the protein moiety of CYP2B4 was fully modified by tBPA with a stoichiometry of one adduct per apoprotein. No further modification of the CYP2B4 apoprotein was observed even after extended periods of incubation (≥ 6×t1/2) under turnover conditions. This stoichiometric modification of the apoprotein is indicative of a single amino acid target for covalent modification by tBPA. A more accurate mass of this adduct was determined to be 174.1±0.2 by measuring the mass of the GSH adduct formed by CYP2B1 [44].
Figure 2.
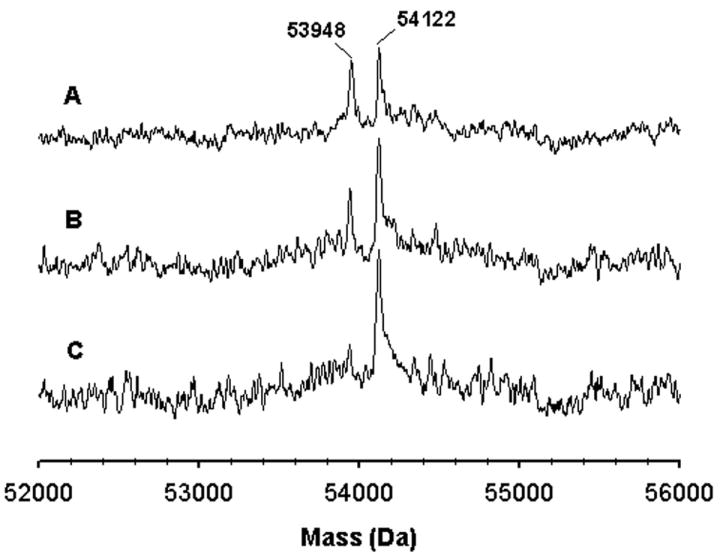
Time-dependent formation of the protein adduct of CYP2B4 with tBPA. The molecular mass of CYP2B4 was analyzed using ESI-LC/MS. The three traces represent the samples obtained at 5 (A), 10 (B), and 15 min (C) after the addition of NADPH to the primary reaction mixture. This figure is reproduced from Figure 3 of ref. [45] with permission from Molecular Pharmacology.
Examination of the integrity of the heme moiety in the modified CYP2B1, 2B4 and 2B6 by HPLC analyses revealed that no heme adducts were formed during the inactivation of CYP2B1 and CYP2B4. However, approximately 3% of the native heme was modified by tBPA to give a heme adduct with CYP2B6 (unpublished data). The identity of the heme adduct formed with CYP2B6 remains to be determined. It is clear that tBPA inactivates CYP2B1 and CYP2B4 exclusively by covalent modification of a single amino acid residue, and inactivates CYP2B6 primarily through amino acid modification with a very minor contribution from heme modification.
Obviously, the key to understanding the inhibitory action of tBPA lies in the identification of the modified amino acid residue. Our previous studies showed that 2EN covalently modified the peptide 290-314 of CYP2B1 where Thr302 is located. Even though Thr302 is a likely target, the specific residue modified by 2EN has not yet been unequivocally identified. With better sample preparation and data mining techniques with SEQUEST, we were able to identify that Thr302 is covalently modified by tBPA in CYP2B1, CYP2B4, and CYP2B6 [45, 46]. As shown in Figure 3, the peptide 294SLFFAGTETTSTTLR308 of CYP2B4 has a mass increase of 174 Da in the modified protein compared with the theoretical mass based on its amino acid sequence. Peptide sequencing of this peptide by LC-MS/MS unequivocally identified Thr302 as the only modified residue. Additional residues other than Thr302 may also be listed as candidates for covalent modification from SEQUEST searches. For example, Thr100 of CYP2B1 and Lys274 of CYP2B6 were listed as potential sites of covalent modifications. Even though the SEQUEST search yielded less favorable Xcorr and Probability scores for Thr100 of CYP2B1 and Lys274 of CYP2B6, we could not rule them out solely based on the scores of the SEQUEST search. However, these two residues were ruled out as the sites for covalent modification of the P450s in subsequent studies using site-directed mutagenesis since mutation of these two residues to aliphatic residues did not prevent covalent modifications by tBPA (unpublished data). Mutation of Thr302 to valine in CYP2B6 did prevent mechanism-based inactivation of CYP2B6 by tBPA, confirming that Thr302 is the only residue susceptible to tBPA modification. Therefore, we strongly recommend that caution be exercised in using only SEQUEST search results to identify modified residues. The suggested site(s) should be verified by alternative methods.
Figure 3.

LC-MS/MS analysis of the tBPA-modified peptide 294SLFFAGTETTSTTLR308 showing that Thr302 is the tBPA-modified residue. Thr302 is marked with an asterisk. The predicted fragment ion series (b and y ions) for the singly charged ion at m/z 1805.4 are denoted as unlabeled and the fragment ions observed for the modified peptide are indicated as labeled. The observed fragment ions are MS/MS spectra of the doubly charged precursor ion at m/z 903.7 obtained in positive mode using the Xcalibur software. This figure was reproduced from Figure 6 of ref. [45] with permission from Molecular Pharmacology.
The covalent modification of Thr302 by tBPA was further established by crystallographic studies of tBPA-modified CYP2B4. Taking advantages of the stoichiometric and essentially complete modification of CYP2B4 by tBPA and the availability of highly expressed recombinant CYP2B4 and cytochrome P450 reductase (CPR), we were able to prepare and purify tBPA-modified CYP2B4 in sufficient quantity ( >1000 nmoles per reaction) for crystallographic studies in collaboration with Dr. Jim Halpert’s laboratory at UCSD. The initial results showed that the tBPA-modified CYP2B4 crystals diffracted at ~3.2-3.5 Å and a (2Fo-Fc) map clearly displayed electron densities near the Thr302 that resembled those expected for tBPA (unpublished data). The crystal structure of the tBPA-modified CYP2B4 is currently being refined to higher resolution. Taken together, these results lead us to conclude that Thr302 is the only amino acid residue in CYP2B4 that is covalently modified by tBPA.
Consequences of the covalent modification of the Thr302 residue in CYP2Bs
tBPA appears to be an ideal probe to examine the consequences of covalent modification of the Thr302 residue since it modifies only Thr302 in CYP2B1 and CYP2B4 with a stoichiometry of 1 and the Thr302-modified enzymes can be readily purified from the reconstituted system for further characterization. As detailed below, biophysical characterization of tBPA-modified CYP2B4 revealed that covalent modification of Thr302 affects the electronic state of the heme, substrate binding in the active site, and the electron transfer from CPR to the P450.
Covalent modification of the Thr302 residue by tBPA perturbs the electronic state of the heme [45]. Similar to that of the unmodified CYP2B4, the optical spectrum of the Thr302-modified CYP2B4 lacks the charge transfer band (CT) at 645 nm, indicating that the ferric heme of the modified CYP2B4 is predominantly in the low-spin state. However, compared to the unmodified CYP2B4, the Soret band of the Thr302-modified CYP2B4 is red-shifted by 5 nm to 422 nm [45]. This significant shift in the Soret band indicates that the micro-environment of the heme has been altered due to the covalent formation of an adduct between tBPA and the Thr302 residue. This change in the heme environment may reflect changes in hydrophobicity and hydration in the active site. The increase in the low-spin character of the heme in the Thr302-modified CYP2B4 was further confirmed by EPR; the g-tensors of the Thr302-modified CYP2B4 are further split to give g-values at 2.491, 2.247 and 1.890 compared with the g-tensors of 2.432, 2.257, 1.917 for the unmodified CYP2B4 and 2.411, 2.247, 1.920 for the unmodified CYP2B4 in the presence of 1 mM tBPA (unpublished data). The EPR results also showed that the Thr302-modified CYP2B4 contains essentially no unmodified CYP2B4 (unpublished data), which is consistent with the results of mass spectral analyses as shown in Figure 2. The ability to produce essentially 100% labeled CYP2B4 enzyme greatly simplifies the interpretation of the data with respect to the effect of this covalent modification on P450 catalysis since it removes the possibility of any complications due to the presence of any significant amount of unmodified enzymes.
The most important impact resulting from the covalent modification of Thr302 by tBPA is that the adduct interferes with substrate binding to the active site and therefore has significant effects on the catalysis [45, 47]. Three different experimental approaches support this hypothesis. First, the tBPA-modified CYP2B4 did not exhibit a typical substrate-induced low-to-high spin shift, or Type I spectral change as judged by the lack of changes in the optical spectrum upon addition of 1 mM benzphetamine to the modified enzyme [45]. Second, the rate of electron transfer from CPR to the tBPA-modified CYP2B4 was not stimulated by the presence of 1 mM benzphetamine, whereas the rate of electron transfer was enhanced by ~20-fold in the unmodified CYP2B4 under the same conditions. Moreover, the rates of electron transfer from CPR to both the unmodified and modified CYP2B4 were comparable in the absence of 1 mM benzphetamine. These results strongly suggest that benzphetamine is not bound in the active site of the modified enzyme in such a way to raise the redox potential of the heme and hence to enhance the electron transfer from CPR to P450s. Third, additional evidence supporting this hypothesis comes from studies of the Thr302-modified CYP2B4 using resonance Raman (RR) spectroscopy [47]. The high-frequency RR spectrum of the Thr302-modified CYP2B4 confirmed that the ferric heme of the modified CYP2B4 is in low-spin state as evidenced by the presence of the spin-state marker bands at 1502 (ν3) and 1583 (ν2) cm-1. Addition of the substrate benzphetamine to the unmodified CYP2B4 led to an increase in the intensity of a high-spin band at 1487 cm-1 at the expense of the low-spin band at 1502 cm-1, typical of a substrate-induced spin conversion. However, addition of benzphetamine to the Thr302-modified CYP2B4 did not enhance the high-spin band at 1487 cm-1 and nor did it affect the low-spin ν3 band. The lack of RR spectral changes provided additional evidence that benzphetamine is not bound in the proper orientation in the active site to influence the electronic state of the heme. The RR spectrum of the CO complex of the ferrous Thr302-modified CYP2B4 showed a relatively large 6 cm-1 downshift of the ν(Fe-CO) stretching mode, with a corresponding ν(C-O) stretching mode upshifted by 9 cm-1 to 1959 cm-1. Both shifts imply substantial decreases in the polarity of the distal heme pocket and weakening of any hydrogen bonding interactions and provide the first direct spectroscopic evidence that the tBPA adduct on the Thr302 is positioned such that it can impact the disposition of exogenous ligands.
As already documented above, tBPA is highly efficient in covalently modifying Thr302. However, the impact of this modification on the catalytic activities of CYP2B4 varies significantly with substrates. With three substrates tested, i.e. 7-ethoxy-4-trifluoromethylcoumarin (7-EFC), benzphetamine and testosterone, the remaining activities of the Thr302-modified enzymes ranged from ~9% for the hydroxylation of testosterone to 30% for the O-deethylation of 7-EFC when compared with the activities of the unmodified enzyme. The activities remaining appear to correlate reasonably with the size of substrate. The more bulky substrates tend to have lower activities (unpublished data).
Molecular basis for the mechanism-based inactivation of CYP2Bs by tBPA
Many acetylenic compounds are known to be mechanism-based inhibitors of P450s (see Table 1), and aryl acetylenes like tBPA and 2EN seems to be the most potent. What is the molecular basis for this potency? In order to answer this question, we used molecular modeling to investigate the binding of tBPA in the active site of the CYP2Bs.
Docking of tBPA into a homology model of CYP2B1 which was constructed based on the closed conformation of CYP2B4 (PDB ID: 1SUO) revealed that tBPA is tucked into a well defined binding pocket surrounded by the heme and several hydrophobic residues. The residues within 4 Å of tBPA include Ile101, Phe115, Phe297, Ala298, Glu301, Thr302, V367 and Ile477. The tert-butyl group is in van der Waals contact with Phe115, Phe297, V367, and Ile477, while the acetylenic group is in van der Waals contact with Ala298 and Thr302. tBPA is oriented in such a conformation that its terminal carbon, denoted as C1, is 3.1 Å away from the heme iron (Fe) and also 3.3 Å away from the Oγ of Thr302 with an angle ∠Fe-C1-Oγ of 138.7°. Surprisingly, all of the poses from 100 runs of docking yielded only one cluster, the lowest-energy pose of which is shown in Figure 4A. This pose is ostensibly the most favorable conformation for the bound tBPA. On the basis of this unique binding mode of tBPA in the active site of CYP2B1, we hypothesized that the close proximity of the terminal carbon of tBPA to both the heme iron and to the Oγ atom of Thr302 is the primary reason for the very efficient covalent modification of Thr302 by the reactive intermediate of tBPA.
Figure 4.


Molecular modeling showing the lowest-energy pose for tBPA bound in the active site of CYP2B1 (A) and a comparison of tBPA binding in the active site of CYP2B1 with that in CYP2B6 (B). tBPA was docked into the homology model of CYP2B1 and the crystal structure of CYP2B6 using Autodock 4.0 software [53] and the poses with the lowest binding energy are shown. (A) the lowest-energy pose of tBPA in CYP2B1. tBPA is depicted as a green stick and the residues within 4 Å of tBPA are shown as wires; (B). CYP2B1 is superimposed onto CYP2B6 by structural alignment of the Cα of protein backbones of the two proteins. tBPA is depicted in green and orange sticks for CYP2B1 and CYP2B6, respectively. Only the residues within 4 Å of tBPA are shown in wires, green for CYP2B1 and red for CYP2B6.
It is well documented that acetylenic compounds may inactivate P450s by either protein alkylation or heme alkylation [48-50]. Oxygenation of the internal carbon of the acetylenic group results in N-alkylation of the prosthetic heme, whereas oxygenation of the terminal carbon usually leads to protein alkylation via a ketene intermediate. Evidence supporting the formation of a ketene intermediate includes formation of the acetic acid product resulting from hydrolysis of the ketene during the inactivation of P450 by acetylenes [48, 51]. Covalent modification of Thr302 by tBPA is most likely to occur through an ester bond as shown in Scheme I. The initial oxygenation of the terminal carbon leads to formation of a ketene intermediate which is followed by nucleophilic attack by the Oγ atom of Thr302 on the ketene to form an adduct with Thr302, resulting in an increase in the mass of the apoprotein of 174.1 Da. This increase in mass is in total agreement with our analyses of the masses of tBPA-GSH adducts and tBPA-apoprotein adduct by LC-MS. The snug binding of tBPA in the active site of the protein as shown in Figure 4A may help to avoid the oxygenation of the internal carbon of tBPA thereby preventing the covalent modification of the heme.
Scheme I.
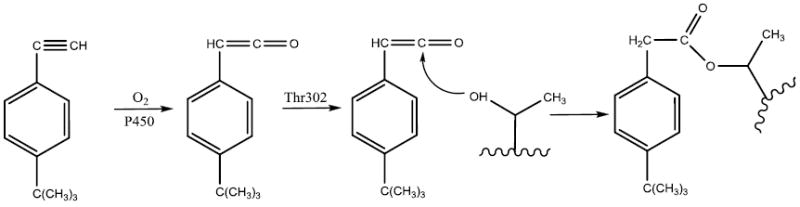
Proposed mechanism for the formation of tBPA-apoprotein adduct.
Docking of tBPA into the crystal structure of CYP2B4 yielded very similar results to those found with CYP2B1 (data not shown). Once again, tBPA is bound in a well defined pocket and surrounded by the same number of residues as in CYP2B1. In addition, Ile363 is in van der Waals contact with the acetylenic group, which may provide additional binding energy to position tBPA in the active site. The geometry of the bound tBPA is very similar to that in CYP2B1; the bond distances between Fe-C1 and C1-Oγ are 2.8 and 3.7 Å respectively and ∠Fe-C1-Oγ is 138.9°. Consistent with experimental results obtained for CYP2B1 and 2B4, this conformation would be expected to result in oxygenation of the C1 carbon, leading to covalent adduction to the apoproteins rather than the heme.
The crystal structure of the K262R variant of CYP2B6 was recently resolved and it showed that the overall structure of CYP2B6 is very similar to that of CYP2B4 [52]. Based on this crystal structure, the mutation of K262→R occurs on the periphery of CYP2B6. Thus we would suggest that its active site architecture is representative of that of the wild type CYP2B6 enzyme and that this crystal structure could be used to investigate the binding mode of tBPA in the active site of CYP2B6. As shown in Figure 4B, tBPA is bound in CYP2B6 in a similar conformation as in CYP2B1 and 2B4 and the residues within 4 Å of the bound tBPA include Ile101, Ile114, Phe115, Phe297, Ala298, Lue363 and Val367 of CYP2B6. Close examination revealed some subtle differences. The C1 carbon of tBPA is 2.5 Å from the heme iron, but 4.3 Å from the Oγ of Thr302. Clearly the C1 carbon moves closer to the heme iron but further away from the Oγ atom of Thr302 in CYP2B6. As a result, the angle ∠Fe-C1-Oγ in CYP2B6 is reduced to 128.1°. For comparison, the structure of CYP2B1 is overlapped with that of CYP2B6 in Figure 4B. As shown, the positions of the Oγ atoms of the Thr302 residues in both proteins remain virtually identical, but tBPA is tilted by ~17° to the heme plane in CYP2B1, resulting in a shorter C1-Oγ distance. One of the forces that contribute to this tilting may stem from the close contact of the tert-butyl group of tBPA with Phe115, Phe297, and Ile477 in CYP2B1, particularly with Ile477 that seals the binding pocket.
CONCLUDING REMARKS
tBPA, a seemingly simple and relatively rigid compound, turns out to be an excellent probe to investigate the mechanism by which P450s are inactivated and the structure/function relationships of P450s and offers advantages for studies on mechanism-based inactivation due to its potency and relatively simple structure. The potency permits us to readily inactivate CYP2Bs and covalently modify Thr302 with approximately 1:1 stoichiometry. This modification on a single site, as observed in CYP2B1 and 2B4, makes it straightforward to interpret the data on the consequences of this modification. Kinetic and spectroscopic studies provide evidence that covalent modification of Thr302 by tBPA affects both the electronic state of the heme and substrate binding to the Thr302-modified enzymes. Specifically the covalent formation of an adduct between tBPA and Thr302 via an ester bond leads to an altered low-spin heme with increased hydrophobicity and decreased hydrogen bonding in the distal heme pocket, and the tBPA adduct on Thr302 prevents substrates from binding properly in the active site. We hypothesize that the steric hindrance imposed by the presence of the tBPA-Thr302 adduct blocking the proper orientation of the substrates in the substrate binding site is the main reason for the inhibition of P450 activity. The residual catalytic activities of the Thr302-modified enzymes appear to decrease with increasing size of substrates.
Molecular modeling provides further insights with respect to the structural and functional relationships of the P450s. It appears that the potent inactivation of the CYP2Bs by tBPA is due to its unique binding mode of tBPA in the active site of CYP2Bs. The close proximity of the terminal carbon of tBPA to both the heme Fe and the Oγ atom of Thr302 provides an ideal scenario for the formation of a reactive ketene intermediate and subsequent covalent linkage with the hydroxyl group of the Thr302 side chain. The very small partition ratio illustrates that the ketene intermediate has a low probability of escaping from the active site, which makes tBPA an efficient inhibitor that targets Thr302 and thus minimizes “collateral” damage to other protein targets. This property of acetylenic compounds should be further explored and exploited in designing specific inhibitors of P450s in the future.
Supplementary Material
Acknowledgments
This series of studies using tBPA were supported in part by a grant from the National Institute of Health (CA16945) to P.F.H. We are also extremely grateful to our collaborators, Dr. David Ballou for use of his stopped-flow instrument, and Dr. James Kincaid for the RR studies, Dr. Brian Hoffman for the EPR studies, and Dr. Jim Halpert for the crystallographic studies of the tBPA-modified CYP2B4.
Abbreviations
- 2EN
2-ethynylnaphthelene
- 17EE
17α-ethynylestradiol
- CPR
cytochrome P450 reductase
- CYP
cytochrome P450
- tBA
tert-butylacetylene
- tBMP
tert-butyl-1-methyl-2-propynyl ether
- tBPA
tert-butylphenylacetylene
- EPR
electron paramagnetic resonance
- LC-MS
liquid chromatography-mass spectrometry
- MALDI-MS
matrix-assisted laser desorption ionization-mass spectrometry
- RR
resonance Raman spectroscopy
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 3.Hamdane D, Zhang H, Hollenberg P. Photosynth Res. 2008;98:657–666. doi: 10.1007/s11120-008-9322-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollenberg PF. Drug Metab Rev. 2002;34:17–35. doi: 10.1081/dmr-120001387. [DOI] [PubMed] [Google Scholar]
- 5.Hollenberg PF, Kent UM, Bumpus NN. Chem Res Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- 6.Uetrecht J. Chem Res Toxicol. 2008;21:84–92. doi: 10.1021/tx700186p. [DOI] [PubMed] [Google Scholar]
- 7.Hong Y, Yu B, Sherman M, Yuan YC, Zhou D, Chen S. Mol Endocrinol. 2007;21:401–414. doi: 10.1210/me.2006-0281. [DOI] [PubMed] [Google Scholar]
- 8.Evans TR, Di Salle E, Ornati G, Lassus M, Benedetti MS, Pianezzola E, Coombes RC. Cancer Res. 1992;52:5933–5939. [PubMed] [Google Scholar]
- 9.Thornalley PJ. Anticancer Drugs. 2002;13:331–338. doi: 10.1097/00001813-200204000-00001. [DOI] [PubMed] [Google Scholar]
- 10.von Weymarn LB, Chun JA, Hollenberg PF. Carcinogenesis. 2006;27:782–790. doi: 10.1093/carcin/bgi301. [DOI] [PubMed] [Google Scholar]
- 11.Moreno RL, Goosen T, Kent UM, Chung FL, Hollenberg PF. Arch Biochem Biophys. 2001;391:99–110. doi: 10.1006/abbi.2001.2390. [DOI] [PubMed] [Google Scholar]
- 12.Kent UM, Roberts-Kirchhoff ES, Moon N, Dunham WR, Hollenberg PF. Biochemistry. 2001;40:7253–7261. doi: 10.1021/bi0102712. [DOI] [PubMed] [Google Scholar]
- 13.Guengerich FP. Curr Drug Metab. 2001;2:93–115. doi: 10.2174/1389200013338694. [DOI] [PubMed] [Google Scholar]
- 14.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. J Biol Chem. 1985;260:16122–16130. [PubMed] [Google Scholar]
- 15.Gerber NC, Sligar SG. J Am Chem Soc. 1992;114:8742–8743. [Google Scholar]
- 16.Gerber NC, Sligar SG. J Biol Chem. 1994;269:4260–4266. [PubMed] [Google Scholar]
- 17.Vidakovic M, Sligar SG, Li H, Poulos TL. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 18.Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y. Proc Natl Acad Sci U S A. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinis SA, Atkins WM, Stayton PS, Sligar SG. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- 20.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 21.Raag R, Martinis SA, Sligar SG, Poulos TL. Biochemistry. 1991;30:11420–11429. doi: 10.1021/bi00112a008. [DOI] [PubMed] [Google Scholar]
- 22.Nagano S, Poulos TL. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 23.Kimata Y, Shimada H, Hirose T, Ishimura Y. Biochem Biophys Res Commun. 1995;208:96–102. doi: 10.1006/bbrc.1995.1310. [DOI] [PubMed] [Google Scholar]
- 24.Vaz AD, Pernecky SJ, Raner GM, Coon MJ. Proc Natl Acad Sci U S A. 1996;93:4644–4648. doi: 10.1073/pnas.93.10.4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeLean JE, Moon N, Dunham WR, Coon MJ. Biochem Biophys Res Commun. 2000;276:762–766. doi: 10.1006/bbrc.2000.3539. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda T, Imai Y, Komori M, Nakamura M, Kusunose E, Satouchi K, Kusunose M. J Biochem. 1993;113:7–12. doi: 10.1093/oxfordjournals.jbchem.a124006. [DOI] [PubMed] [Google Scholar]
- 27.Gotoh O. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 28.Roberts ES, Hopkins NE, Alworth WL, Hollenberg PF. Chem Res Toxicol. 1993;6:470–479. doi: 10.1021/tx00034a013. [DOI] [PubMed] [Google Scholar]
- 29.Roberts ES, Hopkins NE, Zaluzec EJ, Gage DA, Alworth WL, Hollenberg PF. Biochemistry. 1994;33:3766–3771. doi: 10.1021/bi00178a037. [DOI] [PubMed] [Google Scholar]
- 30.Roberts ES, Hopkins NE, Zaluzec EJ, Gage DA, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1995;323:295–302. doi: 10.1006/abbi.1995.9961. [DOI] [PubMed] [Google Scholar]
- 31.Roberts ES, Ballou DP, Hopkins NE, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1995;323:303–312. doi: 10.1006/abbi.1995.9960. [DOI] [PubMed] [Google Scholar]
- 32.Roberts ES, Pernecky SJ, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1996;331:170–176. doi: 10.1006/abbi.1996.0295. [DOI] [PubMed] [Google Scholar]
- 33.Roberts ES, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1998;354:295–302. doi: 10.1006/abbi.1998.0679. [DOI] [PubMed] [Google Scholar]
- 34.Guengerich FP. Mol Pharmacol. 1988;33:500–508. [PubMed] [Google Scholar]
- 35.Lin HL, Kent UM, Hollenberg PF. J Pharmacol Exp Ther. 2002;301:160–167. doi: 10.1124/jpet.301.1.160. [DOI] [PubMed] [Google Scholar]
- 36.Kent UM, Mills DE, Rajnarayanan RV, Alworth WL, Hollenberg PF. J Pharmacol Exp Ther. 2002;300:549–558. doi: 10.1124/jpet.300.2.549. [DOI] [PubMed] [Google Scholar]
- 37.Kent UM, Lin HL, Mills DE, Regal KA, Hollenberg PF. Chem Res Toxicol. 2006;19:279–287. doi: 10.1021/tx050256o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kent UM, Sridar C, Spahlinger G, Hollenberg PF. Chem Res Toxicol. 2008;21:1956–1963. doi: 10.1021/tx800138v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blobaum AL, Kent UM, Alworth WL, Hollenberg PF. J Pharmacol Exp Ther. 2004;310:281–290. doi: 10.1124/jpet.104.065508. [DOI] [PubMed] [Google Scholar]
- 40.Blobaum AL, Lu Y, Kent UM, Wang S, Hollenberg PF. Biochemistry. 2004;43:11942–11952. doi: 10.1021/bi048882s. [DOI] [PubMed] [Google Scholar]
- 41.Blobaum AL, Kent UM, Alworth WL, Hollenberg PF. Chem Res Toxicol. 2002;15:1561–1571. doi: 10.1021/tx020052x. [DOI] [PubMed] [Google Scholar]
- 42.Blobaum AL, Harris DL, Hollenberg PF. Biochemistry. 2005;44:3831–3844. doi: 10.1021/bi0478953. [DOI] [PubMed] [Google Scholar]
- 43.Wright AT, Cravatt BF. Chem Biol. 2007;14:1043–1051. doi: 10.1016/j.chembiol.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin HL, Zhang H, Noon KR, Hollenberg PF. J Pharmacol Exp Ther. 2009;331:392–403. doi: 10.1124/jpet.109.158782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H, Lin HL, Walker VJ, Hamdane D, Hollenberg PF. Mol Pharmacol. 2009;76:1011–1018. doi: 10.1124/mol.109.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin HL, Zhang H, Jushchyshyn M, Hollenberg PF. J Pharmacol Exp Ther. 2010;333:663–669. doi: 10.1124/jpet.109.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mak PJ, Zhang H, Hollenberg PF, Kincaid JR. J Am Chem Soc. 2010;132:1494–1495. doi: 10.1021/ja910276s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan WK, Sui Z, Ortiz de Montellano PR. Chem Res Toxicol. 1993;6:38–45. doi: 10.1021/tx00031a006. [DOI] [PubMed] [Google Scholar]
- 49.Ortiz de Montellano P, Kunze KL. J Am Chem Soc. 1980;102:7373–7375. [Google Scholar]
- 50.Ortiz de Montellano PR, Komives EA. J Biol Chem. 1985;260:3330–3336. [PubMed] [Google Scholar]
- 51.Komives EA, Ortiz de Montellano PR. J Biol Chem. 1987;262:9793–9802. [PubMed] [Google Scholar]
- 52.Gay SC, Shah MB, Talakad JC, Maekawa K, Roberts AG, Wilderman PR, Sun L, Yang JY, Huelga SC, Hong WX, Zhang QH, Stout CD, Halpert JR. Mol Pharmacol. 2010;77:529–538. doi: 10.1124/mol.109.062570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris GM, Goodsell DS, Huey R, Olson AJ. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- 54.Fan PW, Gu C, Marsh SA, Stevens JC. Drug Metab Dispos. 2003;31:28–36. doi: 10.1124/dmd.31.1.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.