

Abstract
Staphylococcus aureus is a common etiologic agent of brain abscesses and possesses numerous virulence factors that manipulate host immunity. One example is superantigens (SAG) that clonally expand T cell subsets bearing specific Vβ receptors. Toll-like receptor 2 (TLR2) is one receptor implicated in S. aureus recognition. However, the interplay between TLR2, SAG, and adaptive immunity during brain abscess formation has not yet been investigated and could reveal novel insights into host-pathogen interactions for regulating protective immunity. A comprehensive analysis of abscess-associated T cell populations in TLR2 KO and WT mice was performed following infection with a S. aureus clinical isolate. Both natural killer T (NKT) and γδ T cell infiltrates were increased in brain abscesses of TLR2 KO mice and produced more IL-17 and IFN-γ compared to WT populations, which could have resulted from elevated bacterial burdens observed in these animals. Analysis of SAG-reactive T cells revealed a predominant Vβ8.1,8.2 infiltrate reactive with staphylococcal enterotoxin B (SEB), whereas SEA-reactive Vβ11 T cells were less numerous. Brain abscesses of TLR2 KO mice had fewer Vβ8.1,8.2 and Vβ11 T cells and produced less TNF-α and IFN-γ compared to WT animals. Treatment of primary microglia with purified SEB augmented TNF-α production in response to the TLR2 ligand Pam3Cys, which may serve to amplify proinflammatory cascades during CNS S. aureus infection. Collectively, these studies demonstrate that TLR2 impacts adaptive immunity to S. aureus infection and modulates SAG responses.
Keywords: Toll-like receptor 2, Staphylococcus aureus, superantigen, staphylococcal enterotoxin B, microglia, natural killer T cells, γδ T cells
INTRODUCTION
Brain abscesses continue to be associated with significant morbidity and mortality, despite recent therapeutic advances (Greenberg, 2008). In addition, long-term morbidity issues arise in patients recovering from these infections as a result of the extensive parenchymal damage typically associated with brain abscess formation, which can manifest as seizures, cognitive deficits, and/or hemiparesis (Davis and Baldwin, 1999; Greenberg, 2008; Lu et al., 2006). Due to the emergence of multi-drug resistant isolates such as methicillin-resistant S. aureus (MRSA) and the ubiquitous nature of bacteria, these CNS infections are likely to persist (Jones et al., 2004; Naesens et al., 2009). Indeed, epidemiological studies have identified S. aureus as one of the most common isolates associated with brain abscesses in humans, with streptococcal species being another frequent etiologic agent of infection (Carpenter et al., 2007; Jones et al., 2004; Prasad et al., 2006). This fact substantiates our need to understand the complexities related to host-pathogen immune dynamics in the brain if we are to be successful at developing novel therapeutics to combat these devastating infections.
Brain abscesses form in response to a parenchymal infection with pyogenic bacteria (Mathisen and Johnson, 1997; Townsend and Scheld, 1998). When pathogenic organisms enter the CNS, an acute edematous response ensues typified by localized microglial and astrocyte activation (Baldwin and Kielian, 2004; Kielian, 2004). The infection culminates in the formation of a mature abscess characterized by extensive necrosis and surrounded by a fibrous capsule. Our laboratory has established a mouse model of experimental brain abscess, which has helped define the basic innate immune pathways involved in abscess pathogenesis and served as a tool to query novel therapeutic agents (Kielian et al., 2001; Kielian et al., 2008). Although we better understand innate immune responses elicited during brain abscess development (Garg et al., 2009; Kielian et al., 2004a; Kielian et al., 2007b), relatively little is known about the impact of adaptive immunity in bacterial clearance and tissue injury.
Antigen-specific CD4+ and CD8+ T cells exist at very low frequencies in naïve hosts. After infection, antigen-reactive naïve T cells undergo clonal expansion resulting in a higher frequency of antigen-specific cells with enhanced effector functions. Upon activation, naïve CD4+ T cells differentiate into Th1, Th2, or Th17 cells depending on the cytokine milieu (Bettelli et al., 2007). In general, with regard to infectious diseases, Th1 cells are required for controlling intracellular infections, Th2 cells combat parasitic infections, and Th17 cells have been implicated in regulating infections caused by extracellular pathogens (Dong, 2008). In contrast, activated CD8+ T cells are cytotoxic and mediate their effector functions through the production of cytokines such as TNF-α as well as perforin granules (Seder, 1999). NKT cells possess semi-invariant TCRs, which are more like the conserved pattern recognition receptors of innate immune cells and participate in anti-microbial defense (Balato et al., 2009; Godfrey et al., 2010; Tupin et al., 2007). γδ T cells are a unique subset that have been postulated to link innate and adaptive immunity by using their TCR as a pattern recognition receptor, which recognizes microbial peptide antigens eliciting IL-17 and IFN-γ release (Born et al., 2010; Deknuydt et al., 2009). To date, the relative frequencies of these various T cell populations and their dynamics of entry/egress into brain abscesses have not yet been investigated.
Activation of the adaptive immune response during bacterial infections leads to a skewing of the Vβ TCR repertoire (Li et al., 1999; Papageorgiou and Acharya, 2000). The Vβ repertoire that is expanded can be predicted based upon preferential TCR affinity of microbial superantigens (SAG). Superantigens are bacterial or viral protein toxins with potent immunostimulatory properties. In contrast to conventional TCR antigens, SAG bind to MHC class II molecules on antigen presenting cells outside the binding groove where they directly cross-link T cell subsets expressing preferential Vβ genes (Li et al., 1999; Papageorgiou and Acharya, 2000). This non-classical mode of T cell activation allows SAGs to activate large fractions of the T cell population (i.e. polyclonal response involving as many as 1 in 5 cells), a significantly higher frequency compared to conventional T cell activation (i.e. monoclonal activation; one in 105–106 cells). Superantigen-induced T cell activation leads to the heightened release of proinflammatory cytokines such as TNF-α that play a role in the induction of toxic shock syndromes often associated with bacterial infections. S. aureus strains can produce several well characterized SAGs including staphylococcal enterotoxin A (SEA) and SEB, which preferentially lead to the expansion of Vβ11 and Vβ8.1,8.2 T cell subsets, respectively (Li et al., 1999; Papageorgiou and Acharya, 2000). With regard to human disease, many S. aureus clinical isolates are high toxin producers (including SEA and SEB)(Dinges et al., 2000; Monk et al., 2004), indicating that the evaluation of SAG involvement during CNS S. aureus infection is clinically relevant.
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs) that recognize invariant pathogen motifs and elicit downstream inflammatory mediator production (Akira, 2006). In addition, TLR signaling is critical for the initiation of adaptive immunity in response to infectious pathogens (Akira et al., 2001; Pasare and Medzhitov, 2004). With regard to S. aureus, TLR2 has been identified as a critical sensor of numerous bacterial motifs including lipoteichoic acids, lipoproteins, and peptidoglycan. We and others have previously demonstrated alterations in innate immune parameters in TLR2 KO mice during brain abscess development (Kielian et al., 2005b; Stenzel et al., 2008); however, relatively little information is currently available regarding the impact of TLR2 on adaptive immunity and whether this PRR participates in the response to bacterial SAGs. The current study demonstrates that the loss of TLR2-dependent signaling enhances NKT and γδ T cell recruitment into brain abscesses, which may be an attempt to facilitate S. aureus clearance that is delayed in TLR2 KO mice. In addition, S. aureus infection induced Vβ8.1,8.2 T cell entry into brain abscesses, which are reactive with staphylococcal enterotoxin B (SEB), whereas few Vβ11 T cells were detected. Interestingly, the number of both Vβ subsets was lower in abscesses from TLR2 KO mice and produced less TNF-α and IFN-γ compared to Vβ T cells recovered from WT animals. Finally, SEB was found to augment TNF-α production in response to the TLR2 ligand Pam3Cys, suggesting a potential mechanism for SAG to augment CNS innate immune responses in addition to its ability to clonally expand Vβ8.1,8.2 subsets. Collectively, these findings demonstrate the complexity of the CNS response to a S. aureus clinical isolate and reveal a link between TLR2, SAG, and Vβ8.1,8.2 and Vβ11 T cells during brain abscess development.
MATERIALS AND METHODS
Mouse strains
TLR2 KO mice (generously provided by Dr. Shizuo Akira, Osaka University, Japan) were backcrossed with C57BL/6 mice for a minimum of eight generations prior to use in these studies. Age- and sex-matched C57BL/6 mice (Charles River, Frederick, MD) were used as WT controls. For all brain abscess studies, TLR2 KO and WT mice were used between 10 and 12 weeks of age.
Generation of experimental brain abscesses
Brain abscesses were induced by the stereotactic intracerebral injection of a methicillin-resistant S. aureus (MRSA) USA300 strain encapsulated in agarose beads as previously described (Kielian et al., 2007b). This USA300 isolate was recovered from an otherwise healthy individual that died from a brain abscess (Sifri et al., 2007). Briefly, mice were anesthetized with 2.5% avertin i.p. and a 1 cm longitudinal incision was made in the scalp to expose the underlying skull sutures to facilitate the identification of bregma. A rodent stereotaxic apparatus equipped with a Cunningham mouse adaptor (Stoelting, Kiel, WI) was used to implant S. aureus-encapsulated beads into the striatum using the following coordinates relative to bregma: + 1.0 mm rostral, + 2.0 mm lateral, and −3.0 mm deep from the surface of the brain. A burr hole was made and a 10 μl Hamilton syringe fitted with a 26-gauge needle was used to slowly deliver 2 μl of S. aureus-laden beads (2 × 103 to 5 × 103 colony forming units [CFU]) into the brain parenchyma. The needle remained in place for 2.5 min following injection to minimize bead efflux and potential leakage into the meninges. The superficial skin incision was closed using surgical glue. Animals were closely monitored over the course of each study for clinical indices of infection. The animal use protocol, approved by the University of Nebraska Medical Center Animal Care and Use Committee, is in accord with the National Institutes of Health guidelines for the use of rodents.
Quantitation of abscess-associated T cell subsets
To determine whether the loss of TLR2-dependent signals impacted the influx of various T cell subsets into the infected CNS, abscess-associated T cell subsets were quantitated by FACS analysis as previously described (Carson et al., 1998; Ford et al., 1995; Kielian et al., 2007b; Renno et al., 1995). Briefly, mice were perfused to eliminate leukocytes from the vasculature, whereupon the entire infected hemisphere was collected to recover abscess-associated cells. This approach ensured that equivalent tissue regions were procured from both TLR2 KO and WT mice for downstream comparisons of leukocyte infiltrates. Following vascular perfusion, tissues were minced in HBSS (Mediatech, Herndon, VA) supplemented with 10% FBS (HyClone, Logan, UT) and filtered through a 70 μm nylon mesh cell strainer using a rubber policeman. At this point, an aliquot of tissue homogenate from each animal was collected to quantitate bacterial burdens. Next, the resulting slurry was digested for 30 min at 37° C in HBSS supplemented with 2 mg/ml collagenase type I and 28 U/ml DNAse I (both from Sigma-Aldrich, St. Louis, MO) to obtain a single-cell suspension. Following enzyme neutralization, cells were layered onto a discontinuous Percoll gradient (1.03 to 1.088 g/ml) and centrifuged at 2,400 rpm for 20 min at room-temperature in a swinging bucket rotor. After centrifugation, myelin debris was carefully aspirated and the cell interface collected. Following extensive washes and incubation in Fc Block™ (BD Biosciences, San Diego, CA) to minimize non-specific antibody binding to Fc receptors, cells were stained with directly conjugated antibodies for multi-color FACS including CD4-PECy5, CD8-APC, NK1.1-FITC, and TCR γδ-PE to detect CD4+ Th, CD8+, NKT (CD4+, NK1.1+) and γδ T cells. Recent studies from our laboratory have established that NK1.1+ cells infiltrating brain abscesses represent NKT and not NK cells (Mariani and Kielian, manuscript in preparation). All antibodies were purchased from BD Biosciences. Cells were analyzed using a BD FACSAria with compensation set based on the staining of each individual fluorochrome alone and correction for autofluorescence with unstained cells. During analysis, events were gated based on a total lymphocyte gate. Controls included cells stained with isotype control antibodies to assess the degree of non-specific staining. Results are presented as the percentage of each cell population recovered from brain abscesses of TLR2 KO or WT mice as well as the absolute number of T cells recovered, with normalization to adjust for the recovery of different cell numbers from the two mouse strains.
Quantitation of SAG-reactive Vβ8.1,8.2 and Vβ11 T cells in brain abscesses and intracellular cytokine staining
To assess the number of Vβ8.1,8.2 and Vβ11 T cells reactive with SEB and SEA, respectively, and whether the frequency of either population was altered in TLR2 KO mice, abscess-associated Vβ8.1,8.2 and Vβ11 T cells were collected and evaluated by intracellular cytokine staining. As described above, TLR2 KO and WT mice harboring brain abscesses were perfused to remove leukocytes from the vasculature and the entire infected hemispheres were collected. Brain tissues were manually disassociated as described above, followed by enzyme digestion and purification using a Percoll gradient. Abscess-associated cells were stained with CD4-AlexaFluor700 and CD4+ T cells purified using a FACSAria (BD Biosciences). FACS-purified CD4+ T cells were immediately stimulated with a cocktail of PMA + ionomycin (Leukocyte Activation Cocktail) in the presence of GolgiPlug™ for 4 h, whereupon cells were incubated with Fc Block™ (all from BD Biosciences) to minimize non-specific antibody binding. Subsequently, T cells were stained with directly-conjugated antibodies against Vβ8.1,8.2 and Vβ11 (both conjugated to FITC; BD Biosciences), fixed and permeabilized with BD Cytofix/Cytoperm Fixation and Permeabilization Solution Kit (BD Bioscience), and stained for intracellular TNF-α or IFN-γ using anti-TNF-α eFluor450® (eBioscience, SanDiego, CA) and anti-IFN-γ-PE-Cy7 (BD Biosciences) antibodies. Cells were analyzed using a BD LSRII with compensation set based on the staining of each individual fluorochrome alone and correction for autofluorescence with unstained cells. Controls included cells stained with directly-conjugated isotype control antibodies to assess the degree of non-specific staining. During analysis, events were collected within the CD4+ gate and results are reported as the absolute number of Vβ8.1,8.2 and Vβ11 T cells recovered from brain abscesses of TLR2 KO or WT mice, with normalization to adjust for the recovery of different cell numbers from the two mouse strains.
Quantitation of abscess-associated bacterial burdens
To quantitate the numbers of viable bacteria associated with brain abscesses of TLR2 KO and WT mice, serial 10-fold dilutions of abscess homogenates were plated onto blood agar plates. Titers were calculated by enumerating colony growth and are expressed as colony forming units (CFU) per gram wet tissue weight.
Western blotting
To demonstrate whether the superantigens SEA or SEB were expressed by S. aureus during brain abscess development, Western blots were performed as previously described (Kielian et al., 2005a). A total of 20 μg protein from brain abscess homogenates was run on 10% SDS-PAGE gels and following transfer, blots were probed with antibodies specific for SEA (Sigma) and SEB (Santa Cruz, San Diego, CA). Purified SEA and SEB (both from Sigma) were included in Western blots as positive controls to demonstrate antibody specificity.
Effects of S. aureus superantigens on microglial activation
Primary microglia were isolated from the brains of neonatal C57BL/6 mice (2–4 days of age) as previously described (Esen and Kielian, 2007). To determine the effects of staphylococcal SAG on microglial activation, microglia were exposed to purified SEA or SEB in the presence or absence of the TLR2 ligand Pam3Cys (Invivogen, San Diego, CA) to assess whether SAG exhibited any additive/synergistic actions with a PAMP relevant to S. aureus. In some experiments, microglia were pre-treated with 100 ng/ml recombinant mouse IFN-γ (BD Biosciences) for 24 h prior to SAG exposure to augment MHC Class II expression, to examine whether SAG effects would be enhanced. Microglia were exposed to Pam3Cys ± SEA/SEB for 24 h, whereupon conditioned supernatants were collected for downstream ELISA analysis. The effects of SEA/SEB on microglial viability were evaluated by the ability of mitochondria to convert the substrate (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; MTT) into formazan crystals as previously described (Kielian et al., 2004b).
Enzyme linked immunosorbent assay (ELISA)
Quantitation of TNF-α levels in conditioned supernatants from microglia treated with Pam3Cys ± SEA/SEB was performed using a standard sandwich ELISA kit according to the manufacturer’s instructions (mouse OptEIA, BD Biosciences; lower limit of sensitivity = 15.6 pg/ml).
Statistics
Significant differences in bacterial titers and T cell infiltrates between TLR2 KO and WT mice at a particular time point were determined using a paired Student’s t-test (SPSS Science, Chicago, IL). For studies examining the effects of SEA/SEB on TLR2-mediated signaling in response to Pam3Cys, a one-way analysis of variance (ANOVA) followed by the Holm-Sidak method for multiple pair-wise comparisons was performed. For all analyses, a p-value of less than 0.05 was considered to be statistically significant.
RESULTS
TLR2 loss influences the frequency of specific T cell subsets during S. aureus brain abscess development
To examine whether the loss of TLR2 affected the influx of various T cell subsets into brain abscesses, FACS analysis was performed in TLR2 KO and WT mice. Although a recent study from our laboratory had examined abscess-associated CD4+ and CD8+ T cell infiltrates in TLR2 KO mice, only limited time points were examined and NKT and γδ T cells were not included in the analysis (Nichols et al., 2009). In addition, our earlier report utilized a S. aureus laboratory adapted strain (RN6390) that is easily amenable to genetic manipulation and possesses a deletion in rsbU, which encodes a positive regulator essential for activation of the stress response sigma factor B (Kullik et al., 1998). In contrast, the current study utilized a S. aureus USA300 isolate with important clinical origins (i.e. recovered from an otherwise healthy patient who died from a brain abscess)(Sifri et al., 2007), which is likely more reminiscent of strains causing natural CNS infections. CD4+ T cell infiltrates were first detectable on day 3 and progressively increased until day 10 after S. aureus exposure, representing the most numerous T cell population associated with brain abscesses (Fig. 1A and Supplemental Fig. 1). There were no significant differences in the relative percentages of CD4+ T cells in brain abscesses of TLR2 KO versus WT mice (Fig. 1A). CD8+ T cells were the next most frequent population and were significantly elevated at day 7 post-infection in TLR2 KO animals (Fig. 1B and Supplemental Fig. 1; p < 0.05, paired t-test). Interestingly, both NKT and γδ T cell infiltrates were increased in brain abscesses of TLR2 KO mice at early time points following S. aureus infection (i.e. days 3 and 7; p < 0.05, paired t-test), but were roughly equivalent at later intervals (i.e. days 10 and 14; Fig. 1C and D and data not shown). Similar relationships were observed between TLR2 WT and KO mice when absolute numbers of each T cell population were determined (Supplemental Table 1).
Figure 1. TLR2 loss induces transient increases in NKT, γδ, and CD8+ T cell recruitment into brain abscesses.

Abscess-associated cells from TLR2 KO and WT mice (n = 4–6 per group) were recovered at the indicated time points post-infection and subjected to FACS analysis to identify CD4+ (A), CD8+ (B), NKT (C), and γδ T cell (D) infiltrates. Results are presented as the average percentage of each population recovered from a total of three independent experiments (mean ± SEM). Similar trends were obtained when absolute T cell numbers were determined based on the total number of cells recovered from the brains of TLR2 KO or WT mice. Significant differences in abscess-associated T cell subsets between TLR2 KO and WT mice are denoted by asterisks (*, p < 0.05).
Quantitation of bacterial burdens associated with brain abscesses of TLR2 KO and WT mice revealed an important role for TLR2 in S. aureus clearance. Specifically, bacterial burdens were significantly increased in TLR2 KO animals at day 7 post-infection (p < 0.05, paired t-test) and remained elevated at day 14 compared to WT mice, although S. aureus titers had also begun to decline in the KO group over time (Fig. 2). Collectively, these results demonstrate the heightened accumulation of NKT and γδ T cells during the early stages of brain abscess formation in TLR2 KO mice, two cell types that span innate and adaptive immunity. It is possible that the enhanced recruitment of these cells is an attempt to compensate for the loss of critical signal(s) that would normally be elicited by TLR2 in response to S. aureus challenge.
Figure 2. TLR2 plays an important role in S. aureus clearance from brain abscesses.

Brain abscess tissues from TLR2 KO and WT mice (n = 4–6 per group) were collected at the indicated time points post-infection and bacterial burdens determined by quantitative culture. Results are expressed as the colony forming units (cfu) of S. aureus per gram of wet tissue weight and were combined from four independent experiments. Significant differences in bacterial titers between TLR2 KO and WT mice are denoted by asterisks (*, p < 0.05).
We next characterized cytokine secretion profiles of the various abscess-associated T cell populations to determine whether alterations in key proinflammatory cytokines were apparent between TLR2 KO and WT mice. Intracellular cytokine staining revealed that the majority of CD4+ T cells infiltrating brain abscesses were Th17 and Th1 subtypes producing IL-17 and IFN-γ, respectively (Fig. 3). In addition, a population of IL-17 and IFN-γ double-positive cells was demonstrated (Supplemental Fig. 2). In general, the percentages of IL-17 producing CD4+ and CD8+ T cells were reduced in TLR2 KO mice at days 7 and 10, respectively (Fig. 3A and C; p < 0.05, paired t-test). In contrast, the numbers of cells producing IFN-γ was increased in both CD4+ and CD8+ T cells in TLR2 KO mice at day 10 post-infection compared to WT animals (Fig. 3B and D). Similar to the increase in NKT infiltrates in TLR2 KO mice, a higher percentage of abscess-associated NKT cells from TLR2 KO animals produced IL-17 and IFN-γ compared to NKT cells from WT mice (Fig. 3E and F; Supplemental Fig. 2). These data indicate that IL-17 and IFN-γ production are elevated in select T cell subsets in the absence of TLR2-dependent signaling, which may represent an attempt to augment the CNS immune response to contain bacterial infection.
Figure 3. Loss of TLR2-dependent signaling leads to alterations in T cell cytokine secretion profiles.

Abscess-associated CD4+ (A and B), CD8+ (C and D), and NKT (E and F) cells from TLR2 KO and WT mice (n = 4–6 per group) were recovered at the indicated time points post-infection and sorted by FACS. After sorting, cells were immediately activated with PMA/ionomycin/Golgi Plug for 4 h and processed for intracellular cytokine staining for IL-17 and IFN-γ. Results are presented as the average percentage of each population recovered from a total of three independent experiments (mean ± SEM). Similar trends were obtained when the absolute number of T cells producing cytokines was determined based on the total number of cells recovered from the brains of TLR2 KO or WT mice. Significant differences in cytokine production by abscess-associated T cell subsets from TLR2 KO and WT mice are denoted by asterisks (*, p < 0.05).
CNS S. aureus infection leads to the preferential accumulation of Vβ8.1,8.2 T cells
S. aureus is known to express numerous secreted virulence factors, including superantigens (SAG). Depending on the isolate, S. aureus has been reported to harbor at least 20 distinct SAGs (Fraser and Proft, 2008). Among the best characterized are SEA and SEB, which trigger the clonal expansion of T cells bearing the Vβ11 and Vβ8.1,8.2 TCRs, respectively (Dinges et al., 2000; Foster, 2005). Upwards of 30% T cell expansion can occur following SAG stimulation and two prominent cytokines produced by SAG-reactive Vβ T cells are TNF-α and IFN-γ (Papageorgiou and Acharya, 2000). Currently, the frequency of SEA- and SEB- reactive Vβ T cell subsets associated with CNS S. aureus infection and the relationship between TLR2 and Vβ T cells have not yet been explored. Since CD4+ T cells have been reported to express TLR2 and respond to pathogen-associated molecular patterns relevant to S. aureus (Kabelitz, 2007; Komai-Koma et al., 2004; Reynolds et al., 2010), it is possible that TLR2 deficiency may alter the numbers of SAG-reactive Vβ populations in brain abscesses.
In terms of frequency, abscess-associated Vβ8.1,8.2 T cells were dramatically higher compared to Vβ11 cells (Fig. 4). This finding could be explained by SEB production by S. aureus during brain abscess formation, whereas SEA was not detectable (Fig. 5 and data not shown, respectively). In addition, both Vβ T cell populations were reduced in brain abscesses of TLR2 KO mice (Fig. 4). Because Vβ8.1,8.2 T cells outnumbered Vβ11 cells in brain abscesses, we evaluated the cytokine expression profiles of the former in TLR2 KO and WT mice. Upon re-stimulation with PMA/ionomycin, intracellular cytokine staining demonstrated that the number of abscess-associated Vβ8.1,8.2 T cells producing TNF-α was reduced in TLR2 KO mice compared to WT animals at both 7 and 14 days post-infection (Fig. 6). Similar effects were observed with IFN-γ producing Vβ8.1,8.2 T cells, where the number of cytokine secreting cells was lower in TLR2 KO mice compared to WT animals (Fig. 6). A subpopulation of total CD4+ and Vβ8.1,8.2 T cells were found to produce both TNF-α and IFN-γ (Supplemental Fig. 3). Although the percentages of TNF-α and IFN-γ expressing cells were roughly equivalent between TLR2 WT and KO mice (Supplemental Fig. 3), calculation of absolute cell numbers revealed dramatic decreases in TLR2 KO animals based on the fact that total cellular infiltrates were increased in these mice. Collectively, these results indicate that TLR2 influences Vβ8.1,8.2 T cell frequency and cytokine release during brain abscess development.
Figure 4. SEB-reactive Vβ8.1,8.2 T cells are more frequent in CNS S. aureus infection compared to SEA-reactive Vβ11 T cells.

Abscess-associated cells from TLR2 KO and WT mice (n = 4–6 per group) were recovered at the indicated time points post-infection and stained with CD4-AlexaFluor 700 and Vβ8.1,8.2-FITC (A) or Vβ11-FITC (B) to identify the numbers of infiltrating SEB- and SEA-reactive T cells, respectively. For analysis, the percentages of Vβ8.1,8.2+ or Vβ11+ T cells within the CD4+ gate were evaluated. Results are presented as the absolute number of Vβ T cell subsets combined from a total of three independent experiments.
Figure 5. Staphylococcal enterotoxin B (SEB) is expressed during brain abscess formation.
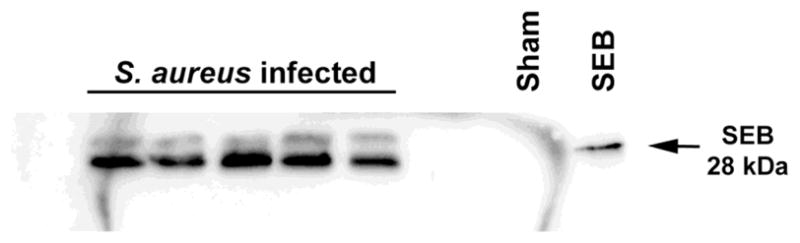
Brain abscess homogenates were prepared from individual animals at day 7 post-infection, whereupon SEB expression was evaluated by Western blotting. A sample collected from a sham injected animal and purified SEB were included as negative and positive controls, respectively.
Figure 6. TLR2-dependent signals influence cytokine release from Vβ8.1,8.2 T cells following S. aureus infection.

Abscess-associated CD4+ T cells from TLR2 KO and WT mice (n = 4–6 per group) were recovered at the indicated time points post-infection by FACS, whereupon cells were immediately activated with PMA/ionomycin/Golgi Plug for 4 h. Cells were stained with Vβ8.1,8.2-FITC and subsequently processed for intracellular cytokine staining for TNF-α and IFN-γ. Results are presented as the absolute number of Vβ8.1,8.2+ cytokine producing T cells combined from a total of three independent experiments.
Staphylococcal enterotoxins augment microglial inflammatory mediator release in response to a TLR2 ligand
Previous reports have demonstrated that SAGs are capable of augmenting TLR4-mediated signaling in response to LPS in dendritic cells and monocytes (Hopkins et al., 2005; Rossi et al., 2004). However, to the best of our knowledge, no studies have investigated the ability of SAG to augment TLR-mediated signaling in response to PAMPs in microglia. This is a relevant question to address since microglia are exposed to both PAMPs and SAG during brain abscess development. Exposure of primary microglia to purified SEB did not elicit TNF-α production at any of the doses examined (Fig. 7A). However, SEB was capable of augmenting TNF-α production in combination with the TLR2 agonist Pam3Cys, compared to cytokine levels observed with Pam3Cys alone (Fig. 7A; p < 0.05, one-way ANOVA). Similar effects were observed following microglial treatment with SEA ± Pam3Cys (data not shown). Importantly, the combination of SEB with Pam3Cys did not induce any evidence of cytotoxicity (Fig. 7B). These findings indicate that besides their ability to induce the clonal expansion of Vβ reactive T cell subsets, SAGs are also capable of enhancing microglial activation in response to TLR2 ligands, which may serve as an amplification loop for antibacterial immunity during S. aureus-induced brain abscess formation.
Figure 7. S. aureus SEB augments microglial TNF-α production in response to the TLR2 ligand Pam3Cys.

Primary microglia were pre-treated for 24 h with 100 ng/ml of recombinant mouse IFN-γ, whereupon cells were exposed to various doses of purified SEB either alone or in combination with the TLR2 ligand Pam3Cys (10 ng/ml). Conditioned medium was collected at 24 h following microglial stimulation and TNF-α expression was quantitated by ELISA. Results presented are representative of three independent replicates for each treatment (mean ± SD) and were replicated in three separate experiments. Significant differences between microglia stimulated with Pam3Cys alone versus Pam3Cys in combination with SEB are denoted by asterisks (*, p < 0.05).
DISCUSSION
Despite advances in defining innate immune pathways elicited during brain abscess development, little information is currently available regarding the functional impact of adaptive immunity in bacterial clearance. Innate immune responses must be tightly regulated to effectively eradicate bacteria while limiting the extent of bystander damage to surrounding brain tissue. This is essential since the CNS is not capable of robust regeneration that would be required to resolve such widespread parenchymal damage characteristic of a brain abscess. It is anticipated that adaptive immune mechanisms are required during the later stages of infection to tailor ongoing innate anti-bacterial responses towards effective pathogen resolution. Therefore, understanding the role that adaptive immunity plays during brain abscess development may identify novel pathways that could be targeted for potential therapeutic interventions.
The loss of TLR2-dependent signaling was found to impact various T cell populations in opposing manners. For example, TLR2 KO mice exhibited enhanced NKT, γδ, and CD8+ T cell infiltrates into brain abscesses, whereas the recruitment of Vβ8.1,8.2 and Vβ11 T cells was reduced compared to WT animals. With regard to NKT cells, these cells become rapidly activated in response to IL-12 even when their invariant TCR is not engaged by a lipid antigen (Tupin et al., 2007). We have previously reported exaggerated IL-12 production in TLR2 KO microglia (Kielian et al., 2005a); therefore, heightened IL-12 release may be one trigger to augment NKT cells in the absence of TLR2 signaling, since IL-12 is a potent activator of these cells (Balato et al., 2009; Godfrey et al., 2010). The ability to become activated in response to the local cytokine milieu makes NKT cells able to rapidly respond to a wide array of infectious pathogens, similar to other cells of the innate immune system. Activated NKT cells produce large amounts of IL-17 and a higher percentage of NKT cells isolated from brain abscesses of TLR2 KO mice were IL-17-positive compared to WT animals. NKT cells can also express Vβ8.2 (Tupin et al., 2007); therefore, they may also be subject to the mitogenic effects of SEB, although this remains speculative. Indeed, a recent study demonstrated that NKT cells may regulate cytokine responses following SEB exposure (Ragin et al., 2006). Finally, enhanced NKT cell accumulation in brain abscesses of TLR2 KO mice may be a consequence of the elevated bacterial burdens seen in these animals. We also observed an increase in γδ T cells in brain abscesses of TLR2 KO mice. Recent studies have demonstrated that γδ T cells can be induced to express MHC Class II and assume antigen presenting cell functions (Born et al., 2010; Brandes et al., 2005; Cheng et al., 2008). Based on this knowledge, it is possible that infiltrating γδ T cells could substitute for the loss of TLR2 signaling by antigen presenting cells; however, this possibility remains highly speculative. Also, the percentages of IL-17 secreting γδ T cells from TLR2 KO mice was elevated compared to WT animals, which may indirectly serve to recruit additional PMN in an attempt to contain S. aureus infection (Witowski et al., 2000; Ye et al., 2001).
One question that arises from this work relates to the major cellular target of TLR2 signaling in vivo. This is a difficult issue to address since there are numerous cell types associated with brain abscesses that express TLR2, including both CNS resident and infiltrating leukocytes. In particular, microglia and astrocytes (CNS intrinsic) as well as neutrophils, macrophages, CD4+ T cells, and even NKT and γδ T cells have recently been reported to express TLR2 (Akira, 2006; Kielian, 2006; Martin et al., 2009; Mokuno et al., 2000). However, a plausible scenario of action can be proposed based on the infiltration kinetics and activation of responding cell types. For example, immediately following S. aureus infection in the CNS, TLR2 is likely pivotal for microglial activation and the production of numerous chemokines and cytokines critical for the recruitment of peripheral immune cells into the site of infection and their subsequent activation (Kielian et al., 2005a). Astrocytes may also participate in this initial recognition event, which are an important source of chemokines following S. aureus infection and can be triggered via TLR2-dependent signaling (Esen et al., 2004). Recent studies from our laboratory using bone marrow chimeras with MyD88 KO mice revealed an essential role for CNS intrinsic cells, presumably microglia and astrocytes, in eliciting a wild type inflammatory response during the early stage of brain abscess development (Garg et al., 2009). However, this phenotype cannot be solely attributed to TLR2 since numerous other TLRs relevant to S. aureus recognition, such as TLR9 and the IL-1R, are both driven by MyD88-dependent pathways and could conceivably participate in the CNS antibacterial response. Nonetheless, this study clearly demonstrated a key role for resident microglia and/or astrocytes in the initial recognition of S. aureus and eliciting downstream inflammatory cascades (Garg et al., 2009).
Within 12–24 h following infection, the importance of TLR2 may shift to include newly recruited immune cells from the peripheral circulation, primarily neutrophils, followed by macrophages that accumulate at significant levels within 48–72 h post-infection (Kielian, 2004; Kielian et al., 2001). At this juncture, TLR2 engagement by S. aureus would provide critical signals for amplification of the CNS immune response targeted at S. aureus clearance. Extensive studies in the experimental brain abscess model have established that the innate immune response is not effective at containing bacterial replication until approximately 5–7 days following S. aureus exposure (Kielian et al., 2001; Kielian et al., 2004a; Kielian et al., 2007a). At this time, T and NKT cell infiltrates are beginning to be prevalent and appear to play an important role in effective bacterial clearance and maintenance of maximal innate immune responses (Mariani and Kielian, manuscript in preparation). In addition, unpublished data from our laboratory has revealed an important role for both CD4+ and NKT cells in effective bacterial clearance and maintaining maximal innate immune responses during the later stages of brain abscess development (Mariani and Kielian, manuscript in preparation). TLR2 may also be engaged on these infiltrating T cell populations to impact later stages of infection. In summary, TLR2 expression is likely important for triggering activation of both CNS resident and the various infiltrating innate and adaptive immune populations throughout the course of brain abscess development. This widespread utilization of TLR2-dependent signaling is evident by the increased mortality rates and altered T cell responses observed in TLR2 KO mice during the current study. A schematic depicting these multiple potential targets of TLR2 signaling are presented in Figure 8.
Figure 8. TLR2 has the potential to impact diverse cell populations throughout the course of CNS bacterial infection.

Immediately following S. aureus invasion in the brain parenchyma, resident microglia and astrocytes recognize the pathogen, in part, via TLR2. TLR2 signaling leads to the downstream production of inflammatory cytokines and chemokines that trigger the egress of neutrophils and monocytes (which differentiate into macrophages) from activated cerebral vasculature into the evolving abscess during the acute stage of infection. Within days, T cell populations begin to accumulate within the abscess proper, many of which have been shown to express TLR2 that could conceivably trigger cellular activation.
Previous studies from our laboratory have evaluated the functional importance of TLR2 in microglial activation to S. aureus comparing responses of TLR2 KO and WT microglia (Kielian et al., 2005a). These experiments revealed that TLR2 loss leads to the exaggerated production of IL-12 cytokine family members in response to intact bacteria; however, this effect was selective in that the expression of numerous other mediators (including chemokines) was not affected. Therefore, our in vitro data with TLR2 KO microglia does not point towards altered chemokine expression (Kielian et al., 2005a); however, it is possible that T cell chemokine production is affected in TLR2 KO mice in vivo to account for the differential recruitment of various T cell populations into brain abscesses of TLR2 KO mice. This possibility is being explored in ongoing studies in our laboratory that will be the subject of a future publication and is beyond the scope of the current report.
The complexity of brain abscess development, both in terms of responding cell types and divergent cytokine production by each, makes is very difficult to assign the role of IL-17 production to any particular cell population. However, when considering the percentages of each cell type associated with brain abscesses, an overall impression in terms of IL-17 can be envisioned. For example, we have previously reported that IL-17 production is higher in brain abscesses of TLR2 KO mice (Kielian et al., 2005b), which corresponds to the heightened numbers of IL-17 producing NKT cells from these animals. In addition, γδ T cell infiltrates were increased in TLR2 KO mice and these cells are also capable of IL-17 production (Cua and Tato, 2010; Martin et al., 2009). However, we did not examine cytokine production in γδ T cells in the current study due to limiting cell numbers recovered from brain abscesses. In addition to IL-17 release from NKT and γδ T cells, IL-17 has been reported to be produced by astrocytes and microglia (Das Sarma et al., 2009; Kang et al., 2010; Tzartos et al., 2008). Therefore, it is possible that all of these mechanisms serve to augment IL-17 production within brain abscesses of TLR2 KO mice. In contrast, the percentages of CD4 and CD8 T cells producing IL-17 were reduced in TLR2 KO animals; however, this was only noted at one time point for each cell population (i.e. days 7 and 14, respectively). Therefore, based on the fact that IL-17 expression is elevated in brain abscesses of TLR2 KO mice, it is evident that this transient reduction in IL-17 production from CD4 and CD8 T cells is not significant enough to impact overall IL-17 levels associated with brain abscesses of TLR2 KO animals.
In contrast to the increased frequency of NKT and γδ T cells infiltrating brain abscesses of TLR2 KO mice, the recruitment of SEA- and SEB-reactive Vβ11 and Vβ8.1,8.2 T cells, respectively, was attenuated compared to WT animals. The mechanism(s) responsible for this finding are not known but could be explained by: 1) reduced chemokine or altered adhesion molecule expression at the blood-brain barrier; 2) limited expansion of Vβ8.1,8.2 and Vβ11 T cells within the infected CNS; or 3) increased apoptosis of Vβ subsets in TLR2 KO mice following SAG expansion. In addition, the frequency of Vβ8.1,8.2 T cells producing either TNF-α or IFN-γ was reduced in TLR2 KO mice, which correlated with the inability to rapidly clear S. aureus from the CNS in these animals. It is also plausible that the loss of TLR2 directly impacts CD4+ T cell behavior since these cells have been reported to express functional TLR2 (Kabelitz, 2007; Komai-Koma et al., 2004). In this regard, the loss of TLR2-dependent signaling may diminish a stimulatory signal required for the clonal expansion of SEB-reactive Vβ8.1,8.2 T cells, resulting in reduced accumulation, as was observed in brain abscesses of TLR2 KO mice in this study. However, this possibility remains highly speculative and awaits future study. Nonetheless, our results do indicate that SEB is capable of augmenting TNF-α production by microglia in combination with the TRL2 ligand Pam3Cys. This may represent a possible mechanism to amplify inflammation during brain abscess development by SAG actions on antigen presenting cells in addition to Vβ8.1,8.2 T cells.
It is important to note that a few findings in the current study differed from a previous report from our laboratory with TLR2 KO mice (Nichols et al., 2009). In particular, our earlier paper demonstrated that Th17 infiltrates were enhanced in TLR2 KO mice, whereas in the current study we did not observe any dramatic differences in CD4+ Th17 cells. However, we show here that NKT and γδ T cells from TLR2 KO mice did produce more IL-17, in agreement with our previously proposed mechanism whereby heightened IL-17 release may represent a compensatory response in an attempt to contain S. aureus infection. This discrepancy is likely explained by the fact that the current studies were performed with a S. aureus clinical isolate that was recovered from a patient who died from a brain abscess, whereas our earlier report utilized a laboratory adapted strain of S. aureus. In fact, this point was noted by Stenzel et al. who used a S. aureus clinical isolate to demonstrate higher mortality rates and bacterial burdens in TLR2 KO mice compared to WT animals (Stenzel et al., 2008). With our use of a new brain abscess clinical isolate, our results are more similar to Stenzel et al. and point to the important issue of distinct S. aureus strains being able to elicit diverse immunological effects. Nonetheless, our studies have extended these initial observations to characterize the effects of TLR2 loss on other T cell populations, SAG-reactive T cell subsets, and their cytokine secretion patterns, where novel differences were identified.
In summary, we demonstrated that TLR2-dependent signals are important for the accumulation of SAG-reactive Vβ8.1,8.2 and Vβ11 T cells into brain abscesses and their subsequent activation. In contrast, TLR2 either delivers a negative regulatory signal to attenuate NKT and γδ T cell influx into the infected CNS, or alternatively, the heightened accumulation of these subsets may have resulted from the inability to effectively clear S. aureus from the brain. The latter may be more plausible as a higher proportion of abscess-associated NKT and γδ T cells infiltrating TLR2 KO mice produced IFN-γ and IL-17, two key anti-bacterial cytokines. Collectively, these studies highlight the impact of TLR2 on adaptive immune responses and point to the important role this pattern recognition receptor plays during CNS bacterial infection.
Supplementary Material
Acknowledgments
This work was supported by the NIH National Institute of Neurological Disorders and Stroke (NINDS) R01 NS055385 to T.K. The authors thank Amanda Angle for excellent technical assistance, Dr. Shizuo Akira for generously providing the TLR2 KO mice, Dr. Costi Sifri for the USA300 isolate and Dr. Charles Kuszynski, Megan Michalak, and Victoria Smith in the UNMC Cell Analysis Facility for assistance with FACS analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. doi: 10.1007/3-540-32636-7_1. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Balato A, Unutmaz D, Gaspari AA. Natural killer T cells: an unconventional T-cell subset with diverse effector and regulatory functions. J Invest Dermatol. 2009;129:1628–1642. doi: 10.1038/jid.2009.30. [DOI] [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- Born WK, Yin Z, Hahn YS, Sun D, O’Brien RL. Analysis of gamma delta T cell functions in the mouse. J Immunol. 2010;184:4055–4061. doi: 10.4049/jimmunol.0903679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science. 2005;309:264–268. doi: 10.1126/science.1110267. [DOI] [PubMed] [Google Scholar]
- Carpenter J, Stapleton S, Holliman R. Retrospective analysis of 49 cases of brain abscess and review of the literature. Eur J Clin Microbiol Infect Dis. 2007;26:1–11. doi: 10.1007/s10096-006-0236-6. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Cheng L, Cui Y, Shao H, Han G, Zhu L, Huang Y, O’Brien RL, Born WK, Kaplan HJ, Sun D. Mouse gammadelta T cells are capable of expressing MHC class II molecules, and of functioning as antigen-presenting cells. J Neuroimmunol. 2008;203:3–11. doi: 10.1016/j.jneuroim.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, Fitzgerald DC, Shindler KS, Rostami A. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2009;6:14. doi: 10.1186/1742-2094-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LE, Baldwin NG. Brain Abscess. Curr Treat Options Neurol. 1999;1:157–166. doi: 10.1007/s11940-999-0015-7. [DOI] [PubMed] [Google Scholar]
- Deknuydt F, Scotet E, Bonneville M. Modulation of inflammation through IL-17 production by gammadelta T cells: mandatory in the mouse, dispensable in humans? Immunol Lett. 2009;127:8–12. doi: 10.1016/j.imlet.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13:16–34. doi: 10.1128/cmr.13.1.16-34.2000. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- Esen N, Kielian T. Effects of low dose GM-CSF on microglial inflammatory profiles to diverse pathogen-associated molecular patterns (PAMPs) J Neuroinflammation. 2007;4:10. doi: 10.1186/1742-2094-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. [DOI] [PubMed] [Google Scholar]
- Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunol Rev. 2008;225:226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- Garg S, Nichols JR, Esen N, Liu S, Phulwani NK, Syed MM, Wood WH, Zhang Y, Becker KG, Aldrich A, Kielian T. MyD88 expression by CNS-resident cells is pivotal for eliciting protective immunity in brain abscesses. ASN Neuro. 2009:1. doi: 10.1042/AN20090004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol. 2010;11:197–206. doi: 10.1038/ni.1841. [DOI] [PubMed] [Google Scholar]
- Greenberg BM. Central nervous system infections in the intensive care unit. Semin Neurol. 2008;28:682–689. doi: 10.1055/s-0028-1105976. [DOI] [PubMed] [Google Scholar]
- Hopkins PA, Fraser JD, Pridmore AC, Russell HH, Read RC, Sriskandan S. Superantigen recognition by HLA class II on monocytes up-regulates toll-like receptor 4 and enhances proinflammatory responses to endotoxin. Blood. 2005;105:3655–3662. doi: 10.1182/blood-2004-07-2523. [DOI] [PubMed] [Google Scholar]
- Jones ME, Draghi DC, Karlowsky JA, Sahm DF, Bradley JS. Prevalence of antimicrobial resistance in bacteria isolated from central nervous system specimens as reported by U.S. hospital laboratories from 2000 to 2002. Ann Clin Microbiol Antimicrob. 2004;3:3. doi: 10.1186/1476-0711-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004a;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005a;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Esen N, Liu S, Phulwani NK, Syed MM, Phillips N, Nishina K, Cheung AL, Schwartzman JD, Ruhe JJ. Minocycline Modulates Neuroinflammation Independently of Its Antimicrobial Activity in Staphylococcus aureus-Induced Brain Abscess. Am J Pathol. 2007a;171:1199–1214. doi: 10.2353/ajpath.2007.070231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-Like Receptor 2 Modulates the Proinflammatory Milieu in Staphylococcus aureus-Induced Brain Abscess. Infect Immun. 2005b;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, McMahon M, Bearden ED, Baldwin AC, Drew PD, Esen N. S. aureus-dependent microglial activation is selectively attenuated by the cyclopentenone prostaglandin 15-deoxy-Delta12,14- prostaglandin J2 (15d-PGJ2) J Neurochem. 2004b;90:1163–1172. doi: 10.1111/j.1471-4159.2004.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, Johnson J. MyD88-Dependent Signals Are Essential for the Host Immune Response in Experimental Brain Abscess. J Immunol. 2007b;178:4528–4537. doi: 10.4049/jimmunol.178.7.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Syed MMd, Liu S, Phillips N, Wagoner G, Drew PD, Esen N. The synthetic PPAR-γ agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. Journal of Immunology. 2008;180:5004–5016. doi: 10.4049/jimmunol.180.7.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029–3034. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullik I, Giachino P, Fuchs T. Deletion of the alternative sigma factor sigmaB in Staphylococcus aureus reveals its function as a global regulator of virulence genes. J Bacteriol. 1998;180:4814–4820. doi: 10.1128/jb.180.18.4814-4820.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Llera A, Malchiodi EL, Mariuzza RA. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- Lu CH, Chang WN, Lui CC. Strategies for the management of bacterial brain abscess. J Clin Neurosci. 2006;13:979–985. doi: 10.1016/j.jocn.2006.01.048. [DOI] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. quiz 780–761. [DOI] [PubMed] [Google Scholar]
- Mokuno Y, Matsuguchi T, Takano M, Nishimura H, Washizu J, Ogawa T, Takeuchi O, Akira S, Nimura Y, Yoshikai Y. Expression of toll-like receptor 2 on gamma delta T cells bearing invariant V gamma 6/V delta 1 induced by Escherichia coli infection in mice. J Immunol. 2000;165:931–940. doi: 10.4049/jimmunol.165.2.931. [DOI] [PubMed] [Google Scholar]
- Monk AB, Curtis S, Paul J, Enright MC. Genetic analysis of Staphylococcus aureus from intravenous drug user lesions. J Med Microbiol. 2004;53:223–227. doi: 10.1099/jmm.0.05408-0. [DOI] [PubMed] [Google Scholar]
- Naesens R, Ronsyn M, Druwe P, Denis O, Ieven M, Jeurissen A. Central nervous system invasion by community-acquired meticillin-resistant Staphylococcus aureus. J Med Microbiol. 2009;58:1247–1251. doi: 10.1099/jmm.0.011130-0. [DOI] [PubMed] [Google Scholar]
- Nichols JR, Aldrich AL, Mariani MM, Vidlak D, Esen N, Kielian T. TLR2 deficiency leads to increased Th17 infiltrates in experimental brain abscesses. J Immunol. 2009;182:7119–7130. doi: 10.4049/jimmunol.0802656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou AC, Acharya KR. Microbial superantigens: from structure to function. Trends Microbiol. 2000;8:369–375. doi: 10.1016/s0966-842x(00)01793-5. [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6:1382–1387. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Mishra AM, Gupta D, Husain N, Husain M, Gupta RK. Analysis of microbial etiology and mortality in patients with brain abscess. J Infect. 2006;53:221–227. doi: 10.1016/j.jinf.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Ragin MJ, Sahu N, August A. Differential regulation of cytokine production by CD1d-restricted NKT cells in response to superantigen staphylococcal enterotoxin B exposure. Infect Immun. 2006;74:282–288. doi: 10.1128/IAI.74.1.282-288.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, Dong C. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi RJ, Muralimohan G, Maxwell JR, Vella AT. Staphylococcal enterotoxins condition cells of the innate immune system for Toll-like receptor 4 stimulation. Int Immunol. 2004;16:1751–1760. doi: 10.1093/intimm/dxh176. [DOI] [PubMed] [Google Scholar]
- Seder R, Mosmann ™. Differentiation of effectos phenotypes of CD4+ and CD8+ T cells. In: Paul W, editor. Fundamental Immunology. Lippencott-Raven; Philadelphia: 1999. pp. 1879–1908. [Google Scholar]
- Sifri CD, Park J, Helm GA, Stemper ME, Shukla SK. Fatal brain abscess due to community-associated methicillin-resistant Staphylococcus aureus strain USA300. Clin Infect Dis. 2007;45:e113–117. doi: 10.1086/522171. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, Deckert M. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol. 2008;172:132–145. doi: 10.2353/ajpath.2008.070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol. 2007;5:405–417. doi: 10.1038/nrmicro1657. [DOI] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jorres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.