

Abstract
Objective
To determine the influence of apoA-I tertiary structure domain properties on the anti-atherogenic properties of the protein. Two chimeric hybrids with the N-terminal domains swapped (human-M apoA-I and mouse-H apoA-I) were expressed in apoA-I-null mice with adeno-associated virus (AAV) and used to study macrophage reverse cholesterol transport (RCT) in vivo.
Methods and Results
The different apoA-I variants were expressed in apoA-I-null mice that were injected with [H3]cholesterol-labeled J774 mouse macrophages to measure RCT. Significantly more cholesterol was removed from the macrophages and deposited in the feces via the RCT pathway in mice expressing mouse-H apoA-I compared to all other groups. Analysis of the individual components of the RCT pathway demonstrated that mouse-H apoA-I promoted ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux more efficiently than all other variants as well as increasing the rate of cholesterol uptake into liver cells.
Conclusion
The structural domain properties of apoA-I affect the ability of the protein to mediate macrophage RCT. Substitution of the N-terminal helix bundle domain in the human apoA-I with the mouse apoA-I counterpart causes a gain of function with respect to macrophage RCT, suggesting that engineering some destabilization into the N-terminal helix bundle domain and/or increasing the hydrophobicity of the C-terminal domain of human apoA-I would enhance the anti-atherogenic properties of the protein.
Keywords: apoA-I, reverse cholesterol transport, high density lipoprotein, macrophage, apolipoprotein
Introduction
Apolipoprotein A-I (apoA-I) is the major protein component of high density lipoprotein (HDL) and its level in plasma is inversely associated with the risk of cardiovascular disease (1–3). The anti-atherogenic properties of apoA-I arise primarily through its role as a mediator of reverse cholesterol transport (RCT), a process by which excess cholesterol is removed from peripheral tissues and transported to the liver for catabolism (4,5). ApoA-I has been shown to promote macrophage RCT in vivo (6).
Human apoA-I is a 243-residue protein made up of repeating amphipathic α-helices (7) and it contains two domains, a N-terminal α-helix bundle spanning residues 1–189 and a separately folded C-terminal domain that spans the remainder of the molecule (8–10). The more hydrophobic C-terminal domain has high lipid affinity compared to the helix bundle domain (11). Mouse apoA-I, which has 65% amino acid identity with human apoA-I, adopts a similar two-domain structure (12). However, compared to the human protein, the N-terminal domain of mouse apoA-I is relatively unstable and has high lipid affinity while the C-terminal domain is more polar and has poor lipid affinity (12).
The differences in tertiary structure domain characteristics between human and mouse apoA-I present the opportunity to understand how the properties of these domains influence the functionality of apoA-I in the RCT pathway. To investigate this question, we generated two domain-swap variants of human and mouse apoA-I and evaluated their abilities to promote macrophage RCT in vivo.
Methods
Preparation of ApoA-I Adeno-Associated Virus (AAV)
To create the cDNA for the domain-swap Human(residues 1–189)Mouse(residues 187–240) apoA-I hereafter referred to as (human-M apoA-I) and Mouse(residues 1–186)Human(residues 190–243) apoA-I hereafter referred to as(mouse-H apoA-I), WT human and WT mouse apoA-I cDNA in a pAAV vector were engineered using the Stratagene “domain-swap” protocol (12). The resulting cDNA were sequenced to confirm the presence of the intended mutation and submitted to the University of Pennsylvania Vector Core for use in creating the liver specific apoA-I AAV, serotype 8 as described before (13–15).
Macrophage RCT Studies
Experiments were performed in male apoA-I-null C57BL/6 mice obtained from Jackson Labs and fed a chow diet. For each experiment, 30 mice (n = 6/group) received intraperitoneal (ip) injection of AAV8 (1 × 1012 GC) of the appropriate AAV. On day 70 post vector injection, each animal received intraperitoneal injections of 3.0 X106 [3H]cholesterol-labeled J774 cells. Blood was collected at 6, 24 and 48 hours. Feces were collected from 0 to 48 h and after exsanguination at 48 h, bile and liver samples were collected as previously described (6,16).
LCAT Activity
The endogenous LCAT cholesterol esterification rate (CER) in whole plasma was measured using a modified Stokke and Norum procedure (17–19). Endogenous LCAT activity was expressed as nanomoles of CE formed per mL of plasma during the 30 min incubation. The exogenous LCAT activity was determined using reconstituted discoidal HDL particles (rHDL) ( prepared as described in Supplementary Materials and Methods) at concentrations from 0–4 μg/mL of cholesterol incubated with recombinant human LCAT at 37 °C for 10–40 min as previously described (19).
Cellular Cholesterol Efflux
After [3H]cholesterol labeling J774 cells in which ABCA1 expression had been upregulated with cAMP in the presence of ACAT inhibitor CP113 818 (2 μg/mL) (20), medium containing the appropriate apoA-I acceptor (0–20 μg/mL) was added. BHK cells in which expression of ABCG1 was induced by treatment with mifepristone were used to measure cholesterol efflux to rHDL particles (20 μg apoA-I/mL)(21). To determine the cholesterol efflux, media were sampled at 4 h, filtered, and counted by liquid scintillation counting to determine the [3H]cholesterol released. [3H] in the media was compared with total [3H] in the cells at time zero to determine the percent release of [3H]cholesterol (20). ABCA1-mediated efflux from J774 cells was calculated by %efflux+cAMP-%efflux-cAMP while ABCG1-mediated flux from BHK cells was calculated by %efflux+mifepristone-%efflux- mifepristone. Km and Vmax values for ABCA1-mediated efflux were calculated by fitting plots of the fractional 4 h lipid efflux against apoA-I concentration to the Michaelis-Menten equation.
Cholesterol Influx to Cells
Rat Fu5AH hepatoma cells were prepared as described previously (22) and incubated with 20% serum containing [3H]cholesterol and [3H]cholesteryl ester that was obtained from mice expressing the apoA-I variants and treated with [3H]cholesterol-labeled macrophages for the RCT assay. After 6 h, the cells were washed three times with PBS, the cell lipids were extracted with isopropyl alcohol as previously described (23) and the levels of [3H] label determined. The contribution of SR-BI to influx of HDL cholesterol was assessed by 2 h pretreatment of Fu5AH cells with Block Lipid Transport-1 (BLT-1) (24) to inhibit the receptor.
Data Analysis
Data are from representative experiments and are expressed as mean ± SD. Statistical tests for significance were done using an unpaired t-test or 1 way Anova followed by a Tukey test for pairwise comparisons.
Additional information about methods is available in the Supplementary Materials at http://atvb.ahahournals.org.
Results
Previous studies of the structures of human and mouse apoA-I, which have a 65% amino acid identity (25), determined that the N- and C-terminal domains of the two proteins had markedly different biophysical properties (12,26). These differences in properties are summarized and explained in the Supplementary Materials. Briefly, the N-terminal helix bundle domain of human apoA-I was relatively stable (free energy of stabilization ΔG = 3.4 ± 0.3 kcal/mol) and exhibited poor lipid binding ability (catalytic efficiency of DMPC vesicle solublization = 0.08 (Supplementary Table 1)), while the C-terminal domain was unstable (unfolded) and exhibited high lipid binding ability (catalytic efficiency of DMPC vesicle solubilization = 0.20) (see Supplementary Materials). Conversely, the N-terminal helix bundle domain of mouse apoA-I was relatively unstable (ΔG = 1.9 ± 0.1 kcal/mol) and showed good lipid binding ability (catalytic efficiency of DMPC vesicle solubilization = 0.30) compared to the human N-terminal domain. The mouse C-terminal domain was disordered and had very poor lipid binding ability due to its highly polar nature. To further study the nature of these differences, two human and mouse domain-swap hybrid molecules, human-M apoA-I and mouse-H apoA-I, were created (12). The combination of the human N-terminal domain and mouse C-terminal domain resulted in a hybrid with a helix bundle of intermediate stability (ΔG = 2.3 ± 0.1 kcal/mol) and relatively poor lipid binding properties (catalytic efficiency of DMPC vesicle solubilization = 0.16). In contrast, the combination of the mouse N-terminal domain and human C-terminal domain resulted in an apoA-I molecule that also had intermediate helix bundle stability(ΔG = 2.4 ± 0.1 kcal/mol) but high lipid binding ability (catalytic efficiency of DMPC vesicle solubilization = 0.36). The differences in tertiary structure domain characteristics between human and mouse apoA-I present the opportunity to explore the influence of the properties of these domains on the functionality of apoA-I in the RCT pathway.
To investigate what effects these mouse/human hybrid apoA-I had on cholesterol metabolism in vivo, apoA-I-null mice (n=6 per group) were injected IP with AAV-expressing WT human apoA-I, WT mouse apoA-I, human-M apoA-I, mouse-H apoA-I or LacZ control at a dose of 1 × 1012 genome copies (GC). Three weeks after injection the WT human apoA-I-expressing mice had modestly higher apoA-I levels and significantly higher levels of HDL cholesterol compared to the WT mouse apoA-I-expressing mice (Table 1). The human-M apoA-I-expressing animals had apoA-I levels higher than both WT groups but HDL cholesterol levels similar to WT mouse apoA-I-expressing mice while the mouse-H apoA-I-expressing mice had significantly lower apoA-I and HDL cholesterol levels. Differences between the groups were not attributable to differential gene expression, as hepatic mRNA levels apoA-I were similar between all groups (Supplementary Figure I).
Table 1.
Plasma ApoA-I and Lipid Levels
| AAVa | ApoA-I (mg/dL) | HDL Cholesterol (mg/dL) | Total Plasma |
|||
|---|---|---|---|---|---|---|
| Cholesterol (mg/dL) | % Cholesteryl ester | Phospholipids (mg/dL) | Triglycerides (mg/dL) | |||
| WT H- apoA-I | 165 ± 3 | 95 ± 11 | 123 ± 14 | 65 ± 5 | 232 ± 30 | 31 ± 6 |
| WT M- apoA-I | 144 ± 6 | 36 ± 5 | 62 ± 7 | 62 ± 2 | 125 ± 12 | 20 ± 3 |
| H-M apoA-I | 196 ± 12 | 39 ± 3 | 61 ± 4 | 48 ± 4 | 158 ± 13 | 40 ± 9 |
| M-H apoA-I | 68 ± 6 | 11 ± 4 | 28 ± 5 | 34 ± 2 | 82 ± 9 | 27 ± 11 |
| LacZ | 0.0 ± 0.0 | 13 ± 3 | 30 ± 5 | 39 ± 2 | 78 ± 11 | 15 ± 6 |
Respective adeno-associated virus injected into apoA-I −/− mice with a dose of 1.0 ×1012 gc. All data taken from week 3 post infection and represent a mean ± S.D. (n=6). Data are representative of three individual experiments.
HDL were isolated via ultracentrifugation and run on a 4–20% non-denaturing gradient gel (Figure 1A). HDL isolated from WT human apoA-I- and human-M apoA-I-expressing mice formed a single HDL species approximately 9 and 8 nm, respectively, in diameter while HDL from WT mouse apoA-I- and mouse-H apoA-I-expressing mice formed particles approximately 9 nm and 12 nm in diameter. HDL isolated from LacZ-expressing mice were primarily 12 nm in diameter with a small amount of HDL that were 9 nm. Gel filtration profiles of serum from apoA-I variant-expressing mice (Supplementary Figure II) showed HDL particle sizes that closely matched the hydrodynamic diameters of the HDL that were isolated and run on 4–20% non-denaturing gradient gels (Figure 1A). Analysis of the apoprotein composition of HDL isolated from mice expressing WT human apoA-I indicated that the primary protein present was apoA-I along with trace amounts of apoA-II and apoA-IV (Figure 1B). For HDL isolated from WT mouse apoA-I-expressing mice the primary apoprotein was apoA-I but approximately 20% was apoE. Much like the WT human apoA-I HDL, apoA-I was the only apoprotein present in HDL isolated from human-M apoA-I-expressing mice. HDL isolated from mouse-H apoA-I-expressing mice had equal amounts of apoA-I and apoE along with traces of apoA-II and apoA-IV, while HDL isolated from LacZ-expressing mice contained primarily apoE along with traces amounts of apoA-II and apoA-IV. The lipid compositions of the isolated HDL (Supplementary Table III) indicate that, on average, the HDL particles from the mice expressing WT mouse apoA-I, mouse-H apoA-I and LacZ contained higher amounts of total cholesterol than the mice expressing WT human apoA-I or human-M apoA-I. This effect is probably due to the presence of the large 12 nm particle that was absent in the WT human apoA-I- and human-M apoA-I-expressing mice. As expected, the relatively large HDL particles with higher cholesterol contents contained lower amounts of surface components (phospholipid and protein).
Fig 1. Particle size and apoprotein composition analysis of HDL from mice expressing the human apoA-I variants.
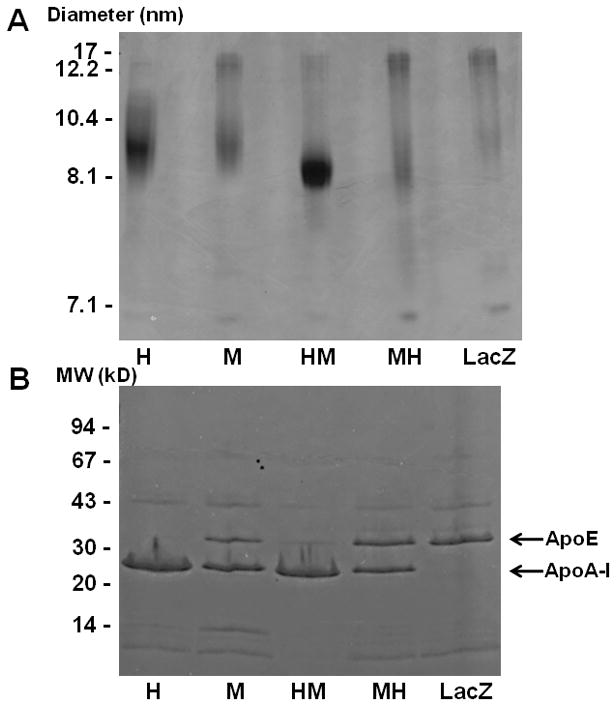
ApoA-I-null mice were infected with AAV and bled at 2,4 and 6 weeks post infection. HDL were isolated from pooled plasma by ultracentrifugation. (A) 10 μg of total HDL protein was loaded on a 4–20% Tris-Glycine non-denaturing gradient gel and stained with Coomassie brilliant blue. (B) To determine the apoprotein composition of the HDL, 2 μg of total protein was analyzed by reducing 8–25% SDS-PAGE. Lanes: (1) WT Human apoA-I, (2) WT Mouse apoA-I, (3) Human-M apoA-I, (4) Mouse-H apoA-I, (5) LacZ.
At week 10 after AAV injection, cholesterol-labled J774 cells were injected for a macrophage RCT study. The [3H]-cholesterol counts in plasma, expressed as a percentage of total labeled cholesterol injected, at each time point were significantly higher for WT human apoA-I than for all other groups (Figure 2A). WT mouse apoA-I and human-M apoA-I had similar levels of [3H]-cholesterol counts in plasma, which were higher than the mouse-H apoA-I and LacZ groups but lower than the WT human A-I group. However, expression of the mouse-H apoA-I variant, which had very low levels of apoA-I and HDL-cholesterol compared to WT human and WT mouse apoA-I (Table 1), induced significantly increased transport of macrophage-derived cholesterol into the feces compared to all other groups (Figure 2B). This was such a striking result that the entire experiment was repeated with an additional 30 mice. The results from the two experiments were very similar, with the counts in the feces from the mouse-H apoA-I-expressing mice being twice that of the counts in the feces of the WT mouse apoA-I-expressing mice in both experiments. [3H]-cholesterol counts in the liver were similar in all groups (data not shown).
Figure 2. Reverse Cholesterol Transport Assay.

Male apoA-I-null mice (n = 6 per group) were injected with AAV (1 X 1012 GC) expressing either WT Human apoA-I, WT Mouse apoA-I, Human-M apoA-I, Mouse-H apoA-I or LacZ 10 weeks prior to the RCT assay. The mice were then injected IP with [3H]cholesterol-labeled, acLDL-loaded J774 macrophages. Data are expressed as percentage ± SD of the [3H]cholesterol tracer relative to total cpm tracer injected. (A) Time course of [3H]cholesterol appearance in plasma. Mice were bled at 2, 6, 24, and 48 hours after injection. (B) Fecal [3H]cholesterol tracer levels (mean ± SD, n = 6). Feces were collected continuously from 0 to 48 hours post-injection. WT Human apoA-I (■), WT Mouse apoA-I (▲), Human-M apoA-I (▼), Mouse-H apoA-I (◇) and LacZ (●). * Indicates p<0.05 compared to WT mouse apoA-I.
To gain further insight into why the mouse-H apoA-I variant had enhanced macrophage RCT compared to all other variants, we investigated several apoA-I-mediated steps in the RCT pathway individually. These steps included ABCA1- and ABCG1-mediated cholesterol efflux from macrophages, the conversion of free cholesterol to cholesteryl ester by LCAT and SR-BI-mediated selective uptake into liver cells of cholesterol and cholesteryl ester from HDL particles (27). To measure ABCA1-mediated cholesterol efflux, J774 macrophages (the cell type used in the macrophage RCT experiments) were labeled with [3H]-cholesterol and then treated with cAMP to up-regulate ABCA1. Lipid efflux was then initiated by the addition of the recombinant apoA-I variants at increasing concentrations (0–20 μg/mL) and the catalytic efficiencies of the cholesterol efflux reactions were determined from the Vmax and Km values (Supplementary Table II). Mouse-H apoA-I was twice as effective at promoting ABCA1-mediated cholesterol efflux compared to the WT human apoA-I protein and 3.5 times as efficient as the WT mouse apoA-I protein, which was the least effective apoA-I variant (Figure 3A). Human-M apoA-I was as effective at promoting ABCA-1-mediated cholesterol efflux as the WT human apoA-I protein and twice as effective as the WT mouse apoA-I protein. The influence of apoA-I structure on ABCG1-mediated cholesterol efflux was measured by incubating BHK cells with discoidal rHDL particles (phospholipid to apoA-I molar ratios of 87±5/1 and hydrodynamic diameters of 9–10 nm (Supplementary Figure V)) containing the apoA-I variants at a concentration of 20 μg/mL of apoA-I. The results in Fig. 3B show that each rHDL was equally proficient at promoting ABCG1-mediated cholesterol efflux.
Figure 3. Influence of apoA-I structure on ABCA1- and ABCG1-mediated cholesterol efflux from cells.

(A) J774 macrophages were labeled with [3H] cholesterol as described under “Materials and Methods.” These labeled cells were then treated with cAMP overnight to up-regulate ABCA1. The lipid efflux was then initiated by the addition of the recombinant apoA-I variants (0–20 μg/mL). After 4 h incubation, the medium was removed, filtered and extracted for lipids and 3H-radioactivity was determined. The catalytic efficiency (Vmax/Km) ((%FC efflux/4h)(μg apoA-I/ml)−1) was calculated from kinetic parameters generated by fitting the fractional efflux at 4 h, measured at various concentrations of apoA-I, to the Michaelis-Menten equation. The Vmax/Km values are plotted as mean ± SD. (B) BHK cells were labeled as described in “Materials and Methods” and treated with mifepristone for 18h to upregulate ABCG1. Cholesterol efflux was initiated by the addition of rHDL (20 μg/mL apoA-I) containing the recombinant apoA-I variants for 4 h. * Indicates p<0.05 compared to WT mouse apoA-I.
The next step in the RCT pathway is the conversion of FC to CE by LCAT in the plasma. The cholesterol esterification rate (CER) was highest for WT human apoA-I and lowest for mouse-H apoA-I with the WT mouse apoA-I and human-M apoA-I demonstrating intermediate activities (Figure 4A). Corresponding to the reduction in the CER, plasma CE levels in the mouse-H apoA-I-expressing mice were significantly reduced compared to both WT apoA-I-expressing groups (Figure 4B). To determine the effects of the apoA-I variants on the kinetic parameters of the LCAT reaction, rHDL ( palmitoyl-oleoyl phosphatidylcholine (POPC):cholesterol:apoA-I molar ratios of 86±5/8±1/1 and hydrodynamic diameters of 9–10 nm) were incubated with recombinant LCAT at 37 °C as described in “Materials and Methods”. The apparent Vmax of WT human apoA-I was twice that of all other variants while the apparent Km of all of the reactions were essentially equal (Supplementary Figure III).
Figure 4. LCAT cholesterol esterification rates.

(A) Cholesterol esterification rate in the plasma of mice expressing the apoA-I variants or LacZ. Whole plasma was radiolabeled with [3H]cholesterol at 4 °C overnight and then incubated at 37 °C for 30 min. LCAT activity was determined as described in “Materials and Methods” and is expressed as nanomoles of CE formed per milliliter of plasma during the 30 min incubation. (B) Percentage of cholesterol as CE in the plasma of mice expressing the apoA-I variants or LacZ. Values are expressed as mean ± SD (n = 6/group). * Indicates p<0.05 compared to WT mouse apoA-I.
The next step in the RCT pathway is SR-BI-mediated selective uptake of cholesterol from HDL in plasma into the liver. Serum samples (which contain [3H]-cholesterol and [3H]-cholesteryl ester) from the 48-hour time point of the RCT experiment in Figure 2A were diluted in media and applied to Fu5AH rat liver hepatoma cells that express high levels of SR-BI. After the 6-hour incubation with or without BLT-1 which inhibits the activity of the SR-BI transporter, the cells were processed as described in Materials and Methods to determine the amount of label that was taken up from the media into the cells (Figure 5). Serum samples from WT human apoA-I- and WT mouse apoA-I-expressing mice were equally effective at promoting total cholesterol uptake and SR-BI-mediated selective cholesterol uptake into the Fu5AH cells. Serum from both hybrid apoA-I-expressing and LacZ-expressing animals promoted significantly more total uptake of cholesterol compared to serum from either WT human apoA-I- or WT mouse apoA-I-expressing animals. Interestingly, BLT-1 did not inhibit uptake of cholesterol from serum expressing either mouse-H apoA-I or LacZ, indicating that SR-BI mediated selective uptake of cholesterol was not a contributing factor for these groups. A similar experiment using rHDL instead of serum demonstrated that the absence of the human C-terminal domain in the apoA-I molecule significantly decreased SR-BI-mediated influx of cholesterol (Supplementary Figure IV).
Figure 5. Influx of cholesterol to hepatic cells from serum of mice expressing apoA-I variants.

[3H]cholesterol- and [3H]cholesteryl ester-labeled sera from the 48 h timepoint of the experiment described in Fig. 2A was diluted to 20% with MEM and incubated with Fu5AH rat hepatoma cells for 6 h with BLT-1 (□)to block SR-BI-mediated influx or without BLT-1 (■). Influx was calculated as described in “Materials and Methods”. * Indicates p<0.05 compared to + BLT-1 group.
Discussion
As mentioned earlier, the structurally distinct N- and C-terminal domains have markedly different biophysical properties in human and mouse apoA-I (see Supplementary Results). Based on these differences, we postulated that a hybrid apoA-I molecule containing the mouse N-terminal and human C-terminal domains may exhibit enhanced atheroprotective properties. To test this hypothesis, the abilities of two domain-swap hybrid apoA-I molecules, human-M apoA-I and mouse-H apoA-I, were compared with the abilities of WT human apoA-I and WT mouse apoA-I to promote macrophage RCT in vivo.
ApoA-I-null mice were injected with AAV expressing either WT human apoA-I, WT mouse apoA-I, human-M apoA-I, mouse-H apoA-I or LacZ. The apoA-I levels for WT human apoA-I-, WT mouse apoA-I- and human-M apoA-I-expressing mice were similar, but the apoA-I levels for mouse-H apoA-I-expressing mice were much lower (Table 1), despite similar hepatic apoA-I mRNA levels for all groups (Supplemental Figure I), indicating that mouse-H apoA-I was catabolized faster than the other apoA-I variants. A potential explanation for the increased rate of mouse-H apoA-I turnover is the presence of a relatively high amount of apoE on mouse-H apoA-I HDL (Figure 1B) which would promote binding to the LDL receptor on hepatocytes. The lower level of apoE on the HDL in animals expressing WT mouse apoA-I caused a smaller reduction in plasma apoA-I concentration relative to WT human apoA-I-expressing mice (Table 1). Once bound to the LDL receptor the mouse-H apoA-I HDL particle would be internalized and catabolized. In contrast, the selective uptake of HDL cholesterol from particles that do not contain apoE occurs via SR-BI which does not lead to catabolism of the apoA-I, maintaining its level in plasma and allowing it to participate in additional rounds of RCT.
All of the apoA-I-expressing mice demonstrated increased macrophage RCT compared to the control LacZ-expressing mice (Figure 2B), confirming the ability of apoA-I to promote RCT. However, the mouse-H apoA-I-expressing mice demonstrated a markedly greater increase in fecal excretion of the tracer compared to the other apoA-I-expressing mice, despite low levels of both apoA-I and HDL-C (RCT levels were increased 2-fold while apoA-I and HDL-C levels were reduced 2.5- and 8.5-fold, respectively, compared to mice expressing WT human apoA-I). The amount of tracer in the plasma (Figure 2A) of mice expressing the various apoA-I variants mirrored the HDL pool size before the injection of the macrophages (Table 1). These results indicate that mouse-H apoA-I is more effective at promoting macrophage RCT than either of the WT apoA-I proteins and that the quality rather than the quantity of the HDL particles is more important in promoting macrophage RCT. The enhanced functionality of the mouse-H apoA-I HDL may be due to a higher fraction of the protein existing as preβ-HDL (lipid-free/poor apoA-I) which participates in ABCA1-mediated cellular cholesterol efflux. To gain greater insight into the mechanistic basis for these effects, we dissected the following 3 steps of the RCT pathway, ABCA1-mediated cholesterol efflux, LCAT-mediated conversion of FC to CE and the uptake of cholesterol into the liver.
The catalytic efficiencies (Vmax/Km) of the apoA-I variants in supporting ABCA1-mediated cholesterol efflux from J774 macrophages were determined and the value for mouse-H apoA-I was two-fold higher than WT human apoA-I and 3.5 fold higher than WT mouse apoA-I (Figure 3A). This increase in catalytic efficiency was likely due to the enhanced ability of mouse-H apoA-I to interact with lipids. When the ability of the apoA-I variants to interact with and solubilize DMPC vesicles was studied, it was determined that mouse-H apoA-I had the highest catalytic efficiency, 16% higher than WT human apoA-I and two fold higher than WT mouse apoA-I (Supplementary Table). According to the exovesiculation model of apoA-I interaction with ABCA1, there are three steps involved in the ABCA1-mediated efflux of cellular cholesterol and phospholipids (28). First, apoA-I binds to ABCA1 and enhances net phospholipid traslocation to the exofacial leaflet which causes unequal lateral packing densities in the two leaflets of the phospholipid bilayer. Second, the resulting membrane strain is relieved by the creation of an exovesiculated lipid domain. This highly curved membrane promotes high affinity binding of apoA-I. Third, the lipid-bound apoA-I then spontaneously solubilizes the exovesiculated domains to form nascent HDL particles. It is this third and final step that is rate limiting for efflux. On this basis, the enhanced ability of mouse-H apoA-I to solubilize lipid and thereby efficiently mediate ABCA1-mediated lipid efflux from macrophages (combined with a likely higher level of the circulating protein being in the lipid-free(poor) state) presumably contributes to this hybrid apoA-I promoting RCT so effectively. The abilities of rHDL particles to promote ABCG1-mediated cholesterol efflux were similar for all of the apoA-I variants (Figure 3B) suggesting that variations in apoA-I domain structure do not affect acceptor particle efficiency when the variants are present in discoidal rHDL particles of similar size (9–10 nm hydrodynamic diameter).
The next step of the RCT pathway, the conversion of FC to CE by LCAT in the plasma, was measured using well established methods (18). The results demonstrated that both hybrid apoA-I variants were unable to activate LCAT to the same extent as the WT proteins (Figure 4). Interestingly, if the CER of the mouse-H apoA-I-expressing sample is normalized to apoA-I concentration, taking into account the low levels of mouse-H apoA-I (Table 1), mouse-H apoA-I is as efficient as WT mouse apoA-I at activating LCAT but still not as efficient as WT human apoA-I. The reductions in CER are likely due to variations in HDL particle size distribution and changes in structure of the apoA-I on the HDL particles, altering the manner in which apoA-I and LCAT interact as LCAT binds to a HDL particle. The relationship between LCAT activity and RCT or cardiovascular disease is uncertain at this time with several studies concluding that higher levels of LCAT activity are associated with increased incidences of cardiovascular disease (29,30), while others describe either a negative or no correlation between LCAT activity and cardiovascular disease or RCT (31–33). In our experiments, the two groups with the lowest CER, mouse-H apoA-I- and LacZ-expressing mice, had the highest and lowest amount of 3H-radioactivity in the feces, respectively (Figure 2B). It follows that LCAT activity does not determine overall macrophage RCT activity (15,33).
The third step of RCT is the uptake of plasma HDL cholesterol into the liver. Serum from WT human apoA-I- and WT mouse apoA-I-expressing animals were equally efficient at promoting total cholesterol uptake into hepatoma cells while serum from human-M apoA-I-, mouse-H apoA-I- and LacZ-expressing mice were all more efficient (Figure 5). A contributing factor to this effect may be the apoE on the HDL from mouse-H apoA-I- and LacZ-expressing mice (Figure 1B) that promotes cholesterol delivery to the liver via the LDL-receptor, which mediates the endocytosis of apoE-containing lipoproteins. The low level of apoE in the HDL from mice expressing WT mouse apoA-I is apparently insufficient to mediate such an effect. Additionally, the lipoproteins in serum from mice expressing mouse-H apoA-I and LacZ have a higher FC to phospholipid ratio than lipoproteins from the serum of the other three groups ( calculated from data in Table 1). This increases the FC concentration gradient between the HDL and cell plasma membrane which can increase the cellular uptake of HDL FC by passive diffusion (34). However, the increased rate of cholesterol uptake from serum that contains human-M apoA-I cannot be explained in this fashion. Interestingly, the inhibition of SR-BI selective uptake with BLT-1 did not affect the uptake of cholesterol from serum containing either mouse-H apoA-I or LacZ, indicating that for these samples SR-BI did not contribute significantly to cholesterol delivery from HDL to the liver cells. Any contribution of SR-BI-mediated selective uptake of HDL cholesterol in these serum samples may be masked by the contribution of lower density lipoproteins; the ratio of HDL cholesterol to total cholesterol ((0.39 – 0.43) calculated from data in Table 1) is relatively low compared to the serum containing WT human apoA-I, WT mouse apoA-I or human-M apoA-I (0.58 – 0.77). The apoB-containing lipoproteins may play a major role in the cholesterol uptake from serum containing mouse-H apoA-I or LacZ. Interestingly, when rHDL were used as a substrate in the same SR-BI-mediated cholesterol uptake system (Supplementary Figure IV) those variants that lacked the human C-terminal domain (WT mouse apoA-I and human-M apoA-I) showed a significant decrease in selective uptake. These results indicate that when all other factors, such as apoB-containing lipoproteins, are removed a more hydrophobic apoA-I C-terminal domain increases SR-BI-mediated selective uptake of HDL cholesterol.
Despite the lower plasma apoA-I and HDL-C levels, expression of mouse-H apoA-I gives rise to more efficient macrophage RCT than does expression of either WT human apoA-I or WT mouse apoA-I. The altered properties of the mouse-H apoA-I hybrid, in particular its increased lipid affinity and the presence of apoE on mouse-H apoA-I HDL (which is mediated by apoE-resident apolipoprotein interactions (35), in this case between mouse apoE and the mouse apoA-I N-terminal helix-bundle domain), led to several major changes in the functionality in vivo of the apoA-I molecule and the HDL particles containing it. It seems likely that this apoA-I variant increases the rate of ABCA1-mediated macrophage cholesterol efflux and nascent HDL particle formation, as well as increasing the rate of cholesterol uptake by liver via the LDL receptor due to the presence of apoE, thereby enhancing the flux of cholesterol along the RCT pathway.
Overall, these results show that the tertiary structure domain properties of apoA-I affect the ability of the protein to mediate macrophage RCT. The fact that substitution of the N-terminal helix bundle domain in the human apoA-I with the mouse apoA-I counterpart causes a gain of function with respect to macrophage RCT suggests that engineering some destabilization into the N-terminal helix bundle domain and/or an increase in hydrophobicity of the C-terminal domain of human apoA-I would enhance the anti-atherogenicity of this protein.
Supplementary Material
Acknowledgments
We thank Dr. John S. Parks of Wake Forest University for generously providing us with recombinant human LCAT.
This work was supported by NIH Grant HL22633.
Footnotes
Disclosures: none
References
- 1.Davidson WS, Thompson TB. The structure of apolipoprotein A-I in high density lipoproteins. J Biol Chem. 2007;282:22249–22253. doi: 10.1074/jbc.R700014200. [DOI] [PubMed] [Google Scholar]
- 2.Thomas MJ, Bhat S, Sorci-Thomas MG. Three-dimensional models of HDL apoA-I: implications for its assembly and function. J Lipid Res. 2008;49:1875–1883. doi: 10.1194/jlr.R800010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090–3100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26:12–19. doi: 10.1161/01.ATV.0000194291.94269.5a. [DOI] [PubMed] [Google Scholar]
- 5.Yancey PG, Bortnick AE, Kellner-Weibel G, De La Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 7.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 8.Saito H, Dhanasekaran P, Nguyen D, Holvoet P, Lund-Katz S, Phillips MC. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J Biol Chem. 2003;278:23227–23232. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 9.Silva RAHG, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A-I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44:2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 10.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito H, Lund-Katz S, Phillips MC. Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Prog Lipid Res. 2004;43:350–380. doi: 10.1016/j.plipres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka M, Koyama M, Dhanasekaran P, Nguyen D, Nickel M, Lund-Katz S, Saito H, Phillips MC. Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry. 2008;47:2172–2180. doi: 10.1021/bi702332b. [DOI] [PubMed] [Google Scholar]
- 13.Tanigawa H, Billheimer JT, Fuki I, Thoyama JC, Rothblat G, Rader DJ. AAV8-mediated overexpression of lecithin-cholesterol acyltransferase fails to promote macrophage reverse cholesterol transport in vivo. Circulation. 2007;116:197–197. doi: 10.1161/CIRCULATIONAHA.108.825109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, Phillips MC. Structural and functional consequences of the milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexander ET, Weibel GL, Joshi MR, Vedhachalam C, de la Llera-Moya M, Rothblat GH, Phillips MC, Rader DJ. Macrophage reverse cholesterol transport in mice expressing apoA-I Milano. Arterioscler Thromb Vasc Biol. 2009;29:1496–1501. doi: 10.1161/ATVBAHA.109.191379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stokke KT, Norum KR. Determination of lecithin - cholesterol acyltransfer in human blood plasma. Scand J Clin Lab Invest. 1971;27:21. doi: 10.3109/00365517109080184. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Thorngate FE, Weisgraber KH, Williams DL, Parks JS. Apolipoprotein E is the major physiological activator of lecithin-cholesterol acyltransferase (LCAT) on apolipoprotein B lipoproteins. Biochemistry. 2005;44:1013–1025. doi: 10.1021/bi0481489. [DOI] [PubMed] [Google Scholar]
- 19.Alexander E, Bhat S, Thomas M, Weinberg R, Cook V, Bharadwaj M, Sorci-Thomas M. Apolipoprotein A-I helix 6 negatively charged residues attenuate lecithin-cholesterol acyltransferase (LCAT) reactivity. Biochemistry. 2005;44:5409–5419. doi: 10.1021/bi047412v. [DOI] [PubMed] [Google Scholar]
- 20.Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat GH, Phillips MC. Influence of apoA-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 21.Sankaranarayanan S, Oram JF, Asztalos BF, Vaughan AM, Lund-Katz S, Adorni MP, Phillips MC, Rothblat GH. Effects of acceptor composition and mechanism of ABCG1-mediated cellular free cholesterol efflux. J Lipid Res. 2008;50:275–284. doi: 10.1194/jlr.M800362-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimetti F, Weibel GK, Duong M, Rothblat GH. Measurement of cholesterol bidirectional flux between cells and lipoproteins. J Lipid Res. 2006;47:605–613. doi: 10.1194/jlr.M500466-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Yancey PG, De la Llera-Moya M, Swarnakar S, Monzo P, Klein SM, Connelly MA, Johnson WJ, Williams DL, Rothblat GH. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275:36596–36604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- 24.Nieland TJF, Penman M, Dori L, Krieger M, Kirchhausen T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci USA. 2002;99:15422–15427. doi: 10.1073/pnas.222421399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 26.Koyama M, Tanaka M, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Interaction between the N- and C-terminal domains modulates the stability and lipid binding of apolipoprotein A-I. Biochemistry. 2009;48:2529–2537. doi: 10.1021/bi802317v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50:S189–S194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282:25123–25130. doi: 10.1074/jbc.M704590200. [DOI] [PubMed] [Google Scholar]
- 29.Wells IC, Peitzmeier G, Vincent JK. Lecithin - Cholesterol Acyltransferase and Lysolecithin in Coronary Atherosclerosis. Exp Mol Pathol. 1986;45:303–310. doi: 10.1016/0014-4800(86)90019-5. [DOI] [PubMed] [Google Scholar]
- 30.Dullaart RPF, Perton F, Sluiter WJ, de Vries R, van Tol A. Plasma Lecithin: Cholesterol Acyltransferase Activity Is Elevated in Metabolic Syndrome and Is an Independent Marker of Increased Carotid Artery Intima Media Thickness. J Clin Endocrinol Metab. 2008;93:4860–4866. doi: 10.1210/jc.2008-1213. [DOI] [PubMed] [Google Scholar]
- 31.Ruhling K, Lang A, Richard F, Van Tol A, Eisele B, Herzberg V, Till U. Net mass transfer of plasma cholesteryl esters and lipid transfer proteins in normolipidemic patients with peripheral vascular disease. Metabolism. 1999;48:1361–1366. doi: 10.1016/s0026-0495(99)90144-0. [DOI] [PubMed] [Google Scholar]
- 32.Solajic-Bozicevic N, Stavljenic-Rukavina A, Sesto M. Lecithin-cholesterol acyltransferase activity in patients with coronary artery disease examined by coronary angiography. Clin Investig. 1994;72:951–956. doi: 10.1007/BF00577734. [DOI] [PubMed] [Google Scholar]
- 33.Tanigawa H, Billheimer JT, Tohyama J, Fuki IV, Ng DS, Rothblat GH, Rader DJ. Lecithin: Cholesterol Acyltransferase expression has minimal effects on macrophage reverse cholesterol transport In vivo. Circulation. 2009;120:160–169. doi: 10.1161/CIRCULATIONAHA.108.825109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson WJ, Mahlberg FH, Rothblat GH, Phillips MC. Cholesterol transport between cells and high-density lipoproteins. Biochim Biophys Acta. 1991;1085:273–298. doi: 10.1016/0005-2760(91)90132-2. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen D, Dhanasekaran P, Phillips MC, Lund-Katz S. Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry. 2009;48:3025–3032. doi: 10.1021/bi9000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.