

Abstract
The design, synthesis, and evaluation of a new series of hexahydrofuropyran-derived HIV-1 protease inhibitors are described. We have designed a stereochemically defined hexahydrofuropyranol-derived urethane as the P2-ligand. The current ligand is designed based upon the X-ray structure of 1a-bound HIV-1 protease. The synthesis of (3aS,4S,7aR)-hexahydro-2H-furo[2,3-b] pyran-4-ol (−)-7 was carried out in optically active form. Incorporation of this ligand provided inhibitor 35a, which has shown excellent enzyme inhibitory activity and antiviral potency. Our structure activity studies have indicated that the stereochemistry and the position of oxygens in the ligand are important to the observed potency of the inhibitor. Inhibitor 35a has maintained excellent potency against multidrug-resistant HIV-1 variants. An active site model of 35a was created based upon the X-ray structure of 1b-bound HIV-1 protease. The model offers molecular insights regarding ligand-binding site interactions of the hexahydrofuropyranol-derived novel P2-ligand.
Introduction
HIV-1 protease inhibitors are critical components of highly active antiretroviral therapy (HAART).1–3 The HAART treatment regimens significantly reduced HIV/AIDS-related mortality.4,5 However, the rapid emergence of drug-resistant HIV-1 strains and the appearance of cross-resistance are severely limiting long-term treatment options.6–8 An estimated 10–25% of newly infected patients harbor at least one viral strain that is resistant to current medications.9–11 In addition, PI regimens suffer from a number of other drawbacks including high pill burden, treatment cost, poor ADMET properties, debilitating side effects, and toxicity issues.12 Therefore, the development of novel PIs with broad-spectrum activity against multidrug-resistant HIV-1 variants remains a major therapeutic objective.13
In our continuing interest to develop novel protease inhibitors (PI) with broad-spectrum activity against multidrug-resistant HIV-1 variants, we have reported a series of PIs including PIs 1a, 1b, 2, and 3.14–16 These inhibitors exhibited excellent antiviral activity against multidrug-resistant HIV-1 variants. Darunavir (TMC-114, Figure 1), has been recently approved by the FDA.17,18 It has displayed a high genetic barrier to resistance and retained high potency against multidrug resistant HIV-1 strains. It has been demonstrated that resistance to 1a is significantly delayed compared to other approved PIs.19–21
Figure 1.

Structures of inhibitors 1–3 and 35a.
Our structure-based design of 1a and other PIs is inspired by the premise that an inhibitor engaged in multiple interactions, especially hydrogen bonding, with the HIV protease backbone atoms, should retain these affinities with mutant strains. 22 As the enzyme backbone conformation is only minimally distorted when mutations occur, backbone atoms-PI interactions are likely maintained therefore sustaining the inhibitor affinity and potency. Inhibitor 1a’s superb resistance profile likely originates from the extensive interactions the inhibitor makes within the HIV protease’s binding site and particularly with the backbone atoms of the enzyme.22–24 Extensive studies of 1a-bound HIV protease crystal structures have consistently revealed tight hydrogen bonding between the inhibitor and the protease backbone.23–25 The stereochemically-defined bis-tetrahydrofuran (bis-THF) P2 ligand in 1 forms a strong hydrogen bonding network between its two cyclic ether oxygens and the backbone amide NH bonds of the protease residues, Asp29 and Asp30.22 These observations likely provide explanations for 1a’s outstanding antiviral activity. Not surprisingly, several other protease inhibitors featuring the bis-THF as the P2 ligand have exhibited equally impressive antiviral activities and resistance profiles.22,26
The bis-THF ligand represents an intriguing pharmacophoric scaffold for the development of PIs to combat drug resistance. To further optimize the bis-THF structural template, we have now investigated ligands that could enhance the backbone-binding as well as improve hydrophobic interactions with the protease active site. The X-ray structure of 1-bound HIV protease has shown a distance of about 3.0 to 3.2 Å between the bis-THF cyclic oxygens and the Asp30 NH amide bond, while a shorter 2.9 Å distance was observed with the Asp29 NH bond.23,25 In order to maximize and promote closer hydrogen bonding with the Asp30 backbone NH bond, we thought a larger ring on the P2 ligand should increase the dihedral angle of the bicyclic acetal, bring the oxygen closer, give more flexibility to the structure, and offer a more optimal alignment of the cyclic oxygen with the Asp30 NH bond. Such factors could realistically promote tighter hydrogen bonding with the Asp30 backbone NH bond. Besides, this extra methylene group in the “inner” ring would also provide more favorable van der Waals interactions within the hydrophobic pocket created by Ile47, Val32, Ile84, Leu76, and Ile50′ residues in the protease S2 subsite. In addition, a larger ring would bring advantageous flexibility to the ligand structure, and could potentially lead to better flexibility and adaptability to protease mutations. Herein, we report the design, synthesis, and biological evaluation of a series of highly potent PIs that combined a (R)-hydroxyethyl sulfonamide isostere with the furopyranol ligand (−)-7. Among all inhibitors of the series, 35a showed the most impressive inhibitory and antiviral activity (Ki = 2.7 pM, IC50 = 0.5 nM respectively). Moreover, inhibitor 35a was evaluated against a panel of multidrug-resistant HIV-1 viruses. It retained potent activity against a variety of multidrug-resistant clinical HIV-1 strains with EC50 values in low nanomolar range, which is superior to other PIs and comparable to 1a. Modeling of 35a based upon the X-ray stcure of 2-bound HIV-1 protease active site has provided critical molecular insight into the ligand-binding site interactions.
Chemistry
The synthesis of enantiomerically pure (3aS,4S,7aR)-hexahydro-2H-furo[2,3-b]pyran-4-ol is shown in Scheme 1. It was achieved starting from known enantiomerically pure lactone 4.27 Lactone 4 was reduced into the corresponding diol using lithium aluminum hydride in 95% yield. Selective monoacetylation at the primary alcohol using AcCl and 2,4,6-collidine at −78 °C,28 and subsequent silylation of the remaining free hydroxyl furnished intermediate 5 in 86% yield (2 steps). Removal of the acetate group, followed by ozonolysis of the olefin, furnished a bicyclic bis-acetal intermediate. Reduction of the hemiacetal moiety using Et3SiH and BF3-Et2O afforded bicyclic intermediate 6 in 55% yield in three steps. Removal of the silyl group with TBAF in THF furnished the desired hexahydrofuropyran-4-ol ligand (−)-7.
Scheme 1.

Synthesis of ligand (−)-7 and its respective enantiomer (+)-7.
To demonstrate the importance of the absolute stereochemistry of the bicyclic structure of ligand (−)-7, its corresponding enantiomer (+)-7 was synthesized starting from intermediate 8 (Scheme 1). Intermediate 8 was synthesized by an enzyme-catalyzed desymmetrization of cyclopentene meso-diacetate followed by a Claisen rearrangement step.27b, 29 The resulting diester was reduced by LAH to provide 8. It was used for the synthesis of (+)-7 and subjected to the same synthetic sequence applied from lactone (−)-4 in the synthesis of (−)-7 (Scheme 1). To examine the importance of each of the two cyclic ether oxygens in the furopyranol ligand (−)-7, we prepared the corresponding cyclohexane and cyclopentane derivatives (Schemes 2 and 3).
Scheme 2.

Synthesis of furocyclohexanol P2 ligand (−)-12
Scheme 3.

Syntheses of ligands (−)-18 and (−)-19.
The synthesis of 4-hydroxy octahydrobenzofuran ligand (−)-12 is shown in Scheme 2. Reaction of diazocyclohexanedione 930 with ethylvinyl ether in presence of a catalytic amount of Rh2(OAc)4 at 23 °C gave derivative 10.31 Hydrogenation of the ketofuran in the presence of Pd/C under H2 (1 atm) furnished the corresponding crude ketone 11 as a 9:1 mixture of diastereoisomers. A one-pot procedure involving L-selectride reduction of the ketone followed by Et3SiH/TMSOTf-promoted reduction of the acetal furnished the racemic alcohol (±)-12 (71% from 10). Enzymatic resolution of (±)-12 using lipase Amano PS-30 provided the desired enantiopure alcohol (−)-12 (98.8% ee by chiral HPLC analysis of the 2,4-dinitrobenzoate derivative), after ca. 55% conversion to the acetate.
The synthesis of cyclopentapyranol ligand is shown in Scheme 3. Pentanone 14 was treated with LDA then reacted with t-butyldimethylsilyloxypropionaldehyde 32 to furnish intermediate 15 (dr 3:1) in 95% yield. A DMSO-TFAA promoted oxidation of the free hydroxy group followed by TFA-promoted cyclocondensation furnished the bicyclic α,β-unsaturated ketone 16. Hydrogenation in presence of 10% Pd/C followed by L-selectride reduction of the ketone gave racemic alcohol (±)-18 as a single diastereomer in 68% yield over 2 steps. Lipase-catalyzed resolution of the alcohol provided enantiomerically pure alcohol (−)18. For the synthesis of a P2 ligand devoid of any cyclic oxygen, known tetrahydroindanone 1733 was similarly hydrogenated in presence of 10% Pd/C to give the corresponding bicyclic ketone. Accordingly, L-selectride-promoted reduction of the ketone provided the corresponding alcohol (dr = 10:1, as observed by 1H and 13C NMR). Lipase-mediated resolution of the major cis-alcohol gave the respective chiral ligand (−)-19 (90% ee determined by chiral HPLC).
Since the introduction of a six-membered ring in the P2 ligand structure may introduce more structural flexibility, we set out to explore ligands in which the cyclic oxygens were moved to adjacent positions. Such ligands would also demonstrate the importance of the oxygen positions in the bicyclic structure of ligand (−)-7. Thus, isomeric ligand 25 was synthesized with the furan oxygen moved to a vicinal position. The synthesis of 4-hydroxyhexahydro-2H-furo[3,4-b]pyran 25 is shown in Scheme 4. Iodoalkoxylation of the 2,5-dihydrofuran 22 using propanediol in the presence of N-iodosuccinimide and catalytic NH4OAc provided iodoalcohol 23. Swern oxidation gave aldehyde 24 in 86% yield. An intramolecular Barbier-type reaction was then conducted using indium in the presence of copper (I) iodide and iodine, to furnish a mixture of diastereoisomeric alcohols.34 Oxidation followed by stereoselective reduction using NaBH4 furnished the racemic cis, cis-bicyclic alcohol (±)-25 as the sole product. Lipase-mediated resolution finally gave the enantiomerically pure alcohol 25.
Scheme 4.

Synthesis of hexahydrofuro[3,4-b]pyran-4-ol ligand 25.
To ascertain the importance of the position of the urethane in (−)-7, we have synthesized hexahydrofuropyran-5-ol ligand 30 shown in Scheme 5. The free hydroxyl on the pyran ring was moved to the C3 position. The synthesis was accomplished starting from enantiomerically pure bis-THF ligand 27 synthesized by us previously.35 Dess-Martin oxidation of 27 provided the corresponding ketone. Homologation of the resulting ketone using trimethylsilyldiazomethane in the presence of AlMe3 followed by treatment of the crude mixture with TBAF and acetic acid provided the furanopyranone 29. Stereoselective reduction of ketone 29 using L-selectride furnished alcohol 30 as a mixture of inseparable diastereoisomers (dr = 5:1). Both isomers were separated after formation of the corresponding activated mixed carbonate 31g.
Scheme 5.

Synthesis of hexahydrofuro[2,3-b]pyran-5-ol ligand 30
The synthesis of the protease inhibitors was accomplished in a two-step sequence shown in Schemes 6 and 7. Each ligand alcohol synthesized above was reacted with 4-nitrophenyl chloroformate in presence of pyridine to form mixed activated carbonates 31a–g in 70–99% yield. The synthesis of the corresponding protease inhibitors was achieved by coupling the mixed activated carbonates with previously reported hydroxyethylsulfonamide isosteres 32–34 (Scheme 7).15 The syntheses of various HIV-PI containing the Tp-THF (−)-7, were achieved by respectively treating the Boc-protected isosteres 32–34 with TFA in CH2Cl2 and subsequently, by coupling the resulting free amine isosteres with activated mixed carbonate 31a in THF/CH3CN in presence of Et3N. The corresponding inhibitors 35a, 36, and 37 were obtained in good yields (Scheme 7). Inhibitors 35b–g were made in a similar manner.
Scheme 6.

Synthesis of activated mixed carbonates 31a–g
Scheme 7.

Syntheses of inhibitors 35a–g, 36 and 37.
Results and Discussion
As mentioned above, our preliminary modeling suggested that a hexahydrofuropyranol (−)-7 ligand may interact with backbone atoms and residues in the protease S2-site. All inhibitors in Table 1 were evaluated in enzyme inhibitory assays following a protocol described by Toth and Marshall.36 Inhibitors that showed potent Ki values, were further evaluated through in vitro antiviral assays. As can be seen, inhibitor 35a, with Tp-THF (−)-7 exhibited an enzyme Ki value of 2.7 pM. Antiviral activity of 35a and other inhibitors were determined in MT-2 human-T-lymphoid cells exposed to HIV-1LAI.19 As shown, 35a has shown remarkable antiviral potency(IC50 = 0.5 nM), comparable to PIs 1a and 1b.
Table 1.
Enzymatic Inhibitory and Antiviral Activity of Compounds 35a–g, 36, and 37.
| Entry | Inhibitor | Ki (nM) | IC50 (μM)a |
|---|---|---|---|
| 1 |
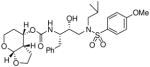 35a |
0.0027 | 0.0005 |
| 2 |
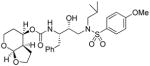 35b |
0.068 | 0.019 |
| 3 |
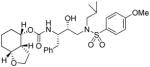 35c |
0.005 | 0.008 |
| 4 |
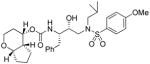 35d |
1.43 | -- |
| 5 |
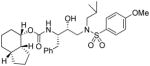 35e |
9 | >1 μM |
| 6 |
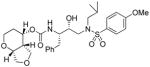 35f |
5.3 | >1 μM |
| 7 |
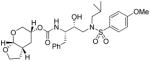 35g |
0.11 | -- |
| 8 |
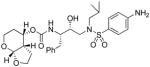 36 |
0.010 | 0.0065 |
| 9 |
 37 |
0.085 | 0.0045 |
Values are means of at least two experiments.
Human T-lymphoid (MT-2) cells (2 × 103) were exposed to 100 TCID50s of HIV-1LAI and cultured in the presence of each PI, and IC50 values were determined using the MTT assay. The IC50 values of amprenavir (APV), saquinavir (SQV), and indinavir (IDV) were 0.03 μM, 0.015 μM, and 0.03 μM, respectively.
The bicyclic ring stereochemistry of the P2 ligand proved to be important as inhibitor 35b, with enantiomeric ligand (+)-7 displayed a significant reduction in enzyme inhibitory potency (>20-fold increase in Ki) as well as antiviral activity (ID50 = 19 nM).
To probe the importance of the cyclic ether oxygens in the bicyclic structure of (−)-7, inhibitors 35c–e were synthesized and evaluated. As shown, inhibitor 35c, with a cyclohexane ring in place of the tetrahydropyran ring, only displayed a 2-fold reduction in Ki-values but a 16-fold decrease in antiviral activity compared to inhibitor 35a. A more dramatic loss of enzymatic potency was observed with compound 35d with a cyclopentane ring in place of a THF ring in the P2 ligand. The Ki value dropped to 1.43 nM. Inhibitor 35e, which lacks both cyclic ether oxygens, displayed even lower Ki and no appreciable antiviral activity. Those results clearly demonstrated the critical role of both cyclic ether oxygens in ligand (−)-7. Furthermore, the difference of activity observed between 35a and 35c, suggests that the O7 oxygen on the THF-ring of (−)-7 exerts a stronger interaction with the enzyme compared to the pyran oxygen. Inhibitor 35f, in which the THF-oxygen of the P2 ligand is located at a vicinal position, also exhibited a substantial loss of potency (i.e. Ki = 5.3 nM) and no antiviral activity. These results corroborated our previous observations with the bis-THF ligand in PIs 1–2. The THF-oxygen in (−)-7 likely has a stronger hydrogen bonding interaction with the Asp29 backbone NH, and may form a weak hydrogen bond with Asp30, in the S2 subsite of the HIV protease. We have investigated the position of the urethane oxygen on the bicyclic ligand in inhibitor 35g. This has resulted in a substantial loss of protease inhibitory activity. Furthermore, we have examined the potency enhancing effect of the Tp-THF ligand with various hydroxyethyl sulfonamide isosteres to give inhibitor 36 and 37. The 4-methoxy sulfonamide derivative 35a appears to be the most potent inhibitor in the series comparable to inhibitor 2. However, the 4-amino derivative 36 exhibited very comparable enzyme inhibitory and antiviral potency similar to 1a.
We have examined inhibitor 35a for is activity against a panel of multidrug-resistant HIV-1 variants and compared it with that of other clinically available PIs including 1a. The results are shown in Table 2. All inhibitors showed high antiviral activity against an HIV-1 clinical strain isolated from a drug-naïve patient (wild-type).19 Compound 35a displayed the most potent activity with an IC50 of 1.9 nM. When tested against multidrug-resistant HIV-1 virus, compound 35a retained impressively high activity to all variants with IC50 values ranging from 2.6–27.5 nM. In contrast, other inhibitors, except 1a, exhibited substantial loss of activity. Interestingly, 1a and 35a showed similar fold-change of IC50 against most multidrug-resistant HIV strains. The results indicated that 35a is highly active against multidrug-resistant HIV-1 variants. This inhibitor outperformed the clinically available PIs with exceedingly high antiviral activity and compared well with 1a, which currently stands as the leading PI for the treatment of drug-resistant HIV infection.
Table 2.
Comparison of the antiviral activity of 35a and other PIs against multidrug resistant clinical isolates in PHA-PBMs cells
| Virus | EC50 (μM) |
|||
|---|---|---|---|---|
| 35a | ATV | LPV | DRV | |
| HIV-1ERS104pre (X4) | 0.0019 ± 0.0015 | 0.0027 ± 0.0006 | 0.031 ± 0.004 | 0.004 ± 0.001 |
| HIV-1MDR/B (X4) | 0.0145 ± 0.0001 (8) | 0.470 ± 0.007 (174) | >1 (>32) | 0.034 ± 0.008 (9) |
| HIV-1MDR/C (X4) | 0.0037 ± 0.0018 (2) | 0.039 ± 0.003 (14) | 0.437 ± 0.004 (14) | 0.009 ± 0.005 (2) |
| HIV-1MDR/G (X4) | 0.0026 ± 0.0004 (1) | 0.019 ± 0.008 (7) | 0.181 ± 0.023 (6) | 0.026 ± 0.009 (7) |
| HIV-1MDR/TM (X4) | 0.0275 ± 0.0055 (14) | 0.075 ± 0.003 (28) | 0.423 ± 0.082 (14) | 0.022 ± 0.015 (6) |
| HIV-1MDR/MM (R5) | 0.0050 ± 0.0023 (3) | 0.205 ± 0.024 (76) | 0.762 ± 0.115 (25) | 0.017 ± 0.005 (4) |
| HIV-1MDR/JSL (R5) | 0.0275 ± 0.0009 (14) | 0.293 ± 0.099 (109) | >1 (>32) | 0.023 ± 0.005 (6) |
The amino acid substitutions identified in the protease-encoding region of HIV-1ERS104pre, HIV-1B, HIV-1C, HIV-1G, HIV-1TM, HIV-1MM, HIV-1JSL compared to the consensus type B sequence cited from the Los Alamos database include L63P; L10I, K14R, L33I, M36I, M46I, F53I, K55R, I62V, L63P, A71V, G73S, V82A, L90M, I93L; L10I, I15V, K20R, L24I, M36I, M46L, I54V, I62V, L63P, K70Q, V82A, L89M; L10I, V11I, T12E, I15V, L19I, R41K, M46L, L63P, A71T, V82A, L90M; L10I, K14R, R41K, M46L, I54V, L63P, A71V, V82A, L90M; I93L; L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, Q92K; and L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, V82A, respectively. HIV-1ERS104pre served as a source of wild-type HIV-1. The EC50 values were determined by using PHA-PBMs as target cells and the inhibition of p24 Gag protein production by each drug was used as an endpoint. The numbers in parentheses represent the fold changes of EC50 values for each isolate compared to the EC50 values for wild-type HIV-1ERS104pre. All assays were conducted in duplicate, and the data shown represent mean values (± 1 standard deviations) derived from the results of two or three independent experiments.
In order to obtain molecular insights into the enzyme-inhibitor interactions of 35a in the protease active site, an active model of 35a was created. A stereoview of the overlaid structure of 35a with the X-ray structure of inhibitor 1b-bound HIV-1 protease is shown in Figure 2. Inhibitor 35a was modeled starting from the X-ray crystal structure of 1b. The conformation of 35a was optimized using the MMFF94 force field,37 as implemented in Molecular Operating Environment (version 2009.10, Chemical Computing Group, Montreal). The modeled structure maintains the important binding interactions (hydroxyl group with Asp25 and Asp25′ carboxylates; cyclic ether oxygens with Asp29 and Asp30 backbone NH groups; methoxy oxygen with the Asp30′ backbone NH bond; carbonyl oxygen and sulfonamide oxygen with a water molecule binding to Ile50 and Ile50′) that are observed in the crystal structure of 1b-bound HIV-1 protease.
Figure 2.
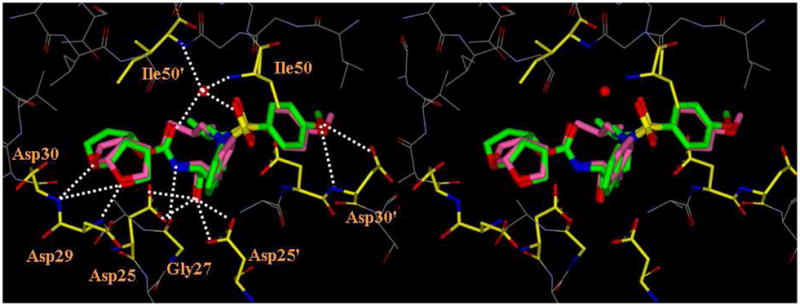
Stereoview of inhibitor 35a, modeled into the active site of HIV-1 protease, and superimposed on the X-ray crystal structure of 1b (PDB code 3I7E).
Conclusions
In summary, we have reported the structure-based design of novel HIV-1 protease inhibitors incorporating a stereochemically defined 4-hexahydrofuropyranol-derived urethanes as the P2-ligand. The inhibitors were designed to make extensive interactions including hydrogen bonding with the protein backbone of the HIV-1 protease active site. The synthesis of (3aS,4S,7aR)-hexahydro-2H-furo[2,3-b] pyran-4-ol [(−)-7, Tp-THF)] was carried out in optically active form using (3aR,6aS)-3,3a,6,6a-tetrahydro-2H-cyclopenta[b]furan-2-one as the starting material. Inhibitor 35a has shown excellent enzyme inhibitory activity and antiviral potency comparable to approved PI, 1a. Furthermore, it has shown excellent activity against multi-PI-resistant variants, superior to other FDA approved inhibitors examined. The data is comparable to 1a. We have carried out a detailed structure activity studies, which indicated that the stereochemistry of the Tp-THF ligand and position of its oxygens are critical to the ligand’s high enzyme affinity. An active model of 35a was created based upon the X-ray crystal structure of 1b-bound HIV-1 protease. The overlaid structures revealed that both oxygens of the hexahydro-Tp-THF ligand can interact with the Asp29 and Asp30 backbone NH’s similar to the bis-THF ligand oxygens. Furthermore, the extra methylene unit in the Tp-THF ligand appears to fill in the hydrophobic pocket in the S2-site more effectively compared to the bis-THF in 1a. The design of an inhibitor targeting the protein backbone may serve as an important guide to combat drug resistance. Further design and chemical modifications are currently underway.
Experimental Section
General Experimental Methods
All anhydrous solvents were obtained according to the following procedures: diethyl ether and tetrahydrofuran (THF) were distilled from sodium/benzophenone under argon; toluene, methanol, acetonitrile, and dichloromethane from calcium hydride and benzene from sodium. Other solvents were used without purification. All moisture-sensitive reactions were carried out in flame-dried flasks under argon atmosphere. Reactions were monitored by thin layer chromatography (TLC) using Silicycle 60A-F254 silica gel pre-coated plates. Flash column chromatography was performed using Silicycle 230–400 mesh silica gel. Yields refer to chromatographically and spectroscopically pure compounds. Optical rotations were recorded on a Perkin Elmer 341 polarimeter. 1H NMR and 13C NMR spectra were recorded on a Varian Inova-300 (300 and 75 MHz), Bruker Avance ARX-400 (400 and 100 MHz) or DRX-500 (500 and 125 MHz). High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. The purity of all test compounds was determined by HRMS and HPLC analysis in the different solvent systems. All test compounds showed ≥95% purity.
(1S,2R)-2-[1-(tert-Butyldimethylsilyloxy)-cyclopent-3-en-2-yl]ethyl acetate (5)
To a stirred suspension of lithium aluminum hydride (93 mg, 2.45 mmol) in dry Et2O (6 mL) was added dropwise a solution of (−)-(1S,5R)-2-oxabicyclo[3.3.0]oct-6-en-3-one (4) (150 mg, 1.19 mmol) in Et2O (4 mL + 1 mL rinse) at 0 °C under argon. The reaction mixture was vigorously stirred at this temperature for 1.5 h. Water (0.1 mL) was then carefully added followed by addition of 3M NaOH (0.1 mL) then water (0.3 mL). The solution was stirred until formation of a white precipitate was complete. EtOAc (3 mL) then Na2SO4 were added and the resulting suspension-was filtered out. The amorphous solid was washed several time with EtOAc (5 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude oil was purified by flash chromatography on silica gel using hexanes/EtOAc (1:1) as the eluent to give the resulting diol (145 mg, 95%) as a colorless oil. TLC: Rf = 0.28 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 300 MHz) δ 5.74 (m, 1H), 5.56 (m, 1H), 4.48 (dt, J = 2.4, 6.6 Hz, 1H), 3.84 (m, 1H), 3.71 (ddd, J = 3.6, 8.7, 10.0 Hz, 1H), 2.75 (m, 1H), 2.67 (m, 1H), 2.36 (d, J = 17.1 Hz, 1H), 1.98-1.75 (m, 1H).
To a stirred solution of the diol (76 mg, 0.59 mmol) in CH2Cl2 (3 mL) was added 2,4,6-collidine (1.2 mmol, 155 μL) followed by acetyl chloride (50 μL, 0.71 mmol) at −78 °C under argon. The resulting solution was stirred at this temperature for 5 h at which point additional acetyl chloride (0.25 μL, 0.24 mmol) was added. The solution was stirred for 2 h then sat. aq. NaHCO3 solution was added. The two layers were separated and the aqueous layer was washed with CH2Cl2 (3 × 5 mL). The combined organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The crude oil was purified by flash chromatography on silica gel using hexanes/EtOAc (6:1 then 4:1) as the eluent to give the monoacetate (88 mg, 87%) as a colorless oil. TLC: Rf = 0.26 (hexanes/EtOAc = 2:1); 1H NMR (CDCl3, 300 MHz) δ 5.80-5.72 (m, 1H), 5.64-5.58 (m, 1H), 4.40 (dt, J = 2.4, 5.6 Hz, 1H), 4.20 (t, J = 7.2 Hz, 2H), 2.74-2.56 (m, 2H), 2.33 (d, J = 17.1 Hz, 1H), 2.06 (s, 3H), 2.04-1.88 (m, 1H), 1.87-1.73 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 171.1, 132.4, 128.4, 72.7, 63.9, 47.2, 42.1, 26.8, 21.0. HRMS-ESI (m/z): [M + H]+ calcd for C9H15O3 171.1021; found 171.1020.
To a stirred solution of the above acetate (54 mg, 0.32 mmol) and 2,6-lutidine (74 μL, 0.63 mmol) in CH2Cl2 (1 mL) was added tert-butyldimethylsilyltrifluoromethanesulfonate (125 mg, 108 μL) at −78 °C under argon. The mixture was stirred for 10 min at which point reaction completion was observed. Sat. aq. NaHCO3 solution (1 mL) and additional CH2Cl2 (2 mL) were added. The two layers were separated and the aqueous layer was further extracted with CH2Cl2 (2 × 2 mL). The combined organic layer was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. The crude oil was purified by column chromatography on silica gel using hexanes/EtOAc (20:1) as the eluent to afford silylated product 5 (90 mg, > 99%) as a colorless oil. TLC: Rf = 0.68 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 5.68 (s, 2H), 4.45 (dt, J = 5.1, 6.3 Hz, 1H), 4.14 (t, J = 6.9 Hz, 2H), 2.67-2.55 (m, 1H), 2.47 (dd, J = 6.9, 15.4 Hz, 1H), 2.23 (dd, J = 4.8, 15.4 Hz, 1H), 2.04 (s, 3H), 2.01-1.85 (m, 1H), 1.72-1.56 (m, 1H), 0.88 (s, 9H), 0.06 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 171.2, 132.7, 128.4, 73.6, 63.8, 45.9, 41.0, 27.4, 25.8, 21.0, 18.1, −4.6, −5.0.
(4S,4aS,7aR)-4-(TERT-Butyldimethylsilyloxy)-hexahydrofuro-[2,3-b]pyrane (6)
To a stirred solution of 5 (76 mg, 0.27 mmol) in MeOH (2 mL) was added K2CO3 (37 mg, 0.27 mmol). The solution was stirred at 23 °C for 2 h then sat. aq. NH4Cl solution (2 mL) was added to the mixture. EtOAc was added and the two layers were separated. The aqueous layer was extracted with EtOAc (4 × 3 mL). The combined organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The resulting oil was purified by flash chromatography on silica gel using hexanes/EtOAc (7:1) as the eluent to give the corresponding alcohol (64 mg, 98%) as a colorless oil. This intermediate was used immediately for the subsequent reaction. TLC: Rf = 0.29 (hexanes/EtOAc = 5:1); 1H NMR (CDCl3, 300 MHz) δ 5.72.5.62 (m, 2H), 4.52 (dt, J = 6.0, 6.9 Hz, 1H), 3.74-3.60 (m, 2H), 2.80-2.68 (m, 1H), 2.49 (ddt, J = 1.8, 7.2, 16.3 Hz, 1H), 2.34-2.29 (m, 1H), 2.06 (br. s, 1H), 1.90-1.62 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 132.9, 128.3, 74.0, 61.1, 46.5, 40.6, 31.2, 25.8, 18.2, −4.7, −5.0.
A stream of ozonized oxygen was bubbled through a solution of the above alcohol (63.8mg, 0.26 mmol) in CH2Cl2 (15 mL) at −78 °C until the blue color persisted (5 min). After the solution was flushed with nitrogen, Me2S (0.5 mL) was added. The solution was warmed to 0 °C and stirred over a 2 h period following which anhydrous Na2SO4 was added. The solution was left at room temperature overnight then filtered and concentrated in vacuo. The resulting solid was quickly passed through a short column of silica gel using hexanes/EtOAc (3:1) as the eluent to afford the hemiacetal (99 mg) as a white-solid mixture of isomers which was submitted directly to the next step. TLC: Rf = 0.26 (hexanes/EtOAc = 3:1). To an ice-cold solution of the crude diacetal (ca. 0.25 mmol) and Et3SiH (0.16 mL, 1.0 mmol) in CH2Cl2 (3 mL) under argon, was slowly added BF3-Et2O (60 μL, 0.5 mmol). The mixture was stirred at 0 °C for 10 min. Sat. aq. NaHCO3 solution (2 mL) and additional CH2Cl2 were added. The two phases were separated and the aqueous layer was further extracted with CH2Cl2 (3 × 2 mL). The combined organic layer was washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. The crude oil was purified by column chromatography on silica gel using hexanes/EtOAc (7:1) as the eluent to give bicyclic acetal 6 (38 mg, 55% 3 steps) as a amorphous solid. TLC: Rf = 0.50 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 4.95 (d, J = 3.4 Hz, 1H), 4.24-4.08 (m, 2H), 3.92 (dt, J = 8.1, 9.1 Hz, 1H), 3.85 (ddd, J = 2.0, 4.5, 12.2 Hz, 1H), 3.30 (dt, J = 2.0, 12.3 Hz, 1H), 2.39 (m, 1H), 2.07 (tt, J = 9.4, 12.0 Hz, 1H), 1.91-1.66 (m, 2H), 1.58-1.48 (m, 1H), 0.89 (s, 9H), 0.07 (s, 3H), 0.067 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 101.2, 68.4, 67.8, 61.1, 47.2, 30.3, 25.7, 22.4, 18.2, −4.6, −4.8.
(3aS,4S,7aR)-Hexahydro-2H-furo[2,3-b]pyran-4ol (−)-7
Bicyclic compound 6 (36 mg, 0.139 mmol) was dissolved in THF (1 mL) and tetrabutylammonium fluoride (1M solution THF, 0.21 mL, 0.21 mmol) was added to the solution. The mixture was stirred for 2 h at 23 °C. Sat. aq. NH4Cl solution was added (2 mL), followed by EtOAc (2 mL). The two phases were separated and the aqueous layer was further extracted with EtOAc (4 × 3 mL). The combined organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The resulting compound was purified by flash chromatography on silica gel using hexanes/EtOAc (1:2 then 1:3) as the eluent to afford pure alcohol (−)-7 (19 mg, 94%) as a amorphous solid. TLC: Rf = 0.15 (hexanes/EtOAc = 1:3); (c 1.06, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 4.99 (d, J = 2.7 Hz, 1H), 4.25-4.16 (m, 2H), 3.96 (q, J = 7.5 Hz, 1H), 3.90 (ddd, J = 2.4, 4.8, 12.3 Hz, 1H), 3.34 (td, J = 3.0, 11.7 Hz, 1H), 2.58-2.45 (m, 1H), 2.14-1.98 (m, 1H), 1.96-1.82 (m, 1H), 1.80-1.62 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 101.4, 68.4, 67.5, 61.0, 46.3, 29.4, 21.8. HRMS-CI (m/z): [M + H]+ calcd for C9H15O3 127.0759; found 127.0757.
(3aR,4R,7aS)-Hexahydro-2H-furo[2,3-b]pyran-4-ol (+)-7
Cyclopentenediol 8 was prepared as described previously.27b The same synthetic sequence was the applied on diol as for the synthesis of (−)-7. Ligand (+)-7 was obtained in high enantiomeric purity (99% ee, , c 0.22, CHCl3).
2-Ethoxy-2,3,6,7-tetrahydrobenzofuran-4(5H)-one (10)
To a stirred solution of 2-diazo-1,3-cyclohexanedione (300 mg, 2.17 mmol) in freshly distilled ethyl vinyl ether (5 mL) was added [Rh2(OAc)4] (10 mg, 0.02 mmol). The mixture was stirred at room temperature for 5 h, after which the reaction was diluted with Et2O and few drops of pyridine were added. A red precipitate formed. The solution was filtered on a short pad of silica, flushing with Et2O/THF (4:1) as eluent. After evaporation, the residue was purified by column chromatography on silica gel using hexanes/CH2Cl2/THF (8:1:1) as the eluent to furnish benzofuranone derivative 17 (347 mg, 88%). TLC: Rf = 0.29 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 5.72 (dd, J = 3.3, 7.4 Hz, 1H), 3.88 (m, 1H), 3.62 (m, 1H), 2.92 (ddt, J = 2.2, 7.4, 15.8 Hz, 1H), 2.70-2.62 (m, 1H), 2.52-2.37 (m, 2H), 2.33 (t, J = 6.5 Hz, 2H), 2.12-1.95 (m, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3, 100 Hz) δ 195.2, 175.7, 112.3, 108.5, 65.0, 36.3, 32.7, 23.8, 21.5, 14.9.
2-Ethoxyhexahydrobenzofuran-4(2 H)-one (11)
To a solution of the ketone 10 (140 mg, 0.77 mmol) in EtOAc (9 mL) was added 5% Pd/C (128 mg, 60 μmol) and the mixture was stirred under H2 (1 atm) for 1.5 h at room temperature. The mixture was then filtered on Celite and the pad washed with EtOAc. Evaporation of the solvent furnished the corresponding crude ketone 11 as an essentially pure mixture of diastereoisomers (130 mg, dr = 9:1). The ketone was directly submitted to the next step without purification. TLC Major isomer: Rf = 0.35 (hexanes:EtOAc = 2:1).
cis-Octahydrobenzofuran-4-ol [(±)-12]
A solution of ketone 11 (130 mg, ca. 0.7 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C under Ar. L-Selectride (1M solution, 0.9 mL, 0.9 mmol) was slowly added to the solution over 5 min and the reaction mixture was stirred for 1.5 h at −78 °C. Upon complete conversion, Et3SiH (0.6 mL, 437 mg, 3.7 mmol) was added followed by dropwise addition of TMSOTf (380 μL, 466 mg, 2.1 mmol). The solution was stirred for 2.5 h while slowly warming to 0 °C. The reaction was quenched by addition of saturated aq. NaHCO3 solution (5 mL). The two phases were separated and the aqueous phase was extracted with Et2O (5x). The combined organic layer was washed with brine, dried (MgSO4), and evaporated under vacuum. The residue was purified by column chromatography on silica gel using hexanes:EtOAc (3:1 to 2:1) as the eluent to yield the desired alcohol (±)-12 (78 mg, 71% over 2 steps) as a colorless oil. TLC: Rf = 0.25 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 400 MHz) δ 4.01 (dt, J = 4.6, 8.8 Hz, 1H), 3.88-3.82 (m, 2H), 3.78 (dt, J = 7.1, 8.7 Hz, 1H), 2.31 (m, 1H), 2.12-1.90 (m, 2H), 1.74-1.50 (m, 5H), 1.32-1.22 (m, 1H); 13C NMR (CDCl3, 100 Hz) δ 77.6, 69.1, 66.7, 43.2, 30.2, 26.9, 25.9, 16.2.
(3aS,4S,7aR)-Octahydrobenzofuran-4-ol [(−)-12]
Racemic alcohol 12 (70 mg, 0.5 mmol) was dissolved in THF (5 mL), vinyl acetate (120 μL, 1.25 mmol) was added. Amano lipase PS-30 (30 mg) was added and the resulting suspension was stirred at 15–17 °C. After 48 h, 30 mg additional enzyme was added and the mixture was left for additional 48 h until which ca. 54 % conv. was reached (NMR and GC). The resulting suspension was diluted with Et2O and filtered on celite, the filter cake rinsed with Et2O. After evaporation of the remaining solvent, the residue was purified by column chromatography using hexanes/EtOAc (5:1, 3:1 then 2:1) as the eluent to yield acetyl furanol 13 (38 mg, 41%) and the desired enantioenriched (−)-hexahydrobenzofuranol (−)-12 (24 mg, 35%). The enantiomeric excess of the 2,4-dinitrobenzoate derivative of (−)-12 was determined to be 98.8% ee by chiral HPLC, Column ChiralPak IA, hexane/isopropanol (90/10 to 50/50, 40 min), 1 mL/min, 35 °C, λ = 254 nm, Rt Major = 16.54 min, Rt minor = 37.1 min.
2-[3-(tert-Butyldimethylsilyl)oxy)-1-hydroxypropyl]cyclopentanone (15)
A solution of lithium diisopropylamide (14 mmol), freshly prepared by adding nBuLi (1.6 M solution in hexanes, 8.75 mL, 14 mmol) to diisopropylamine (1.97 mL, 1.42 g, 14 mmol) in THF (30 mL) at 0 °C under argon followed by stirring for 30 min, was cooled to −78 °C and cyclopentanone 14 (1.12 mL, 1.07 g, 12.7 mmol) in THF (5 mL) was added dropwise over 10 min. After stirring at −78 °C for 1.5 h, 3-tert-butyldimethylsilyloxy-propionaldehyde (1.55 g, 8.2 mmol) in THF (20 mL) was added dropwise over 5 min. The mixture was stirred for an additional 2 h and the reaction was quenched by addition of saturated aqueous NH4Cl solution (10 mL). Following dilution with Et2O, the two phases were separated, and the aqueous phase was extracted with Et2O (2×). The combined organic phase was washed with brine, dried (MgSO4), filtered, and evaporated. The residue was quickly purified by column chromatography on silica gel using hexanes/EtOAc (20:1 to 10:1) as the eluent to give 15 as a 3:1 mixture of diastereoisomers (2.13 g, 95%). Light yellow oil. TLC: Rf = 0.37 and 0.23 (hexanes/EtOAc = 5:1); 1H NMR (CDCl3, 400 MHz) δ 4.27 (dt, J = 3.1, 9.3 Hz, 0.3H), 4.10 (s, 1H), 3.91 (m, 1H), 3.87 (m, 0.3H), 3.85-3.75 (m, 2.6H), 2.38-2.30 (m, 6.5H), 1.80-1.56 (m, 5.2H), 0.88 (brs, 12H), 0.06 (s, 2H), 0.05 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 222.8, 220.4, 70.4, 70.2, 62.6, 60.5, 54.5, 53.9, 39.1, 38.7, 37.0, 36.6, 26.4, 25.9, 25.8, 23.5, 20.7, 20.5, 18.2, −5.5, −5.6; HRMS-CI (m/z): [M - OH]+ calcd for C14H27O2Si 255.1780; found 255.1785.
2,3,6,7-Tetrahydrocyclopenta[b]pyran-4(5H)-one (16)
To a solution of DMSO (425 μL, 468 mg, 6 mmol) in CH2Cl2 (3 mL) was added (CF3CO)2O (406 μL, 609 mg, 2.9 mmol) dropwise at −78 °C under argon. The resulting mixture was stirred at that temperature for 45 min then a pre-cooled solution of ketone 15 (272 mg, 1 mmol) in CH2Cl2 (3 mL) was added. The reaction mixture was stirred at −78 °C for 30 min, then at −15 °C for 15 min and cooled back to −78 °C. Et3N (1.25 mL, 911 mg, 9 mmol) was added and the mixture was stirred at −78 °C for 45 min. The reaction was quenched by addition of sat. aq. NH4Cl solution and the mixture warmed to room temperature. The two phases were separated and the aqueous phase was extracted with CH2Cl2 (3×) then EtOAc (1×). The combined organic phase was washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using hexanes/EtOAc (20:1 then 15:1 with a few drops of acetic acid) as the eluent to give the corresponding diketone (221 mg, 82%) as a light orange oil. TLC: Rf = 0.37 (hexanes/EtOAc = 10:1); 1H NMR (CDCl3, 400 MHz) δ 12.7 (br.s., 1H), 3.90 (t, J = 6.2 Hz, 0.66H), 3.89 (t, J = 6.5 Hz, 2H), 3.46 (t, J = 7.8 Hz, 0.33H), 2.86 (dt, J = 3.0, 6.2 Hz, 0.66H), 2.58 (t, J = 7.2 Hz, 2H), 2.45 (t, J = 6.5 hz, 2H), 2.40 (t, J = 7.9 Hz, 2H), 2.31-2.19 (m, 0.66H), 2.10-1.97 (m, 0.66H), 1.95-1.82 (m, 2H), 0.86 (s, 9H), 0.86 (s, 3H), 0.04 (s, 1H), 0.03 (s, 1H), 0.03 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 212.9, 206.1, 203.6, 175.4, 110.9, 62.4, 59.6, 58.5, 45.6, 38.7, 37.8, 37.0, 25.7, 25.6, 25.0, 20.6, 20.3, 18.1, −5.6; HRMS-CI (m/z): [M + H]+ calcd for C14H26O3Si 271.1729; found 271.1733.
A solution of this diketone (54 mg, 0.2 mmol) was dissolved in CH2Cl2 (2 mL) and cooled to 0 °C under argon. Trifluoroacetic acid (90 μL, 134 mg, 1.2 mmol) was then added dropwise. The mixture was stirred at 0 °C for 30 min then warmed to room temperature and stirred for 4 h. As completion was reached, solid NaHCO3 (ca. 150 mg) was then added and the mixture diluted with EtOAc. After stirring for 10 min, the suspension was filtered on a small celite pad. The solvent was evaporated under reduced pressure and the residue purified by column chromatography on silica gel using hexanes/EtOAc (4:1) as the eluent to furnish α,β-unsaturated ketone 16 (26 mg, 94%) as a colorless oil. TLC: Rf = 0.23 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 400 MHz) δ 4.49 (t, J = 6.9 Hz, 2H), 2.59-2.45 (m, 6H), 1.89 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 189.6, 178.5, 114.5, 69.5, 35.4, 32.6, 25.6, 19.0.
Octahydrocyclopenta[b]pyran-4-ol [(±)-18]
A solution of α,β-unsaturated ketone 16 (109 mg, 0.79 mmol) in EtOAc (6 mL) was added with 10% Pd/C (50 mg, 0.047 mmol) and carefully placed under H2 (1 atm). The mixture was stirred at room temperature for 12 h. The suspension was then filtered over a Celite pad, the pad washed with EtOAc, and the resulting solution evaporated under reduced pressure. The essentially pure ketone (81 mg) was directly carried out to the next step without further purification. TLC: Rf = 0.37 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 400 MHz) δ 4.22-4.15 (m, 2H), 3.69 (td, J = 2.8, 12.0 Hz, 1H), 2.71 (ddd, J = 7.2, 12.3, 15.7 Hz, 1H), 2.48 (dt, J = 4.0, 9.0 Hz, 1H), 2.23 (ddt, J = 1.4, 2.8, 15.7 Hz, 1H), 2.00-1.80 (m, 5H), 1.71-1.63 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 210.2, 82.8, 65.9, 55.1, 38.5, 33.3, 28.4, 22.8.
The ketone was diluted in CH2Cl2 (5 mL) under argon and cooled to −78 °C. L-Selectride (1M solution in THF, 0.80 mL, 0.8 mmol) was added dropwise and the resulting mixture was stirred at this temperature for 2 h. Hydrogen peroxide (30% aqueous solution, 3 mL) and 3N NaOH aqueous solution were added and the mixture was warmed to 23 °C, and stirred for 5 h. The phases were separated and the aqueous phase extracted with CH2Cl2 (4×). The combined organic phase was washed with brine, dried (Mg2SO4), filtered, and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using hexanes/EtOAc (4:1 then 1.5:1) as the eluent to yield cis-bicyclic alcohol (±)-18 (77 mg, 68% 2 steps) as a colorless oil. TLC: Rf = 0.13 (hexanes/EtOAc = 2:1); 1H NMR (CDCl3, 400 MHz) δ 4.11 (dt, J = 5.6, 11.1 Hz, 1H), 3.91 (ddd, J = 2.0, 4.5, 11.7 Hz, 1H), 3.84-3.81 (m, 1H), 3.33 (dt, J = 2.3, 11.9 Hz, 1H), 2.17-2.08 (m, 1H), 1.92-1.81 (m, 1H), 1.79-1.55 (m, 7H); 13C NMR (CDCl3, 125 MHz) δ 80.5, 68.3, 65.4, 47.0, 32.6, 29.7, 21.6, 21.3.
(4S,4aS,7aS)-Octahydrocyclopenta[b]pyran-4-ol ((−)-18)
Racemic alcohol (±)-18 (68 mg, 0.48 mmol) was dissolved in THF (5 mL) and vinyl acetate (225 μL, 2.4 mmol) was added. Amano lipase PS-30 (30 mg) was added and the resulting suspension was stirred at 15–20 °C. The mixture was left stirring for >48 h until around 50 % conversion was reached (as seen by NMR). The resulting suspension was diluted with Et2O and filtered on celite, the filter cake rinsed with Et2O. After evaporation of the remaining solvent, the residue was purified by column chromatography using hexanes/EtOAc (5:1, 3:1 then 1.5:1) to yield the desired enantioenriched pyranol (−)-18 (25 mg, 37%). (c 1.32, CHCl3). An enantiopurity of 94.1% ee for the alcohol was measured by chiral HPLC analysis of the corresponding activated carbonate 31d: Column ChiralPak IA, 0.7 mL/min, Hexanes/IPA (98:2 to 85:15, from 0 to 45 min), λ = 210 nm, T = 30 °C, Rt minor = 22.4 min, Rt Major = 23.3 min.
(±)-endo-cis-Bicyclo[4.3.0]nonan-2-ol [(±)-19]
Enone 1733 (106 mg, 0.77 mol) was dissolved in THF (10 mL), the flask was purged with argon. Pd/C 10% (60 mg, 0.06 mmol) was added to the solution and the resulting suspension was stirred under hydrogen (1 atm). TLC monitoring first shows isomerization of the enone, through migration of the olefin to the internal position, followed by slow formation of the reduced cis-product. After 12 h, the solution was filtered on a pad of celite and the solvent removed in vacuo. The residue was purifed by flash column chromatography on silica gel using hexanes/EtOAc (30:1 to 10:1) to give the reduced ketone (98 mg, 92%). TLC: Rf = 0.65 (hexanes/EtOAc = 5:1); 1H NMR (CDCl3, 400 MHz) δ 2.62-2.54 (m, 1H), 2.48-2.38 (m, 1H), 2.38-2.23 (m, 2H), 2.08-1.98 (m, 1H), 1.94-1.30 (m, 9H); 13C NMR (CDCl3, 100 MHz) δ 214.6, 53.1, 42.9, 39.6, 31.0, 27.2, 26.6, 23.8, 23.0. A solution of the ketone (135 mg, 0.98 mmol) in CH2Cl2 (3 mL) was cooled to −78 °C under argon. L-Selectride (1M solution THF, 1.2 mL) was added dropwise to the solution and the reaction mixture was stirred at −78 °C for 1 h. Hydrogen peroxide solution (30% solution, 1.5 mL) then NaOH (3M solution, 1.5 mL) were added and the reaction was warmed to 23 °C, and stirred for 1 h. After dilution with water (2 mL) then addition of Na2SO3 saturated aqueous solution (3 mL), the aqueous phase was successively extracted with CH2Cl2 (4×). The combined organic phase was dried (Na2SO4), filtered, and evaporated in vacuo. The residue was purified by column chromatography on silica gel using hexanes/EtOAc (6:1) to yield racemic alcohol (±)-19 (92 mg, 66%) as a colorless oil. TLC: Rf = 0.25 (hexanes/EtOAc = 5:1); 1H NMR (CDCl3, 500 MHz) δ 3.96 (m, 1H), 2.26-2.17 (m, 1H), 1.93 (m, 1H), 1.79-1.53 (m, 7H), 1.47-1.15 (5 H), 0.96 (dq, J = 3.3, 13.0 Hz, 1H); 13C NMR (CDCl3, 125 MHz) δ 71.6, 46.4, 40.1, 31.5, 29.5, 27.0, 23.9, 21.4, 21.2; HRMS-EI (m/z): [M - OH]− calcd for C9H15 122.1096, found 122.1097.
(−)-(1R,2S,6R)-Bicyclo[4.3.0]nonan-2-ol [(−)-19]
Racemic 19 (86 mg, 0.62 mmol) was dissolved in THF (5 mL), vinyl acetate (0.5 mL) was added. Amano lipase PS-30 (60 mg) was added and the resulting suspension was stirred at 23 °C until 50% conv. was reached (NMR) in ca. 6 h. The resulting suspension was diluted with Et2O and filtered on celite, the filter cake rinsed with Et2O. After evaporation of the remaining solvent, the residue was purified by column chromatography using hexanes/EtOAc (8:1, 6:1 then 4:1) to yield acetate 21 and the desired enantioenriched (−)-indanol (−)-19 (38.5 mg, 45% yield). (c 1.02, CHCl3), ( (c 1.0, CHCl3).38 The enantiomeric excess of the 2,4-dinitrobenzoate derivative was determined to be 89.9% ee by chiral HPLC, Column ChiralPak IA, hexane/isopropanol (100/0 to 90/10, 15min; 90/10 to 80/20, 15 min), 1 mL/min, Rt minor = 16.58 min, Rt Major = 19.5 min.
3-((4-Iodotetrahydrofuran-3-yl)oxy)propan-1-ol (23)
To a solution of freshly distilled 2,5-dihydrofuran (700 mg, 0.740 mL, 10 mmol), in a mixture of dry 1,3-propanediol/dimethoxyethane (1:1, 5 mL) at 0 °C under argon, were successively added NH4OAc (77 mg, 1 mmol), followed by N-iodosuccinimide (11 mmol, 2.47 g). The mixture was warmed to 23 °C and stirred for 12 h protected from light. The reaction was quenched by addition of sat. aq. Na2SO3 then diluted with water. The mixture was extracted with Et2O/EtOAc (1:1). The combined organic phase was dried (Na2SO4), filtered, and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using hexanes/EtOAc (4:1, 3:1 then 2.5:1) to give iodoalcohol 23 (1.2g, 45%) as a pale yellow oil. TLC: Rf = 0.3 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 4.33 (m, 1H), 4.29-4.19 (m, 3H), 4.04 (dd, J = 2.2, 9.8 Hz, 1H), 3.79 (dd, J = 1.5, 9.8 Hz, 1H), 3.76-3.69 (m, 3H), 3.60 (m, 1H), 1.81 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 88.2, 76.1, 71.8, 67.9, 60.6, 32.3, 23.4.
3-((4-Iodotetrahydrofuran-3-yl)oxy)propanal (24)
Oxalyl chloride (580 mg, 392 μL, 4.6 mmol) was diluted in CH2Cl2 (12 mL) under argon and the solution was cooled to −78 °C. Dry DMSO (715 mg, 650 μL, 9.15 mmol) in CH2Cl2 (3 mL) was added to the cold solution dropwise and the mixture was stirred for 30 min. A solution of alcohol 23 (500 mg, 1.83 mmol) in CH2Cl2 (4 mL) was then added slowly, and the mixture was kept stirring for an additional hour at −78 °C.Et3N (1.3 g, 1.8 mL, 12.8 mmol) was then introduced, the white suspension was stirred at −78 °C for 20 min and slowly warmed to rt. A 0.5 M phosphate buffer solution pH 5.5 (20 mL) was added, the two phases were separated and the resulting aqueous phase was extracted with Et2O (4×). The combined organic phase was dried (MgSO4), filtered, and evaporated. The residue was purified by flash column chromatography using hexanes/EtOAc (6:1 to 4:1) to yield the desired aldehyde 24 (433 mg, 86%) as a light yellow oil. TLC: Rf = 0.76 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 300 MHz) δ 9.77 (t, J = 1.3 Hz, 1H), 4.35 (m, 1H), 4.30-4.19 (m, 3H), 4.04 (dd, J = 2.3, 9.8 Hz, 1H), 3.92 (ddd, J = 5.3, 6.7, 9.5 Hz, 1H), 3.77 (dd, J = 1.7, 10.1 Hz, 1H), 3.75 (ddd, J = 5.2, 6.2, 9.5 Hz, 1H), 2.69 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 200.1, 88.3, 76.1, 71.8, 63.1, 43.7, 23.3.
Hexahydro-2H-furo[3,4-b]pyran-4-ol ((±)-25)
To a solution of aldehyde 24 (100 mg, 0.37 mmol) in DME (10 mL) was successively added indium (60 mg, 0.55 mmol), CuI (48 mg, 0.25 mmol), and a catalytic amount of iodine (10 mg, 0.037 mmol). After stirring the suspension for 5 min, water (4 mL) was added and the mixture was stirred at room temperature for 4 h. The suspension was filtered on a celite pad, washing the pad with THF. The solvent was reduced under vacuum and the resulting aqueous phase acidified with 1M HCl and saturated with NaCl. The aqueous phase was extracted with EtOAc and the combined organic phase was dried over MgSO4. After filtration, and evaporation, the crude was purified by flash column chromatography on silica gel using hexanes/EtOAc (1:1 to 1:5) to provide the bicyclic alcohol (±)-25 (25 mg, 47%) as a mixture of diastereoisomers. TLC: Rf = 0.28 (EtOAc 100%). Pyridinium chlorochromate (74 mg, 0.346 mmol) was added to a suspension of flame-dried 4Å MS in CH2Cl2 (2 mL) at room temperature under argon. A solution of the above alcohol (25 mg, 0.173 mmol) in CH2Cl2 (1.5 mL) was transferred to the suspension at 0 °C and the solution was stirred for 1 h at 0 °C. The reaction was quenched by addition of isopropanol and the mixture was filtered on a silica pad flushing with Et2O. After evaporation of the solvent, the corresponding ketone thus obtained was used directly to the next step. TLC: Rf = 0.45 (hexanes/EtOAc = 1:1); The ketone was re-dissolved in EtOH (1.5 mL), the solution was cooled to −20 °C and NaBH4 (25 mg, 0.66 mmol) was added at once. After stirring at this temperature for 30 min, the reaction was quenched by addition of sat. aq. NH4Cl solution (1.5 mL). The solution was extracted with EtOAc and the combined organic phase dried (Na2SO4), filtered, and evaporated. The corresponding racemic alcohol (±)-25 was purified by flash column chromatography using hexanes/EtOAc (1:1 to 1:5) as the eluent. Colorless oil (12 mg, 50% 2 steps). TLC: Rf = 0.25 (100 % EtOAc). 1H NMR (CDCl3, 300 MHz) δ 4.26 (m, 1H), 4.05 (t, J = 3.0 Hz, 1H), 4.04-3.95 (m, 3H), 3.94-3.85 (m, 2H), 3.40 (dt, J = 2.5, 11.8 Hz, 1H), 2.60 (m, 1H), 1.94 (d, J = 4.0 Hz, 1H), 1.80 (ddt, J = 4.6, 11.5, 12.5 Hz, 1H), 1.74 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 78.3, 74.5, 67.1, 66.4, 65.0, 45.5, 30.0.
To a solution of racemic (±)-25 (10 mg, 0.07 mmol) in dry THF (1 mL) under an argon atmosphere, was added vinyl acetate (60 mg, 65 μL, 0.7 mmol) followed by addition of Immobilized Amano Lipase PS-30 (10 mg) on Celite-545. The mixture was stirred at 15–20 °C for 2 days until >50% conversion could be observed by NMR of aliquots. The resulting suspension was diluted in Et2O and filtered on a small celite pad. The solvents were evaporated and the residue purified by flash chromatography using hexanes/EtOAc (1:1 to 1:5) as the eluent to give enantiomeric alcohol 25 (4.6 mg, 46%) as a colorless oil. An enantiopurity of >99.5% ee for the alcohol was measured by analysis of the corresponding activated carbonate 31f on chiral HPLC (Column ChiralPak IC, hexane/isopropanol 52:48, 1 mL/min, λ = 215 nm, T = 24 °C, Rt minor = 14.4 min, Rt Major = 15.5 min).
(3aR,6aR)-Tetrahydrofuro[2,3-b]furan-3(2H)-one (28)
Enantiomerically pure (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol (bis-THF) 27 (85 mg, 0.65 mmol) was diluted in dry CH2Cl2 (6 mL) under argon, the solution was cooled to 0 °C and anhydrous Na2HPO4 (52 mg, 0.36 mol) was added. Dess-Martin periodinane (360 mg, 0.85 mmol) was added at once at 0 °C and the resulting suspension warmed to 23 °C and stirred for 3 h. The reaction was then quenched by successive addition of sat. aq. NaHCO3 and sat. aq. Na2SO3 solutions (1.5 + 1.5 mL). The phases were separated and the aqueous phase was extracted with CH2Cl2 then EtOAc. The combined organic phases were dried (Na2SO4), filtered on a small pad of silica gel, and evaporated to dryness. The residue was purified by column chromatography on silica gel using hexanes/EtOAc (3:1) to furnish ketone 28 (73 mg, 87%) as a white crystalline solid. TLC: Rf = 0.57 (hexanes/EtOAc = 1:1); Spectral data corresponded to those previously reported in the literature.35
(3aS,7aR)-Tetrahydro-2H-furo[2,3-b]pyran-5(3H)-one (29)
AlMe3 (25% w/w hexanes, 250 μL, 0.6 mmol) was diluted in dry CH2Cl2 (5 mL) under argon and the solution was cooled to −78 °C. A solution of ketone 28 (64 mg, 0.5 mmol) in dry CH2Cl2 (5 mL) was slowly added dropwise. After 10 min, TMSCHN2 (2 M solution in Et2O, 275 μL, 0.55 mmol) was added. The reaction was stirred for 2 h while warmed to −30 °C. Saturated Rochelle’s salts solution (5 mL) was added and the mixture was stirred for 1 h. The phases were separated, the aqueous phase extracted with CH2Cl2, and the combined organic phase was dried (MgSO4). The solution was filtered on a small silica gel pad, flushing with Et2O, and the collected organic phase evaporated. A crude mixture of the desired ketone along with α-silylated derivatives and isomers was then obtained. The mixture was re-dissolved in THF (5 mL). AcOH (6 drops) and TBAF (0.5 mL, 0.5 mmol) were successively added. The resulting mixture was stirred at 23 °C for 3 h and evaporated to dryness. The residue was purified by flash column chromatography on silica gel using hexanes/EtOAc (5:1) as the eluent to give ketone 29 (45 mg, 63%). TLC: Rf = 0.35 (hexanes/EtOAc = 2:1); 1H NMR (CDCl3, 400 MHz) δ 5.49 (d, J = 6.8 Hz, 1H), 4.11 (d, J = 18.2 Hz, 1H), 4.10 (m, 1H), 3.92 (d, J = 18.2 Hz, 1H), 3.74 (dt, J = 6.5, 8.9 Hz, 1H), 2.85 (m, 1H), 2.71 (d, J = 6.3, 15.6 Hz, 1H), 2.48 (d, J = 3.9, 15.6 Hz, 1H), 2.15 (m, 1H), 1.55 (ddt, J = 7.7, 8.9, 12.7 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 210.7, 100.9, 67.5, 67.1, 39.2, 36.2, 31.3.
(3aS,5R,7aR)-Hexahydro-2H-furo[2,3-b]pyran-5-ol (30)
A solution of the ketone 29 (25 mg, 0.173 mmol) dissolved in CH2Cl2 (5 mL) was cooled to −78 °C under argon. L-Selectride (1M in THF, 200 μL, 0.2 mmol) was added dropwise. The solution was stirred at this temperature for 3 h and quenched by addition of sat. aq. NH4Cl solution. The aqueous phase was extracted with EtOAc, the combined organic extract was dried (Na2SO4), filtered, and evaporated. The crude was purified by column chromatography on silica gel using hexanes/EtOAc (2:1, 1:1, then 1:2) to yield alcohol 30 as a 5:1 mixture of diastereoisomers (18 mg, cis major). The stereoisomers were separated in the subsequent synthesis of the mixed activated carbonate 31g. TLC: Rf = 0.25 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 300 MHz) δ 5.08 (d, J = 3.8 Hz, 0.2H), 5.05 (d, J = 3.3 Hz, 1H), 4.16-4.11 (m, 1.2H), 3.95-3.84 (m, 1.6H), 3.81-3.70 (m, 2H), 3.63 (m, 1H), 3.27 (dd, J = 7.9, 11.2 Hz, 0.2H), 2.35-1.70 (m, 6H).
(3aS,4S,7aR)-Hexahydro-2H-furo[2,3-b]pyran-4-yl (4-nitrophenyl) carbonate (31a)
Furopyranol ligand (−)-7 (9 mg, 0.063 mmol) was diluted in CH2Cl2 (0.5 mL) under argon. The solution was cooled to 0 °C and dry pyridine (17 μL, ca. 0.21 mmol) was added 4-nitrophenyl chloroformate (24 mg, 0.12 mmol) was added at once to the solution upon which a white precipitate formed. The reaction was stirred for 2 h while warming to rt. Upon completion, the mixture was concentrated reduced pressure and the residue was purified by column chromatography on silica gel using hexanes/EtOAc (6:1 then 3:1) as the eluent to give the corresponding activated carbonate 31a (18 mg, >99%). TLC: Rf = 0.25 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 300 MHz) δ 8.29 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.7 Hz, 2H), 5.30-5.19 (m, 1 H), 5.07 (d, J = 2.7 Hz, 1H), 4.28 (dt, J = 3 Hz, 1H), 4.04-3.95 (m, 2H), 3.47-3.37 (m, 1H), 2.80-2.68 (m, 1H), 2.30-2.10 (m, 1H), 2.05-1.90 (m, 3H); 13C NMR (CDCl3, 75 MHz) δ 155.3, 151.7, 145.4, 125.3, 121.7, 101.1, 75.4, 68.5, 60.5, 43.2, 25.8, 22.5.
(3aR,4R,7aS)-Hexahydro-2H-furo[2,3-b]pyran-4-yl (4-nitrophenyl) carbonate (31b)
The title compound was obtained from (+)-7 as described for (−)-7 in 86% yield after purification on column chromatography on silica gel using hexanes/EtOAc (6:1 then 3:1). Spectral data were consistent with those recorded for 31a.
(3aR,4S,7aR)-Octahydrobenzofuran-4-yl (4-nitrophenyl) carbonate (31c)
The title compound was obtained from (−)-12 as described for (−)-7 in 83% yield after purification by column chromatography on silica gel using hexanes/EtOAc (8:1 to 6:1). TLC: Rf = 0.7 (hexanes/EtOAc = 3:1); 1H NMR (CDCl3, 400 MHz) δ 8.28 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 5.07 (m, 1H), 4.13-4.05 (m, 2H), 3.90 (q, J = 8.2 Hz, 1H), 2.72 (m, 1H), 2.10-2.00 (m, 2H), 1.90-1.68 (m, 4H), 1.55-1.45 (m, 1H), 1.34-1.23 (m, 1H); 13C NMR (CDCl3, 100 MHz) δ 155.4, 151.9, 145.2, 125.2, 121.7, 77.7, 77.1, 66.5, 41.2, 27.0, 26.2, 25.4, 18.0.
((4S,4aR,7aS)-Octahydrocyclopenta[b]pyran-4-yl) (4-nitrophenyl) carbonate (31d)
The title compound was obtained from (−)-18 as described for (−)-7 in 85% yield after purification by column chromatography on silica gel using hexanes/CH2Cl2/THF (4:1:0 then 4:1:0.1) as the eluent. TLC: Rf = 0.31 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 8.28 (d, J = 9.1 Hz, 2H), 7.38 (d, J = 9.1 Hz, 2H), 5.21 (m, 1H), 4.00 (ddd, J = 1.8, 4.7, 12.0 Hz, 1H), 3.93 (dt, J = 2.5, 2.7 Hz, 1H), 3.43 (dt, J = 2.1, 12.0 Hz, 1H), 2.36 (m, 1H), 2.04-1.82 (m, 4H), 1.82-1.62 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 155.5, 151.9, 145.3, 125.3, 121.8, 80.7, 77.3, 65.0, 43.7, 32.6, 26.3, 22.3, 21.7.
(3aR,4S,7aR)-Octahydro-1H-inden-4-yl (4-nitrophenyl) carbonate (31e)
The title compound was obtained from (−)-19 as described for (−)-7 in 90% yield after purification by column chromatography on silica gel using hexanes/EtOAc (20:1 to 10:1) as the eluent. 1H NMR (CDCl3, 400 MHz) δ 8.27 (d, J = 9.1 Hz, 2H), 7.38 (d, J = 9.1 Hz, 2H), 5.05 (m, 1H), 2.41 (m, 1H), 2.05 (m, 1H), 1.98-1.24 (m, 11H), 1.05 (dq, J = 3.4, 12.7 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 155.7, 151.9, 145.2, 125.2, 121.8, 80.7, 42.8, 40.2, 31.3, 26.6, 25.7, 23.4, 22.4, 21.3.
(4S,4aS,7aR)-Hexahydro-2H-furo[3,4-b]pyran-4-yl (4-nitrophenyl) carbonate (31f)
The title was obtained from (−)-25 as described for (−)-7 in >99% yield following column chromatography purification on silica gel using hexanes/EtOAc (3:1 then 2:1) as the eluent. 1H NMR (CDCl3, 400 MHz) δ 8.29 (d, J = 9.1 Hz, 2H), 7.38 (d, J = 9.1 Hz, 2H), 5.32 (m, 1H), 4.20-3.88 (m, 6H), 3.50 (m, 1H), 2.81 (m, 1H), 2.10-1.90 (m, 2H).
[(3aS,5R,7aR)-Hexahydro-2H-furo[2,3-b]pyran-5-yl]-(4-nitrophenyl) carbonate (31g)
The title compound was obtained from 30 as described for (−)-7 in 70% yield. Purified and separated from the 5-epi diastereoisomer following flash column chromatography on silica gel using hexanes/EtOAc (3:1, 2:1, then 1:1) as the eluent. Amorphous solid (70% from a 5:1 mixture of diastereoisomers). TLC: Rf = 0.16 (hexanes/EtOAc = 2:1); 1H NMR (C6D6, 800 MHz) δ 7.64 (d, J = 9.0 Hz, 2H), 6.69 (d, J = 9.0 Hz, 2H), 4.76 d, J = 3.6 Hz, 1H), 4.35 (m, 1H), 4.02 (dt, J = 3.8, 8.6 Hz, 1H), 3.94 (dt, J = 2.8, 13.0 Hz, 1H), 3.60 (q, J = 8.0 Hz, 1H), 3.12 (dd, J = 2.0, 13.0 Hz), 2.04 (m, 1H), 1.67 (dq, J = 3.1, 15.1 Hz, 1H), 1.50 (m, 1H), 1.46-1.38 (m, 2H); 13C NMR (C6D6, 200 MHz) δ 154.9, 151.9, 145.2, 124.9, 121.2, 100.7, 72.0, 67.4, 63.8, 35.9, 27.9, 27.3.
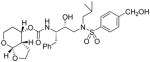
(3aS,4S,7aR)-Hexahydro-2H-furo[2,3-b]pyran-4-yl-(2S,3R)-4-(N-isobutyl-4-methoxyphenyl sulfonamido)-3-hydroxy-1-phenylbutan-2-yl carbamate (35a)
Sulfonamide isostere 32 (42 mg, 0.08 mmol) was dissolved in a 30% TFA solution in CH2Cl2 (3 mL), the solution was stirred at 23 °C for 2 h after which the solvent was evaporated under reduced pressure. The corresponding Boc-deprotected intermediate (0.08 mmol) was then diluted in dry acetonitrile (0.8 mL) at 0 °C under argon and Et3N (0.3 mL, 0.2 mmol) was added. A solution of activated carbonate 31a (18.6 mg, 0.06 mmol) in acetonitrile or THF (0.5 mL) was then added to the mixture. The reaction was stirred at 23 °C until completion was reached (2–3 days). The solution was then evaporated in vacuo and the resulting residue purified by flash chromatography on silica gel using hexanes/EtOAc (2:1 then 1:1) as the eluent to afford the inhibitor 35a as a amorphous solid (19.8 mg, 55%). TLC Rf = 0.35 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 300 MHz) δ 7.71 (d, J = 8.9 Hz, 2H), 7.33-7.17 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.05-4.90 (m, 1H), 4.93 (d, J = 3.6 Hz, 1H), 4.84 (d, J = 8.4 Hz, 1H), 4.15 (dt, J = 2.4, 9.0 Hz, 1H), 3.87 (s, 3H), 3.98-3.76 (m, 4H), 3.31 (t, J = 11.7 Hz, 1H), 3.22-2.90 (m, 4H), 2.90-2.78 (m, 2H), 2.48-2.32 (m, 1H), 1.96-1.25 (m, 5H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.1, 155.5, 137.6, 129.8, 129.4, 128.4, 126.5, 114.3, 101.1, 72.9, 70.2, 68.5, 60.9, 58.9, 55.7, 54.9, 53.8, 43.5, 35.6, 27.3, 26.2, 22.3, 20.2, 19.9. HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8NaS 599.2403, found 599.2406.
(3aS,4S,7aR)-Hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-4-(4-amino-N-isobutylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-yl carbamate (36)
The title compound was obtained from 31a and sulfonamide isostere 33 as described for inhibitor 35a, in 64% yield following purification by flash-chromatography using CHCl3/2% MeOH as the eluent. TLC: Rf = 0.45 (hexanes/EtOAc = 1:3); 1H NMR (CDCl3, 300 MHz) δ 7.55 (d, J = 8.7 Hz, 2H), 7.32-7.16 (m, 5H), 6.67 (d, J = 8.7 Hz, 2H), 4.97 (m, 1H), 4.93 (d, J = 3.4 Hz, 1H), 4.85 (d, J = 8.7 Hz, 1H), 4.20-4.11 (m, 3H), 3.92-3.80 (m, 5H), 3.31 (dt, J = 2.2, 11.9 Hz, 1H), 3.15 (dd, J = 8.1, 15.2 Hz, 1H), 3.05 (dd, J = 4.2, 14.1 Hz, 1H), 3.01-2.80 (m, 3H), 2.75 (dd, J = 6.6, 13.4 Hz, 1H), 2.40 (m, 1H), 1.97-1.60 (m, 4H), 1.46 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 155.5, 150.7, 137.7, 129.5, 129.5, 128.4, 126.5, 126.2, 114.1, 101.1, 72.8, 70.1, 68.5, 60.8, 58.9, 54.8, 53.8, 43.4, 35.5, 27.3, 26.2, 22.2, 20.2, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C28H39N3O7NaS 584.2406; found 584.2402.
(3aS,4S,7aR)-Hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(4-(hydroxymethyl)- N-isobutylphenylsulfonamido)-1-phenylbutan-2-yl carbamate (37)
The title compound was obtained from 31a and sulfonamide isostere 34 as described for inhibitor 35a in 72% yield following purification by flash-chromatography on silica gel using CHCl3/2%MeOH as the eluent. Amorphous solid. TLC: Rf = 0.23 (hexanes/EtOAc = 1:2); 1H NMR (CDCl3, 400 MHz) δ 7.76 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 7.32-7.17 (m, 5H), 4.96 (m, 1H), 4.93 (d, J = 3.2 Hz, 1H), 4.85 (d, J = 8.5 Hz, 1H), 4.80 (s, 2H), 4.15 (t, J = 8.5 Hz, 1H), 3.92-3.80 (m, 4H), 3.70 (s, 1H), 3.31 (t, J = 11.6 Hz, 1H), 3.16 (dd, J = 8.0, 15.0 Hz, 1H), 3.10-2.95 (m, 3H), 2.88-2.76 (m, 2H), 2.41 (m, 1H), 2.04 (m, 1H), 1.95-1.78 (m, 2H), 1.76-1.56 (m, 2H), 1.47 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 155.6, 146.2, 137.6, 137.1, 129.4 128.5, 127.6, 127.1, 126.5, 101.1, 72.8, 70.2, 68.4, 64.2, 60.8, 58.8, 54.9, 53.7, 43.4, 35.5, 27.3, 26.2, 22.2, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8NaS 599.2403, found 599.2414.
(3aR,4R,7aS)-Hexahydro-2H-furo[2,3-b]pyran-4-yl ((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (35b)
The title compound was obtained from 31b and sulfonamide isostere 32 in 65 % yield as described for inhibitor 35a, following purification by column chromatography on silica gel using hexanes/EtOAc (3:1 then 1.5:1) as the eluent. White amorphous solid. TLC: Rf = 0.44 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 7.70 (d, J = 8.9 Hz, 2H), 7.31-7.26 (m, 2H), 7.25-7.20 (m, 3H), 6.98 (d, J = 8.9 Hz, 2H), 5.00 (m, 1H), 4.97 (d, J = 2.7 Hz, 1H), 4.88 (d, J = 8.0 Hz, 1H), 4.17 (t, J = 7.7 Hz, 1H), 3.99-3.72 (m, 6H), 3.87 (s, 3H), 3.31 (dt, J = 1.9, 12.0 Hz, 1H), 3.13 (dd, J = 8.4, 15.0 Hz, 1H), 3.08-2.84 (m, 4H), 2.79 (dd, J = 6.7, 13.4 Hz, 1H), 2.53 (m, 1H), 2.00 (m, 1H), 1.83 (m, 1H), 1.73 (m, 1H), 1.68-1.54 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 163.1, 155.7, 137.7, 129.8, 129.5, 128.5, 126.5, 114.3, 101.2, 72.6, 70.2, 68.4, 60.8, 58.7, 55.6, 55.1, 53.7, 43.6, 35.3, 27.3, 26.2, 22.5, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8NaS 599.2403, found 599.2407.
(3aR,4S,7aR)-Octahydrobenzofuran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl carbamate (35c)
The title compound was obtained from 31c and sulfonamide isostere 32 in 75 % yield as described for inhibitor 35a, following purification by column chromatography on silica gel using hexanes/EtOAc (3:1 then 2.5:1) as the eluent. TLC: Rf = 0.39 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 7.72 (d, J = 8.9 Hz, 2H), 7.311-7.16 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 4.83 (m, 2H), 3.95-3.75 (m, 5H), 3.87 (s, 3H), 3.68 (q, J = 8.1 Hz, 1H), 3.14 (dd, J = 8.4, 15.2 Hz, 1H), 3.08 (dd, J = 4.1, 14.1 Hz, 1H), 3.05-2.99 (m, 1H), 2.96 (dd, J = 8.4, 13.4 Hz, 1H), 2.87-2.75 (m, 2H), 2.35 (m, 1H), 1.83 (m, 1H), 1.70-1.40 (m, 7H), 1.20 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 163.0, 156.1, 137.7, 129.7, 129.5, 129.4, 128.4, 126.4, 114.3, 73.0, 71.8, 66.6, 58.8, 55.6, 54.7, 53.7, 41.2, 35.6, 27.3, 27.2, 27.0, 25.7, 20.1, 19.9, 17.7; HRMS-ESI (m/z): [M + Na]+ calcd for C30H42N2O7NaS 597.2610, found 597.2621.
(4S,4aR,7aS)-Octahydrocyclopenta[b]pyran-4-yl ((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (35d)
The title compound was obtained from 31d and sulfonamide isostere 32 in 81 % yield as described for inhibitor 35a, following purification by column chromatography on silica gel using hexanes/EtOAc (3:1 then 2.5:1) as the eluent. TLC: Rf = 0.58 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 7.70 (d, J = 8.9 Hz, 2H), 7.30-7.17 (m, 5H), 6.96 (d, J = 8.9 Hz, 2H), 4.94 (m, 1H), 4.81 (d, J = 8.1 Hz, 1H), 3.86 (s, 3H), 3.90-3.76 (m, 4H), 3.33 (t, J = 11.9 Hz, 1H), 3.13 (dd, AB, J = 8.3, 15.0 Hz, 1H), 3.08-2.91 (m, 3H), 2.85 (m, 1H), 2.79 (dd, J = 6.8, 13.5 Hz, 1H), 2.04 (m, 1H), 1.81 (m, 2H), 1.76-1.64 (m, 3H), 1.64-1.49 (m, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 163.0, 156.0, 137.7, 129.8, 129.4, 128.4, 126.4, 114.3, 80.5, 72.7, 71.7, 65.2, 58.7, 55.6, 54.8, 53.7, 44.1, 35.6, 32.5, 27.2, 26.6, 22.0, 21.6, 20.1, 19.8; HRMS-ESI (m/z): [M + Na]+ calcd for C30H42N2O7S 597.2610, found 597.2612.
(3aR,4S,7aR)-Octahydro-1H-inden-4-yl-(2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl carbamate (35e)
The title compound was obtained from 31e and sulfonamide isostere 32 as described for inhibitor 35a. Following preliminary purification by flash-chromatography using hexanes/CH2Cl2:THF (8:1:1) as the eluent, the inhibitor was obtained as a mixture of unseparable isomeric compounds. Compound 35e was derivatized into the corresponding N,O-isopropylidene compound by treatment of 35e (20 mg) with 2,2-dimethoxypropane (0.1 mL) and a catalytic amount of pTSA (1.5 mg) in dry CH2Cl2 (1 mL) for 8 h at 23 °C. After neutralization with Et3N, the organic phase was evaporated to dryness. Following a quick silica gel column (hexanes/EtOAc = 8:1), the resulting inhibitor was purified by HPLC: Preparative HPLC column SunfirePM Prep C18 OBD, 30 × 100 mm, Eluent: MeOH/H2O 85:15 (30 min) then 90:10 (15 min), flow 15 mL.min−1, Rt = 42 min. The isopropylidene derivative was then obtained as a colorless oil (24 mg). The product was then taken into MeOH (2 mL), pTSA.H2O (36 μmol, 1.5 mg) was added and the resulting solution was refluxed for 6 h. After neutralization with a few drops of Et3N, the solution was evaporated and the residue purified by column chromatography on silica gel using hexanes/CH2Cl2/THF (8:1:1) to give inhibitor 35e (15 mg, 43% from 31e). TLC: Rf = 0.35 (hexanes//EtOAc = 5:1); 1H NMR (CDCl3, 400 MHz) δ 7.71 (d, J = 8.9 Hz, 2H), 7.32-7.18 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 4.79 (m, 1H), 4.70 (d. J = 8.1 Hz, 1H), 3.90 (m, 1H), 3.87 (s, 3H), 3.81 (m, 1H), 3.18-3.02 (m, 3H), 2.98-2.82 (m, 2H), 2.78 (dd, J = 6.6, 13.2 Hz, 1H), 2.10 (m, 1H), 1.90 (m, 1H), 1.82 (m, 1H), 1.74-1.19 (m, 11H), 0.95 (m, 1H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 163.0, 156.4, 137.7, 129.9, 129.5, 129.4, 128.5, 126.4, 114.3, 74.9, 72.8, 58.8, 55.6, 54.8, 53.8, 43.1, 39.9, 35.7, 31.3, 27.2, 26.9, 26.1, 23.5, 22.2, 21.3, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C31H44N2O6NaS 595.2818, found 595.2816.
(4S,4aS,7aR)-Hexahydro-2H-furo[3,4-b]pyran-4-yl ((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (35f)
The title compound was obtained from 31f and sulfonamide isostere 32 in 75 % yield as described for inhibitor 35a, following purification by column chromatography using hexanes/EtOAc (3:1 then 2.5:1) as the eluent. TLC: Rf = 0.24 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 800 MHz) δ 7.70 (d, J = 8.8 Hz, 2H), 7.30 (m, 2H), 7.24-7.20 (m, 3H), 6.97 (d, J = 8.8 Hz, 2H), 5.05 (m, 1H), 4.83 (d, J = 8.5 Hz, 1H), 4.03 (t, J = 3.2 Hz, 1H), 3.96 (m, 1H), 3.87 (s, 3H), 3.87 (s, 3H), 3.88-3.81 (m, 5H), 3.62 (t, J = 8.3 Hz, 1H), 3.39 (t, J = 11.5 Hz, 1H), 3.14 (dd, J = 8.4, 15.0 Hz, 1H), 3.02 (dd, J = 4.0, 14.1 Hz, 1H), 2.99-2.94 (m, 2H), 2.84 (dd, J = 8.7, 14.1 Hz, 1H), 2.77 (dd, J = 6.6, 13.4 Hz, 1H), 2.51 (m, 1H), 1.81 (m, 1H), 1.78 (dq, J = 4.5, 12.4 Hz, 1H), 1.71 (dd, J = 5.4, 12.4 Hz, 1H), 0.91 (d J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 200 MHz) δ 163.0, 155.5, 137.5, 129.6, 129.45, 129.38, 128.5, 126.6, 114.3, 78.4, 74.4, 72.6, 70.0, 66.1, 64.9, 58.8, 55.6, 54.9, 53.7, 42.7, 35.4, 27.2, 26.9, 20.1, 19.8; HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8S 599.2403, found 599.2397.
(3aS,5R,7aR)-Hexahydro-2H-furo[2,3-b]pyran-5-yl ((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)carbamate (35g)
The title compound was obtained from 31g and sulfonamide isostere 32 in 86 % yield as described for inhibitor 35a, following purification by column chromatography on silica gel using hexanes/EtOAc (gradient 3:1 to 1.5:1) as the eluent. TLC: Rf = 0.33 (hexanes/EtOAc = 1:1); 1H NMR (CDCl3, 400 MHz) δ 7.72 (d, J = 8.9 Hz, 2H), 7.32-7.26 (m, 2H), 7.25-7.17 (m, 3H), 6.98 (d, J = 8.9 Hz, 2H), 4.98 (d, J = 3.5 Hz, 1H), 4.89 (d, J = 8.7 Hz, 1H), 4.54 (m, 1H), 4.11 (dt, J = 3.5, 8.3 Hz, 1H), 3.87 (s, 3H), 3.90-3.77 (m, 4H), 3.74 (m, 1H), 3.56 (d, J = 12.7 Hz, 1H), 3.12 (dd, J = 8.5, 15.1 Hz, 1H), 3.09-2.91 (m, 3H), 2.84 (dd, J = 8.5, 14.1 Hz, 1H), 2.79 (dd, J = 6.8, 13.4 Hz, 1H), 2.08 (m, 1H), 2.04-1.93 (m, 2H), 1.90-1.76 (m, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 163.4, 155.7, 137.6, 129.7, 129.5, 128.5, 126.5, 114.4, 101.0, 72.5, 68.0, 67.1, 65.4, 58.8, 55.6, 54.9, 53.8, 36.2, 35.8, 28.3, 27.8, 27.2, 20.1, 19.9; HRMS-ESI (m/z): [M + Na]+ calcd for C29H40N2O8NaS 599.2403, found 599.2397.
Supplementary Material
Acknowledgments
The research was supported by grants from the National Institutes of Health (GM53386). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho), and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho.
Abbreviations
- bis-THF
bis-tetrahydrofuran
- Cp-THF
cyclopentanyltetrahydrofuran
- Tp-THF
tetrahydropyranyltetrahydrofuran
- PI
protease inhibitor
- HAART
highly active antiretroviral therapy
- APV
amprenavir
- DRV
darunavir
- SQV
saquinavir
- IDV
indinavir
- LPV
lopinavir
- RTV
ritonavir
- ATV
atazanavir
Footnotes
Supporting Information Available: HPLC and HRMS data of inhibitors 35a–g, 36, and 37. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Conway B. HAART in Treatment-experienced Patients in the 21st Century: the Audacity of Hope. Future Virol. 2009;4:39–41. [Google Scholar]
- 2.Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview on the Effectiveness of Triple Combination Therapy in Antiretroviral-naive HIV-1 Infected Adults. AIDS. 2001;15:1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 3.Walensky RP, Paltiel AD, Losina E, Mercincavage LM, Schackman BR, Sax PE, Weinstein MC, Freedberg KA. The Survival Benefits of AIDS Treatment in the United States. J Infec Dis. 2006;194:11–19. doi: 10.1086/505147. [DOI] [PubMed] [Google Scholar]
- 4.Sepkowitz KA. AIDS — The First 20 Years. N Engl J Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 5.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining Morbidity and Mortality among Patients with Advanced Human Immunodeficiency Virus Infection. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Pillay D, Bhaskaran K, Jurriaans S, Prins M, Masquelier B, Dabis F, Gifford R, Nielsen C, Pedersen C, Balotta C, Rezza G, Ortiz M, de Mendoza C, Kucherer C, Poggensee G, Gill J, Porter K. The Impact of Transmitted Drug Resistance on the Natural History of HIV Infection and Response to First-line Therapy. AIDS. 2006;20:21–28. doi: 10.1097/01.aids.0000196172.35056.b7. [DOI] [PubMed] [Google Scholar]
- 8.Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. Factors Associated with Clinical and Virological Failure in Patients Receiving a Triple Therapy Including a Protease Inhibitor. AIDS. 2000;14:141–149. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 9.Little SJ, Holte S, Routy JP, Daar ES, Markowitz M, Collier AC, Koup RA, Mellors JW, Connick E, Conway B, Kilby M, Wang L, Whitcomb JM, Hellmann NS, Richman DD. Antiretroviral-drug Resistance among Patients Recently Infected with HIV. N Engl J Med. 2002;347:385–394. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- 10.Grant RM, Hecht FM, Warmerdam M, Liu L, Liegler T, Petropoulos CJ, Hellmann NS, Chesney M, Busch MP, Kahn JO. Time Trends in Primary HIV-1 Drug Resistance among Recently Infected Persons. J Am Med Assoc. 2002;288:181–188. doi: 10.1001/jama.288.2.181. [DOI] [PubMed] [Google Scholar]
- 11.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D On behalf of the UK Collaborative group on HIV Drug Resistance. Demonstration of Sustained Drug-Resistant Human Immunodeficiency Virus Type 1 Lineages Circulating among Treatment-Naive Individuals. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lucas GM. Antiretroviral Adherence, Drug resistance, Viral Fitness and HIV Disease Progression: a Tangled Web is Woven. J Antimicrob Chemother. 2005;55:413–416. doi: 10.1093/jac/dki042. [DOI] [PubMed] [Google Scholar]
- 13.Spaltenstein A, Kazmierski WM, Miller JF, Samano V. Discovery of Next Generation Inhibitors of HIV Protease. Curr Top Med Chem. 2005;5:1589–1607. doi: 10.2174/156802605775009694. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-Tetrahydrofuran: A Privileged Ligand for Darunavir and a New Generation of HIV-Protease Inhibitors That Combat Drug-Resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky A, Yu, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-based Design of Novel HIV-1 Protease Inhibitors to Combat Drug Resistance. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SGP, Amano M, Mitsuya H. Probing multidrug-resistance/protein-ligand interaction with new oxatricyclic designed ligands in HIV-1 Protease inhibitors. ChemMedChem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.FDA approves Darunavir on June 23, 2006: FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.html.
- 18.On October 21, 2008, FDA granted traditional approval to Prezista (darunavir), co-administered with ritonavir and with other antiretroviral agents, for the treatment of HIV-1 infection in treatment-experienced adult patients. In addition to the traditional approval, a new dosing regimen for treatment-naïve adult patients was approved.
- 19.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI) UIC-94017 (TMC114) with Potent Activity against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor Active against Protease Inhibitor-Resistant Viruses, Including a Broad Range of Clinical Isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefebvre E, Schiffer CA. Resilience to Resistance of HIV-1 Protease Inhibitors: Profile of darunavir. AIDS Rev. 2008;10:131–142. [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 23.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High Resolution Crystal Structures of HIV-1 Protease with a Potent Non-Peptide Inhibitor (UIC-94017) Active against Multi-Drug-Resistant Clinical Strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 24.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Bethune MP, Schiffer CA. Structural and Thermodynamic Basis for the Binding of TMC114 a Next-Generation Human Immunodeficiency Virus Type 1 Protease Inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. Ultra-High Resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. A Novel Bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI), GRL-98065, Is Potent against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob Agents Chemother. 2007;51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Nakashima H, Masayuki S, Taniguchi T, Ogasawara K. Chiral Preparation of Polyoxygenated Cyclopentanoids. Synthesis. 2000;6:817–823. [Google Scholar]; (b) Laumen K, Schneider MP. A Facile Chemoenzymatic Route to Optically Pure Building Blocks for Cyclopentanoid Natural Products. J Chem Soc, Chem Commun. 1986:1298–1299. [Google Scholar]
- 28.Ishihara K, Kurihara H, Yamamoto H. An Extremely Simple, Convenient, and Selective Method for Acylating Primary Alcohols in the Presence of Secondary Alcohols. J Org Chem. 1993;58:3791–3793. [Google Scholar]
- 29.Nara M, Terashima S, Yamada S. Stereochemical Studies - LVII Synthesis of Optically Active Compounds by the Novel Use of Meso-Compounds-1. Efficient Synthesis of Two Structural Types of Optically Pure Prostaglandin Intermediates. Tetrahedron. 1980;36:3161–3170. [Google Scholar]
- 30.Moriarty RM, Bailey BR, III, Prakash O, Prakash I. Capture of Electron-Deficient Species with Aryl Halides. New Syntheses of Hypervalent Iodonium Ylides. J Am Chem Soc. 1985;107:1375–1378. [Google Scholar]
- 31.Müller P, Allenbach YF, Bernardinelli G. On the Enantioselectivity of Transition Metal-Catalyzed 1,3-Cycloadditions of 2-Diazocyclohexane-1,3-diones. Helv Chim Acta. 2003;86:3164–3178. [Google Scholar]
- 32.Ferrie L, Reymond S, Capdevielle P, Cossy J. Formal Chemoselective Synthesis of Leucascandrolide A. Org Lett. 2007;9:2461–2464. doi: 10.1021/ol070670a. [DOI] [PubMed] [Google Scholar]
- 33.Tsantali GG, Takakis IM. Expeditious Copper-Catalyzed Conjugate 1,4-Addition of Bromo[2-(1,3-dioxolan-2-yl)ethyl]magnesium to α,β-Cycloalkenones and Subsequent Tranformations. J Org Chem. 2003;68:6455–6458. doi: 10.1021/jo034350q. [DOI] [PubMed] [Google Scholar]
- 34.Shen ZL, Geo GL, Loh TP. Indium-Copper and Indium-Silver Mediated Barbier Grignard Type Alkylation Reaction of Aldehydes using Unactivated Alkyl Halides in Water. J Org Chem. 2008;73:3922–3924. doi: 10.1021/jo8000589. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh AK, Leschenko S, Noetzel M. Stereoselective Photochemical 1,3-Dioxolane Addition to 5-Alkoxymethyl-2(5H)-furanone: Synthesis of Bis-Tetrahydrofuranyl Ligand for HIV Protease Inhibitor UIC-94017 (TMC-114) J Org Chem. 2004;69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- 36.Toth MV, Marshall GR. A Simple Continuous Fluorometric Assay for HIV Protease. Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 37.Halgren TA. MMFF VII. Characterization of MMFF94, MMFF94s, and Other Widely Available Force Fields for Conformational Energies and for Intermolecular-Interaction Energies and Geometries. J Comput Chem. 1999;20:730–748. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 38.Hodgson DM, Lee GP, Marriot RE, Thompson AJ, Wisedale R, Witherington J. Isomerisations of Cycloalkene- and Bicycloalkene-derived Achiral Epoxides by Enantioselective α-deprotonation. J Chem Soc Perkin Trans. 1998;I:2151–2161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.